
Assessment of a Large-Scale Unbiased Malignant Pleural Effusion Proteomics Study of a Real-Life Cohort
 , , , , , and
, , , , , and 
Abstract
:Simple Summary
Abstract
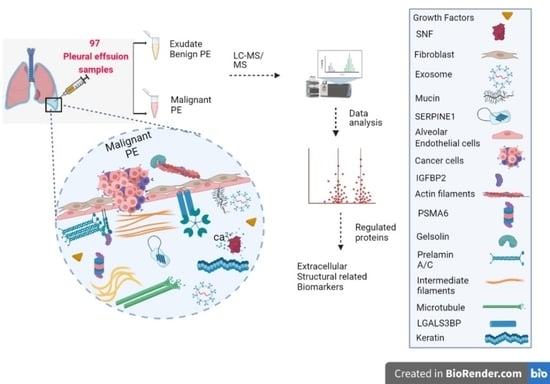
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Peptide Preparation
2.3. Protein Measurements
2.4. Mass Spectrometry Analysis
2.5. Database Search
2.6. Statistical Analysis and Machine Learning
2.7. Functional Analysis of Differentially Regulated Proteins
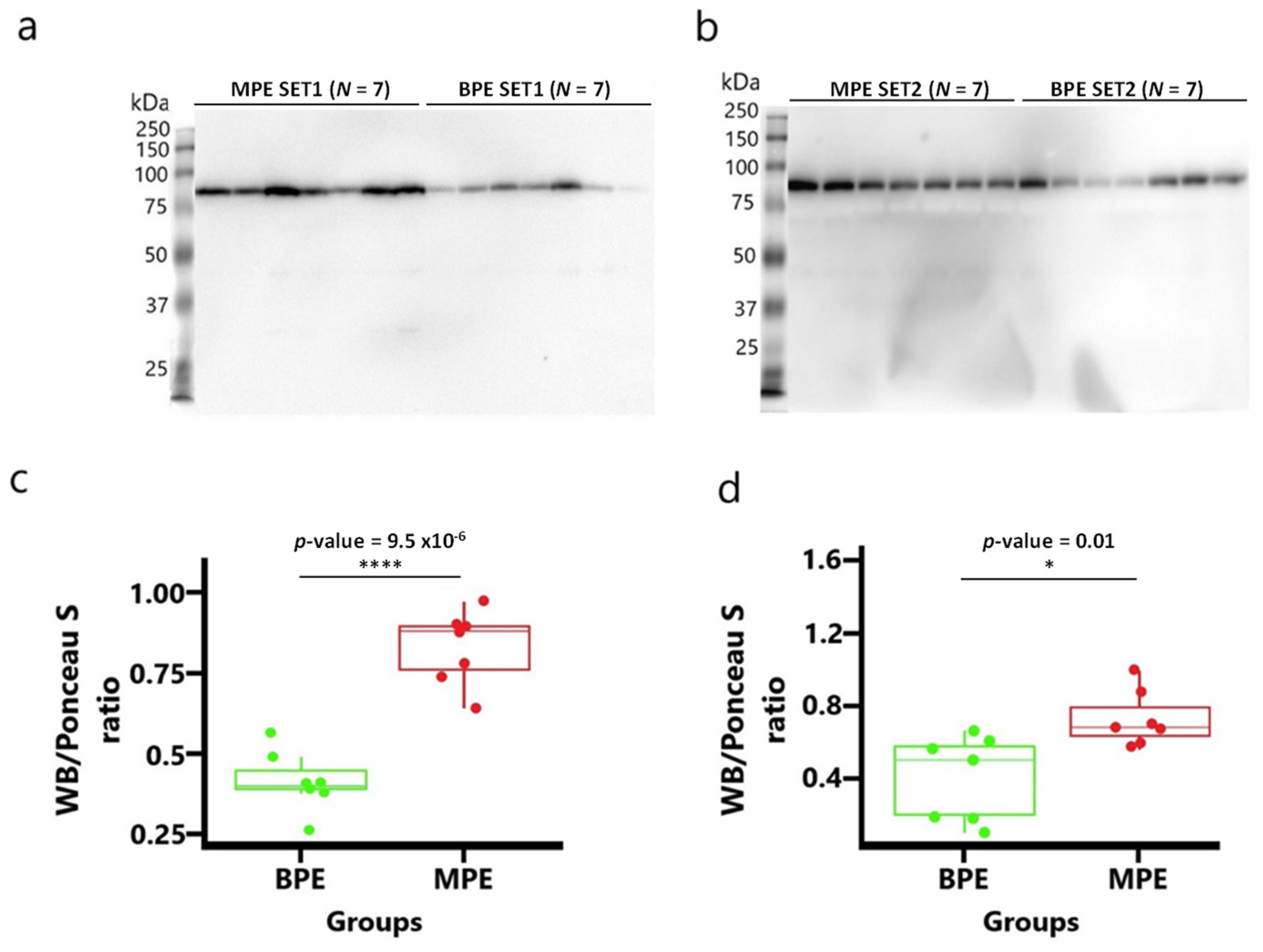
2.8. Western Blotting Analysis
3. Results
3.1. Outline of Study
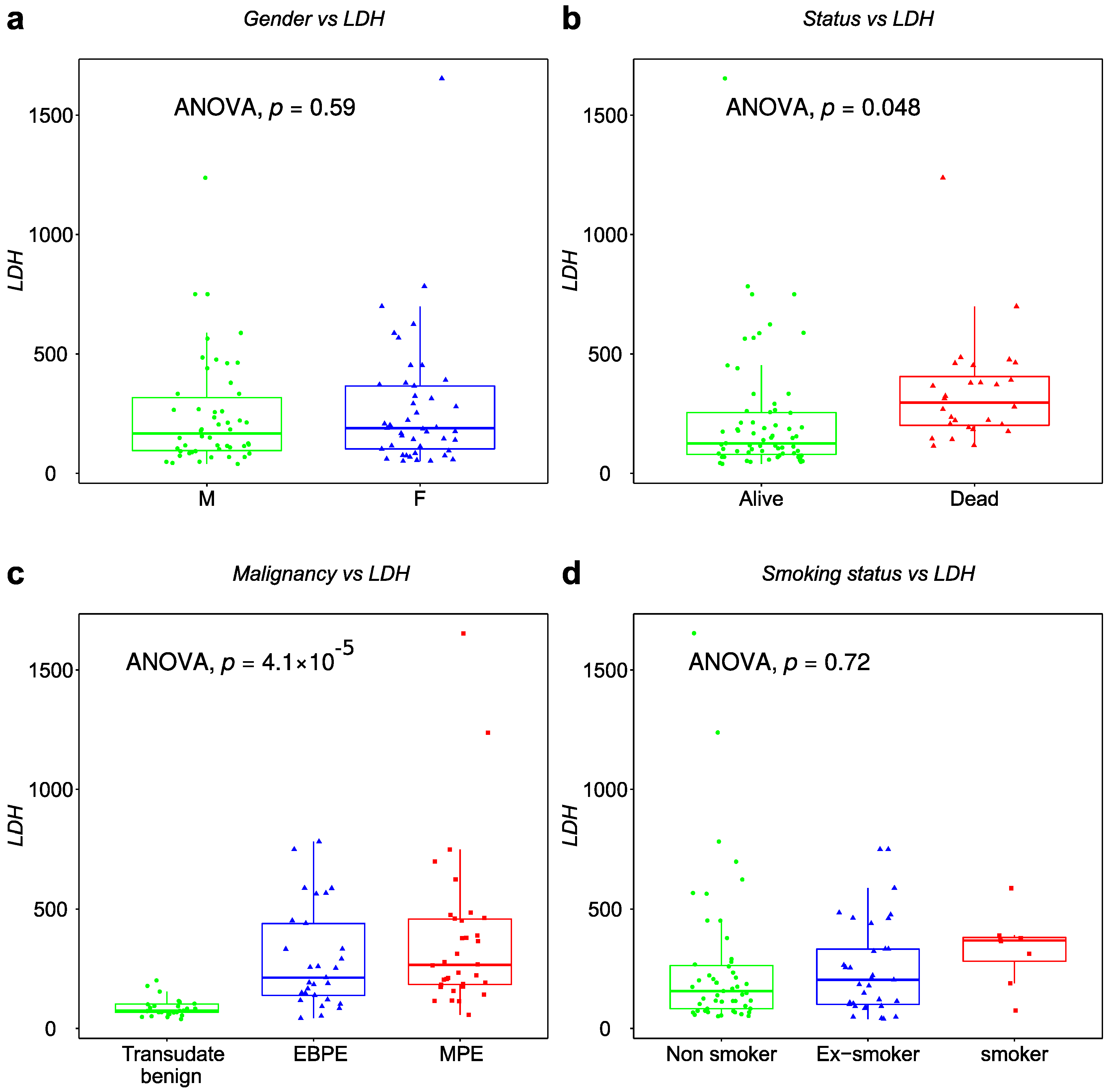
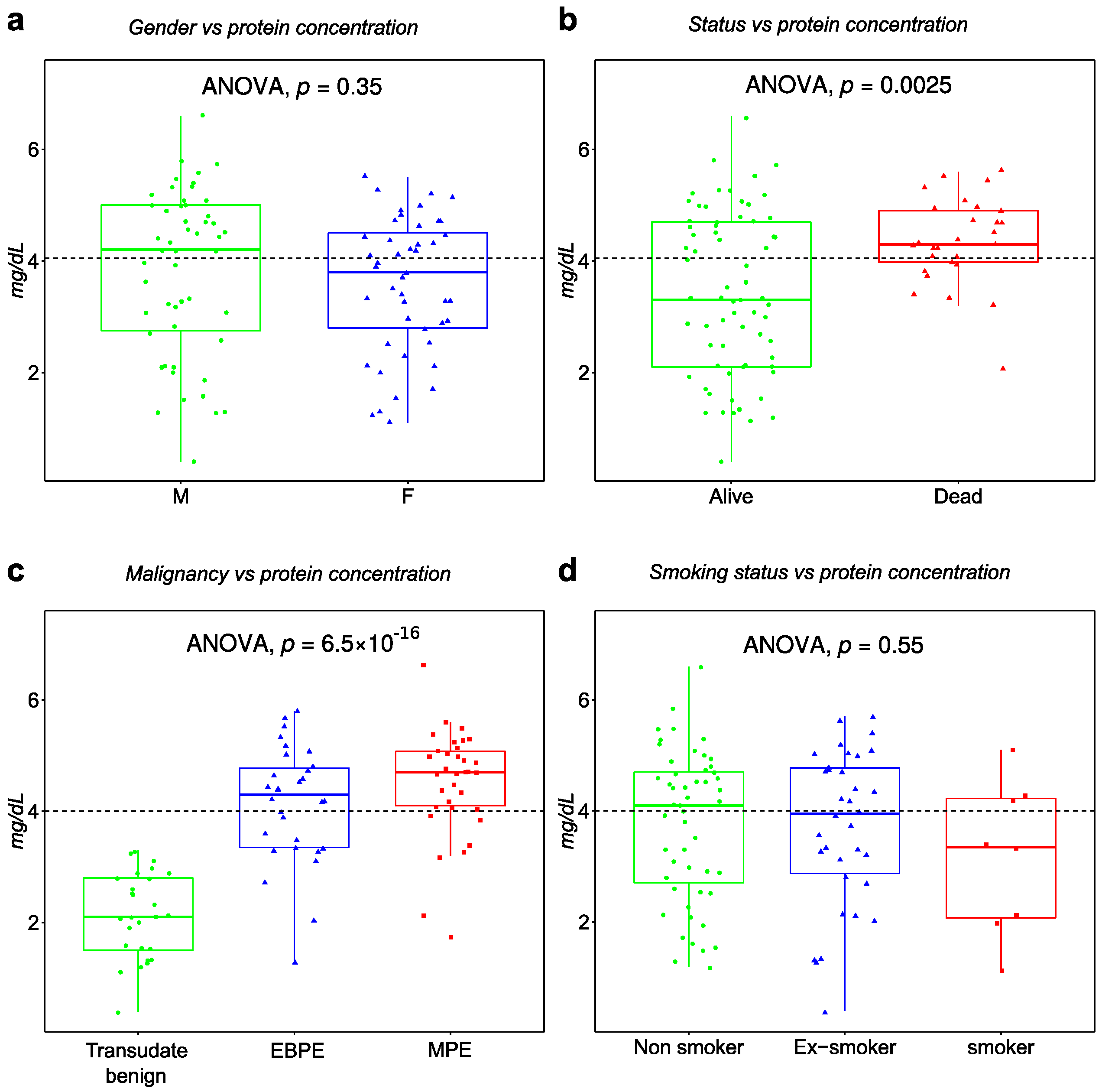
3.2. Demographic and Clinical Characteristics of Patients
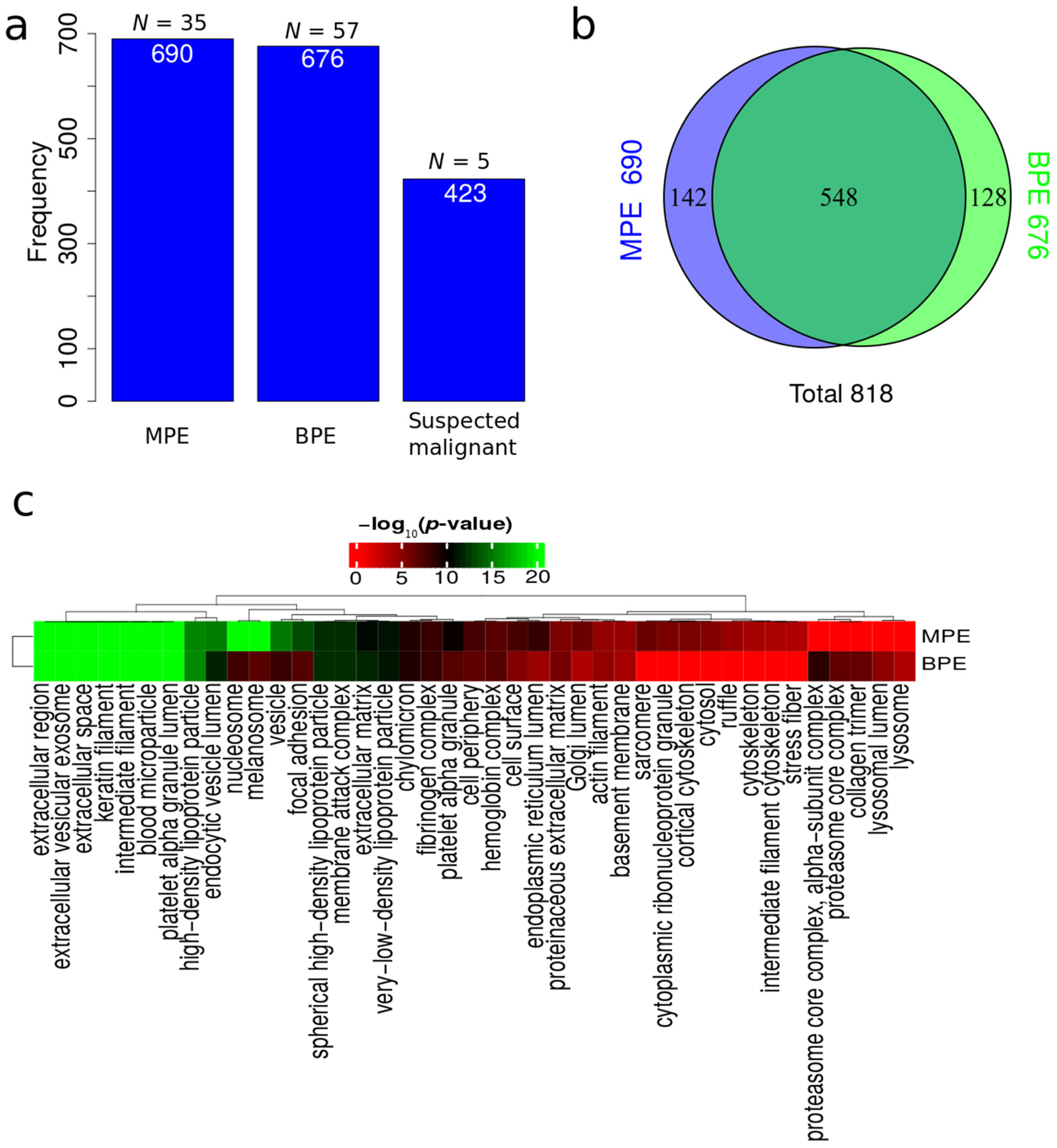
3.3. Protein Identification
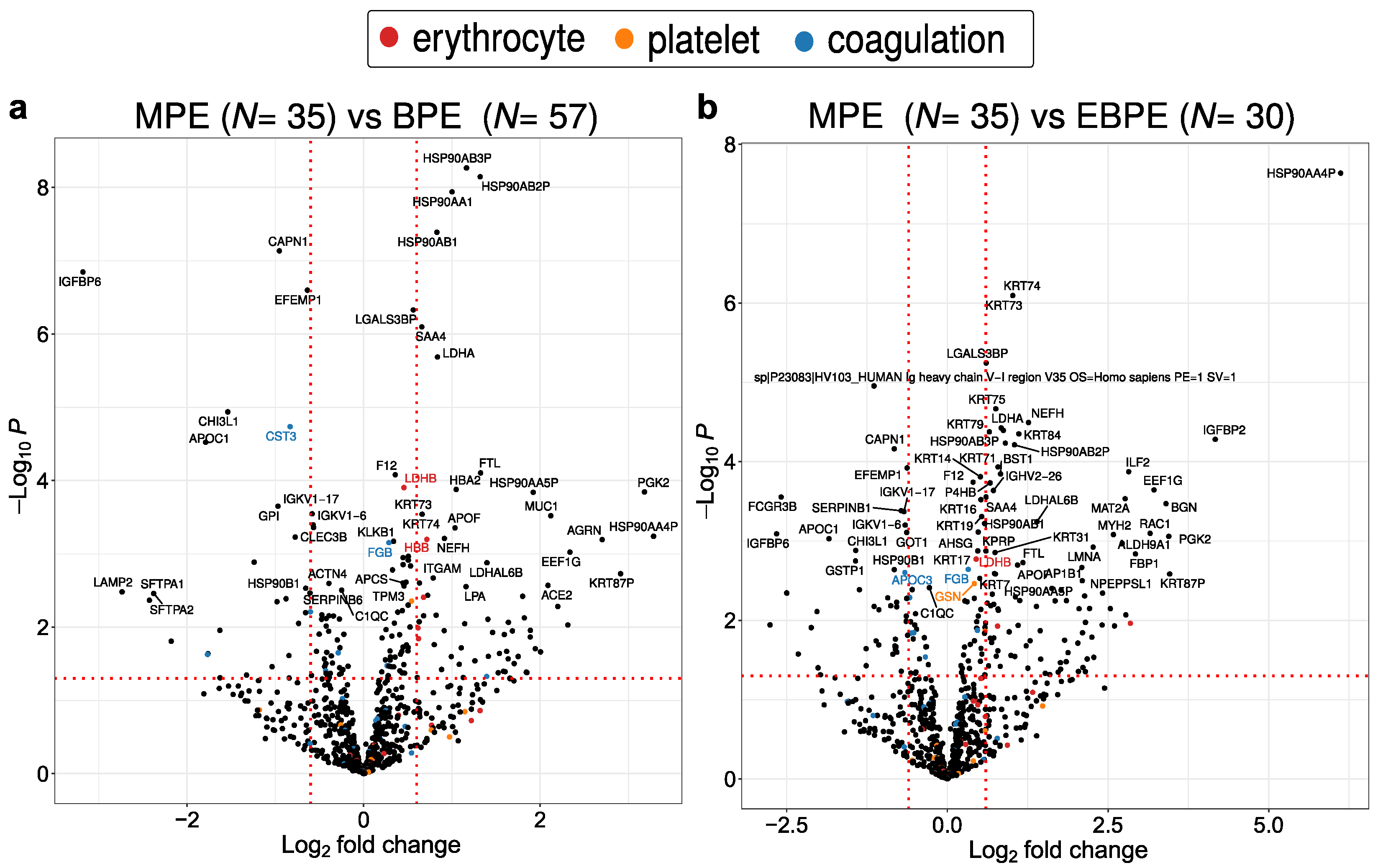
3.4. Protein Dysregulation
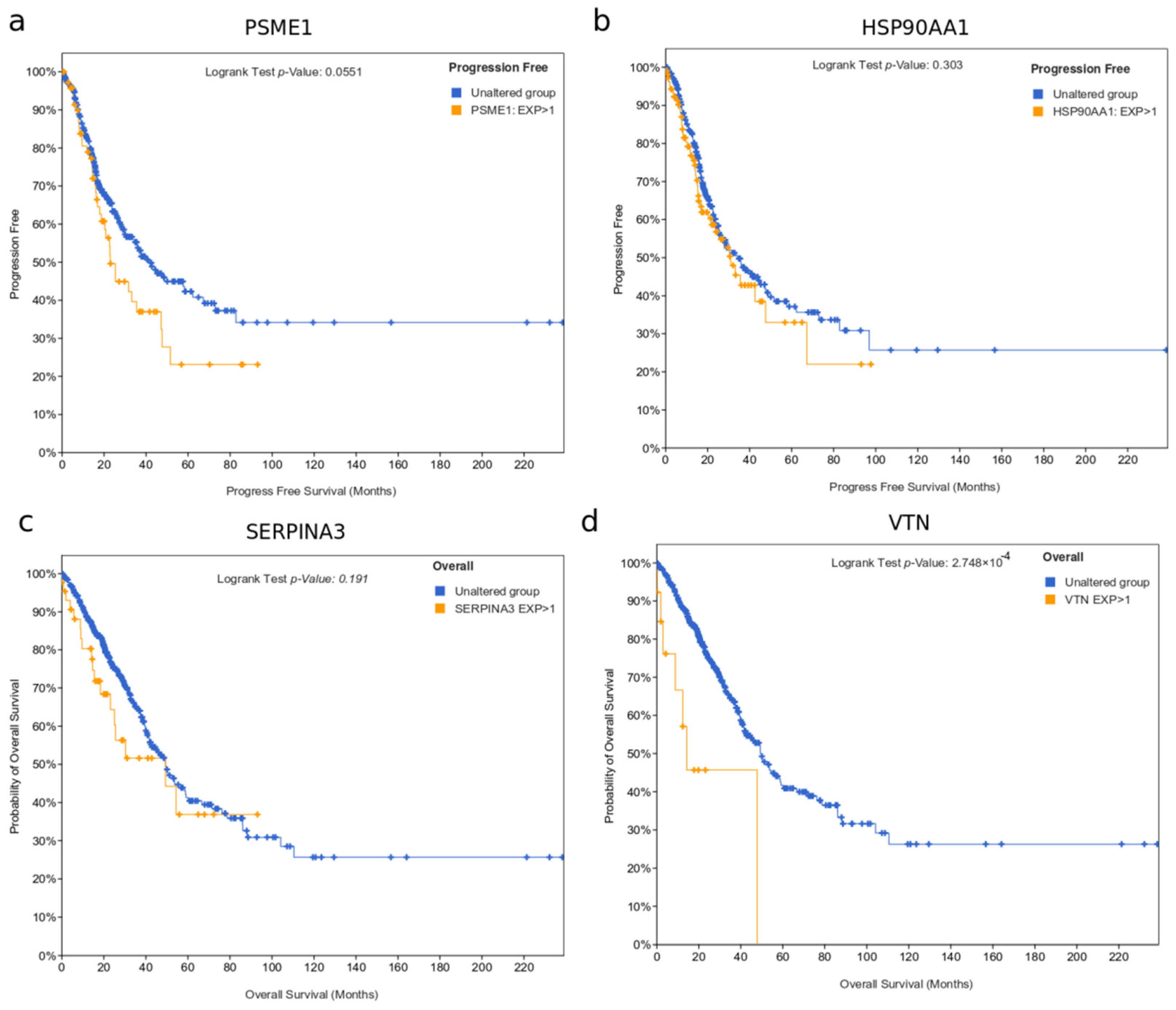
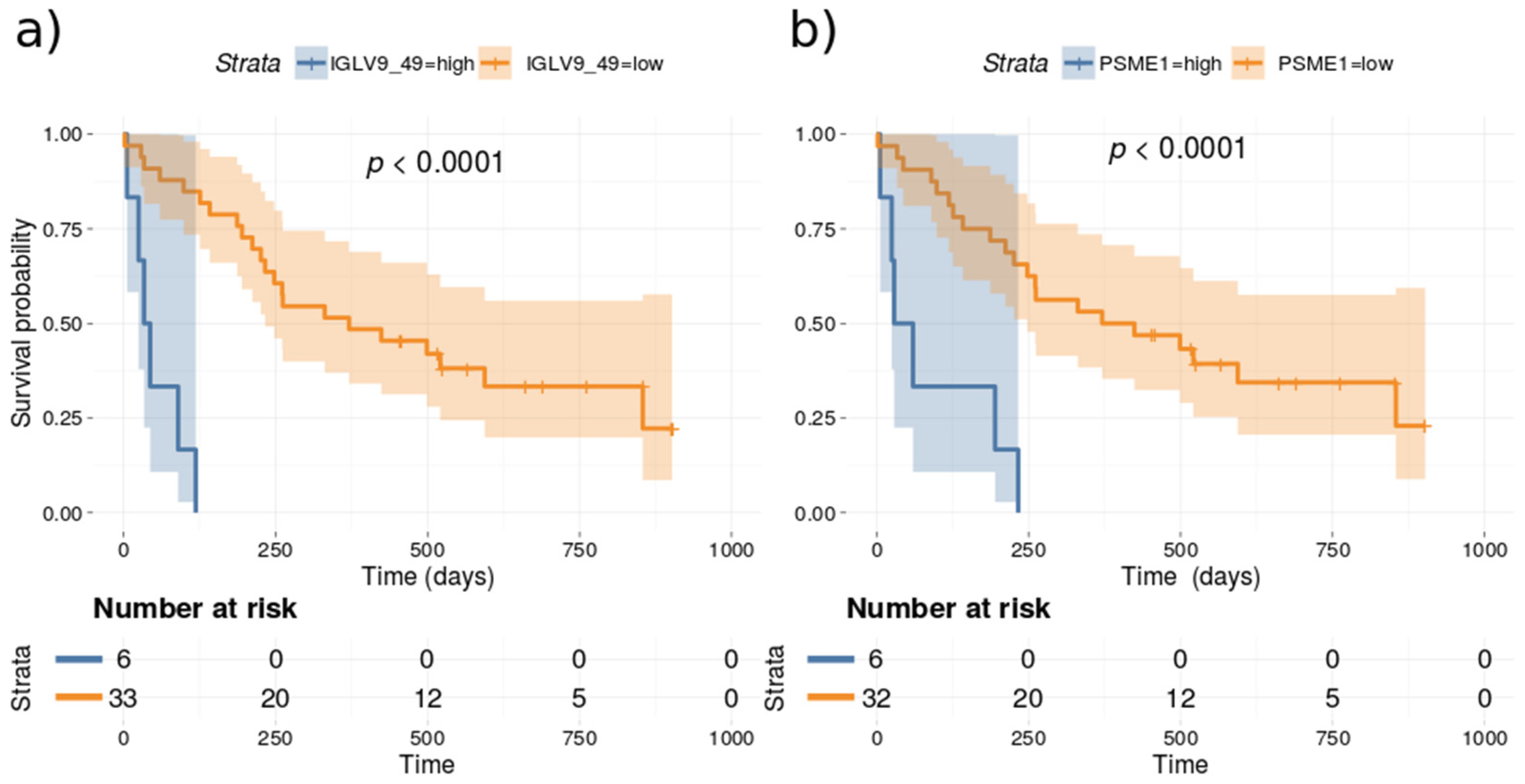
3.5. Survival Analysis Based on Protein Markers
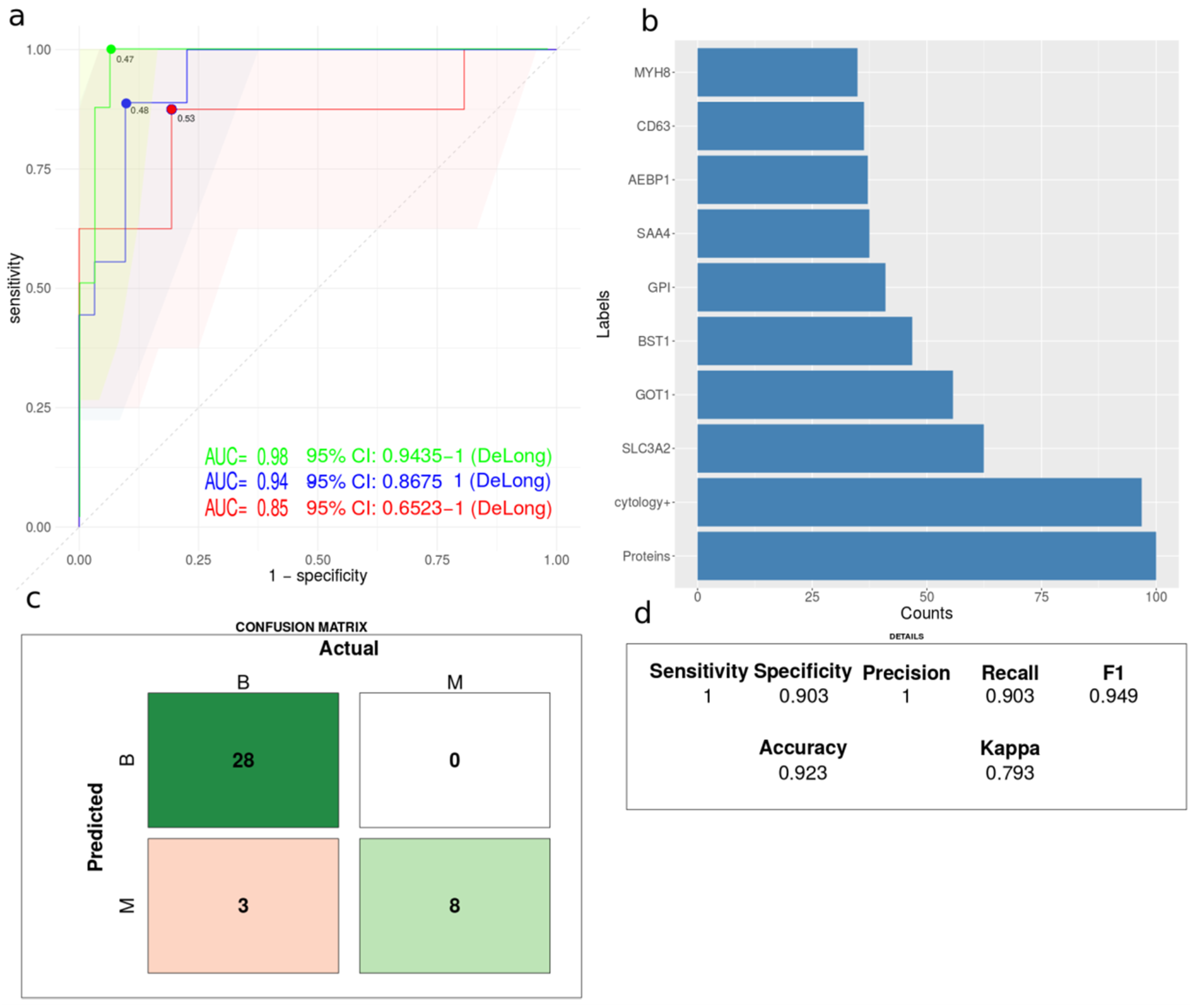
3.6. Comparative Classification Models
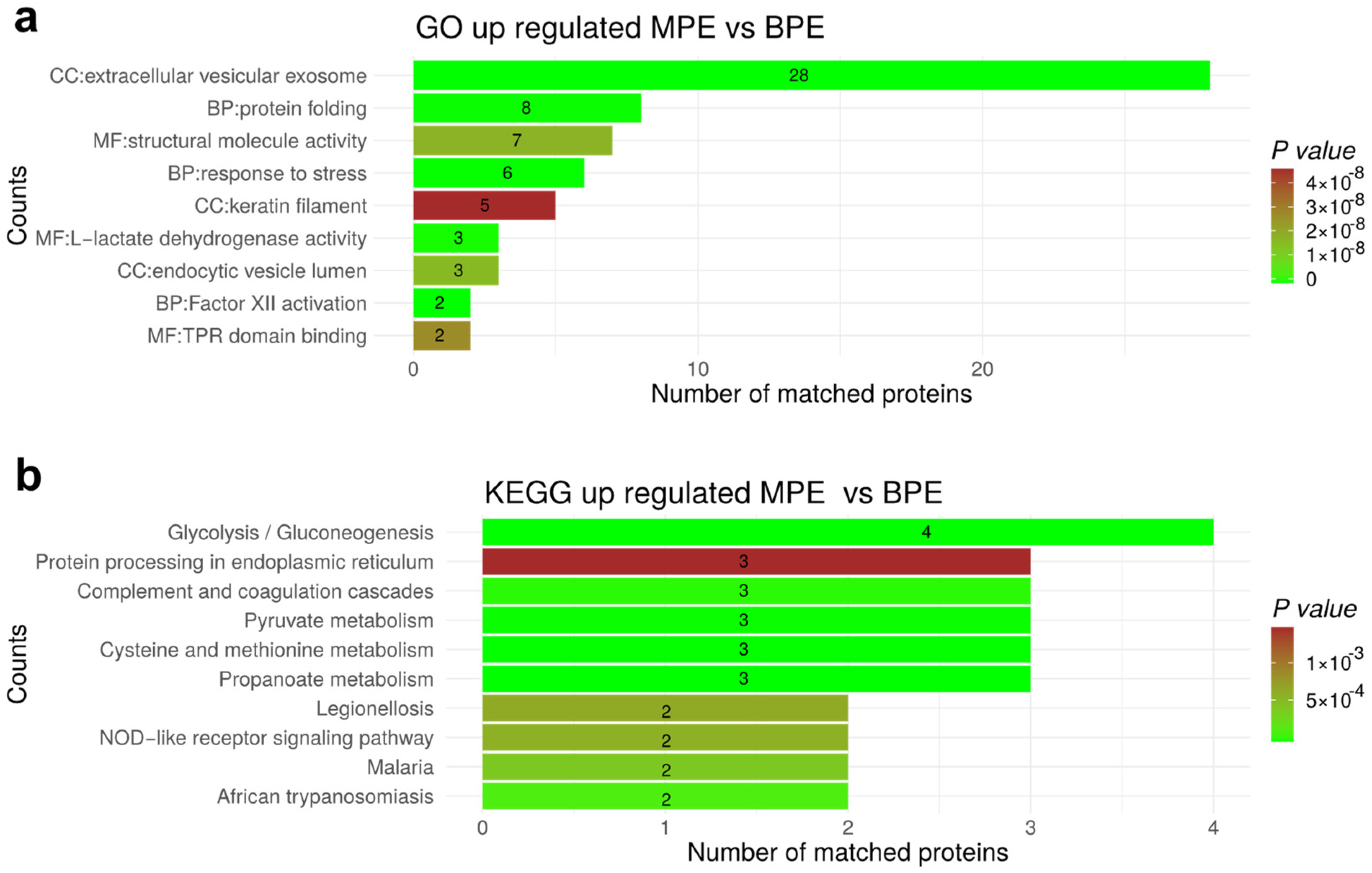
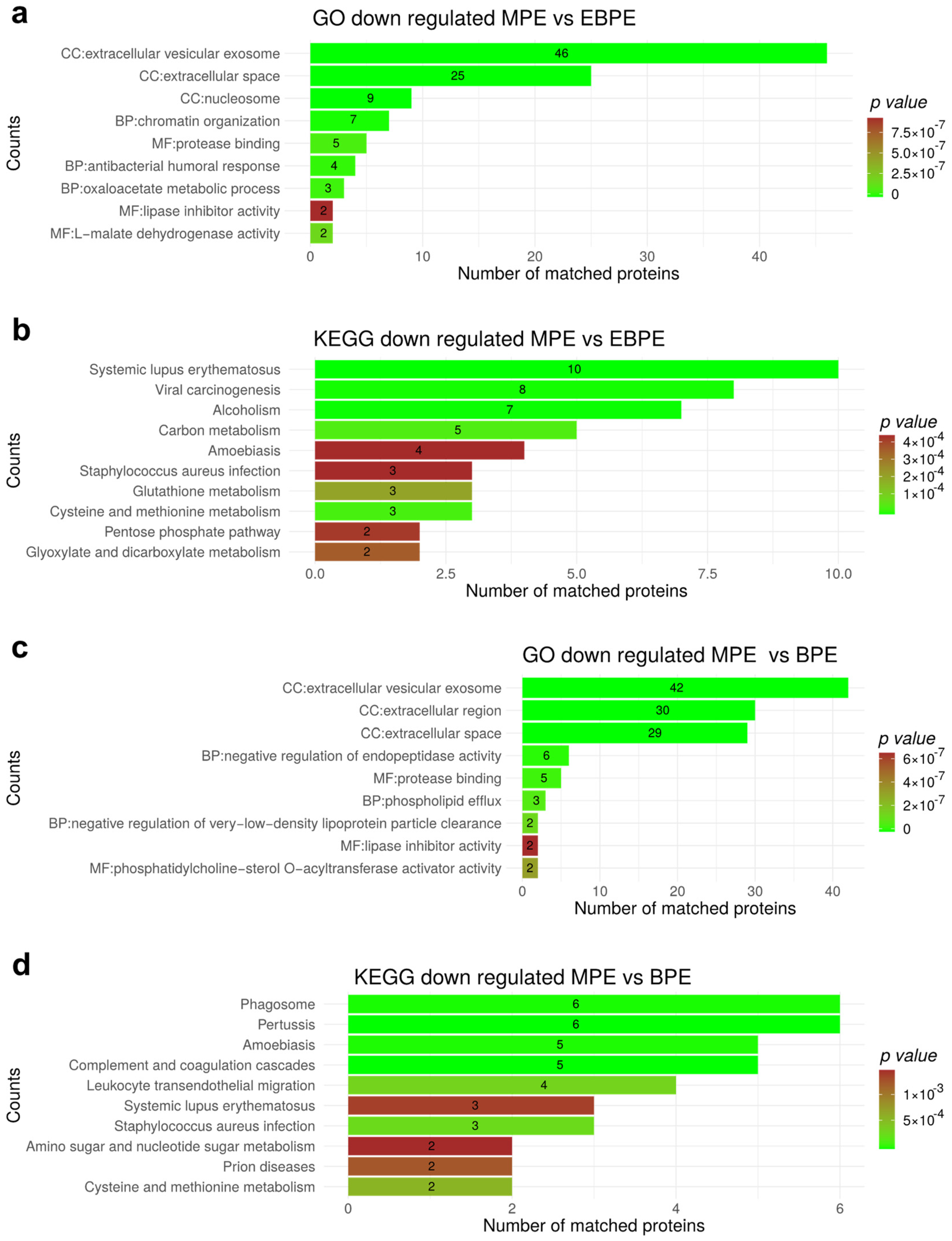
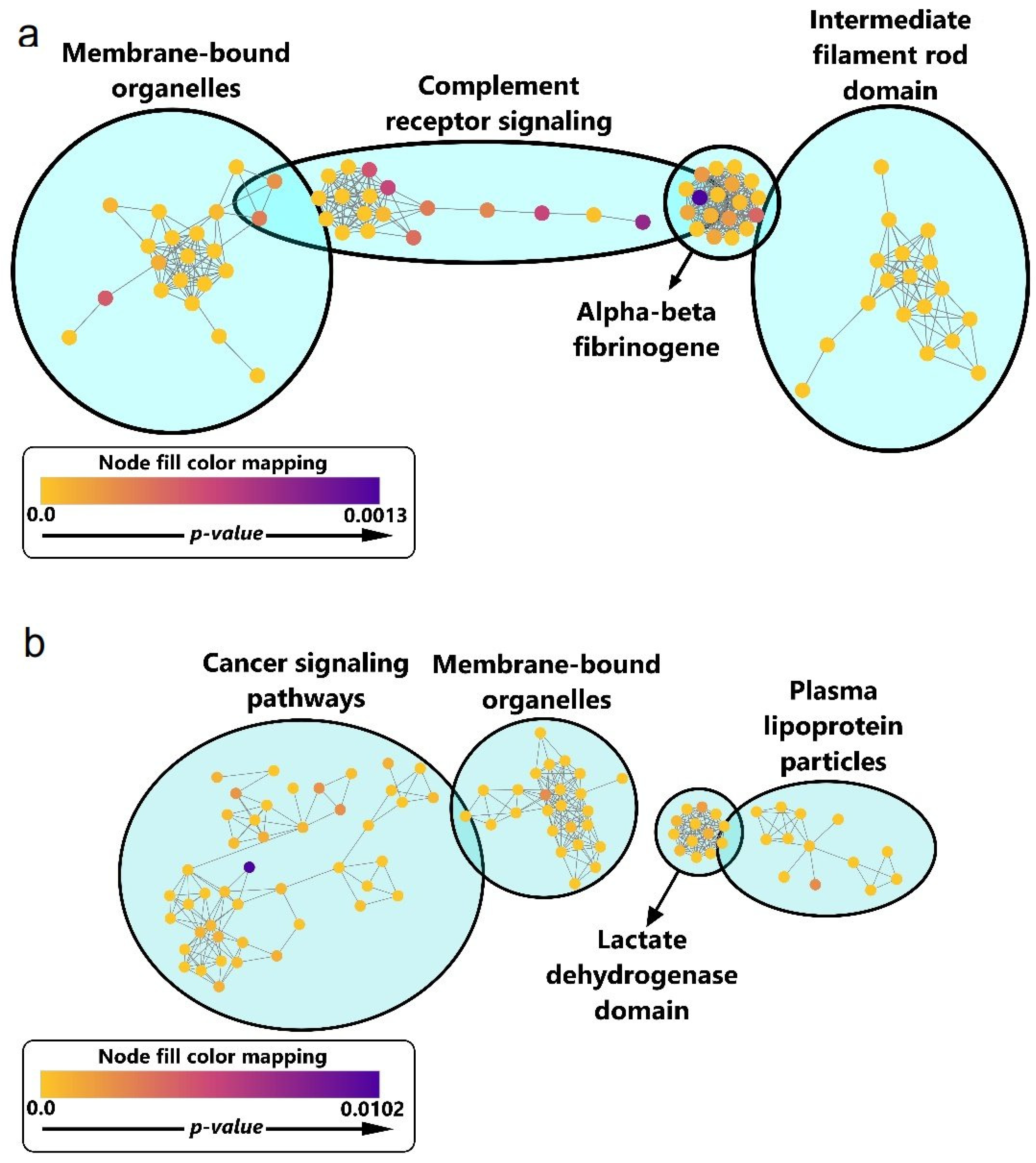
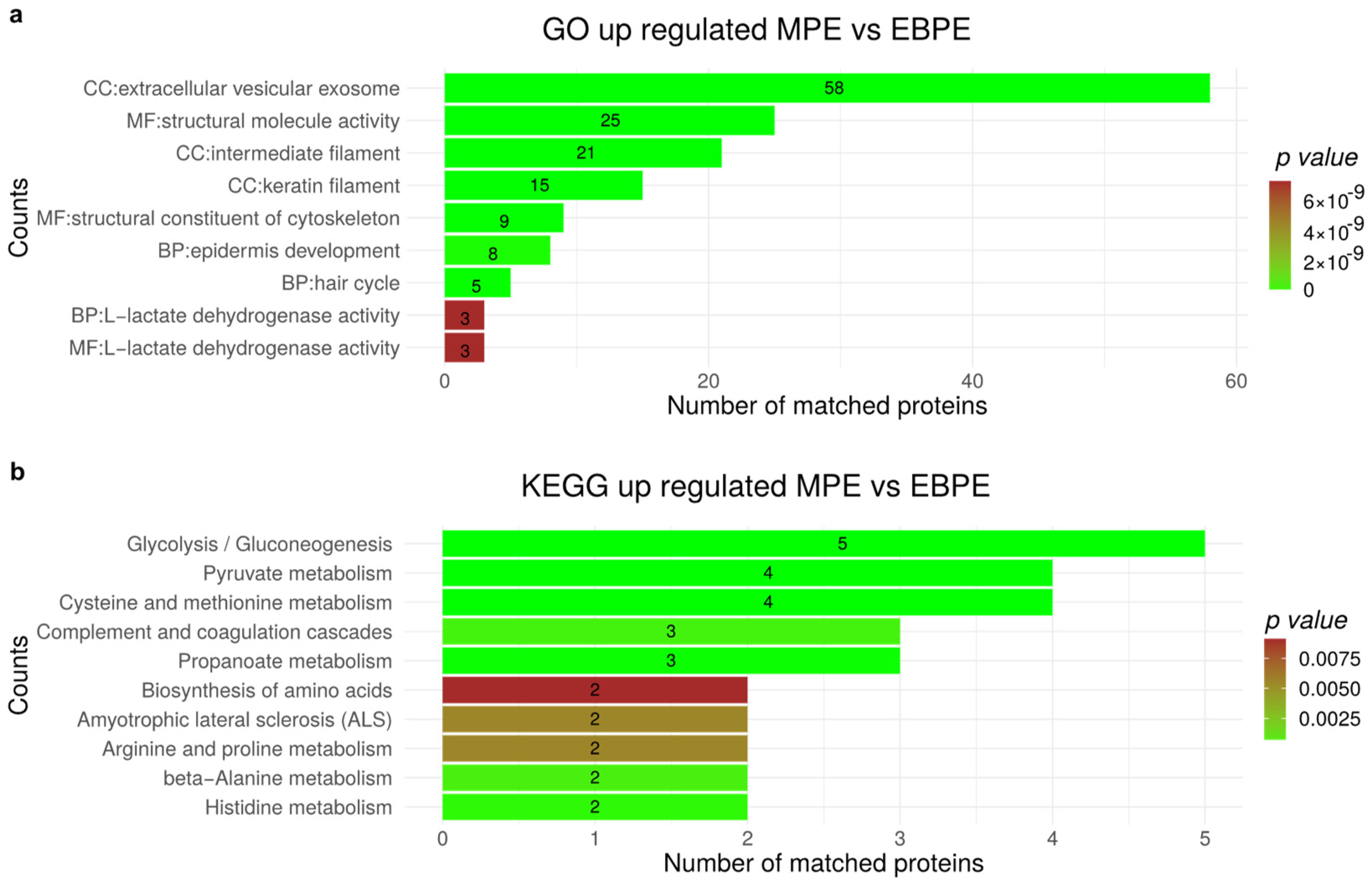
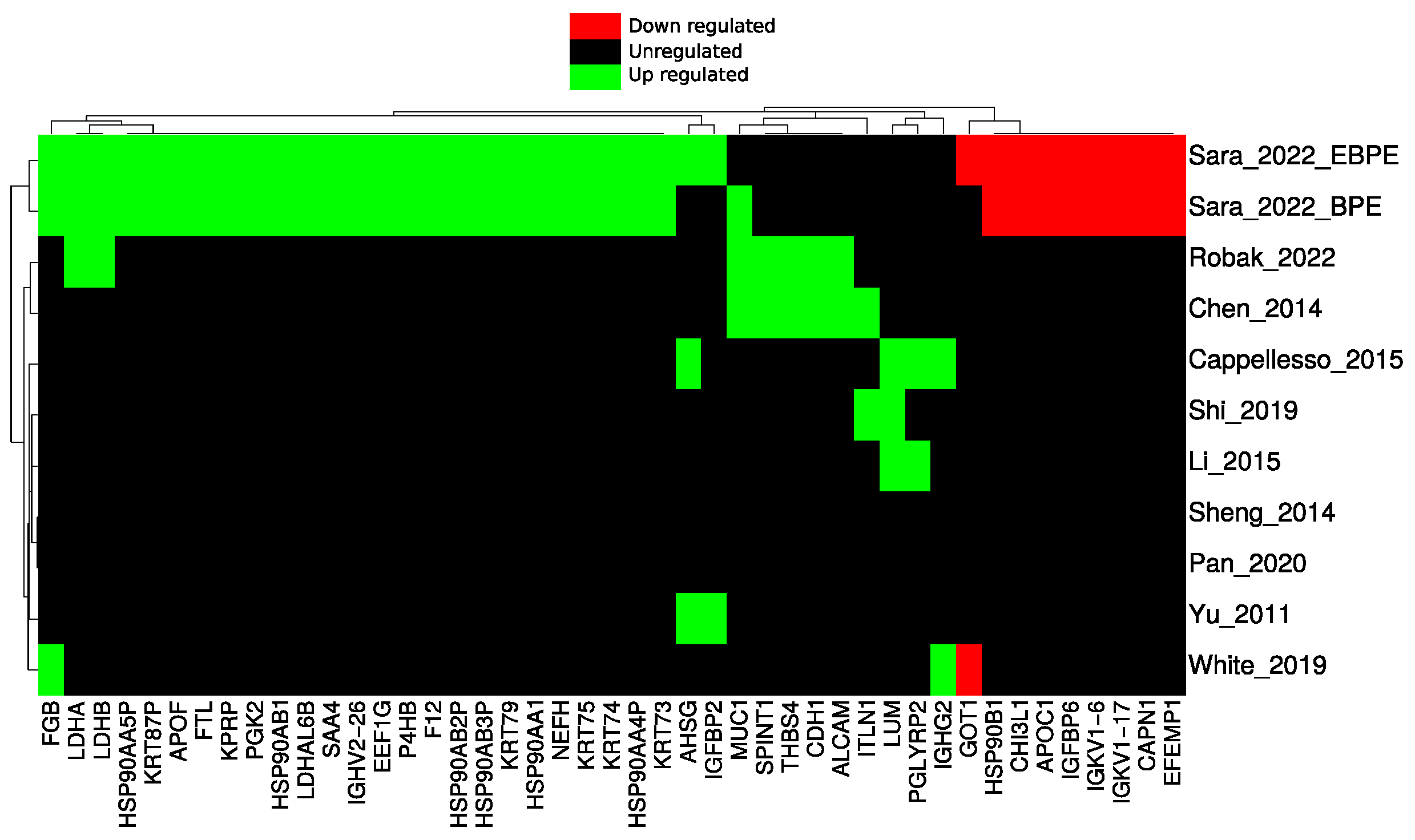
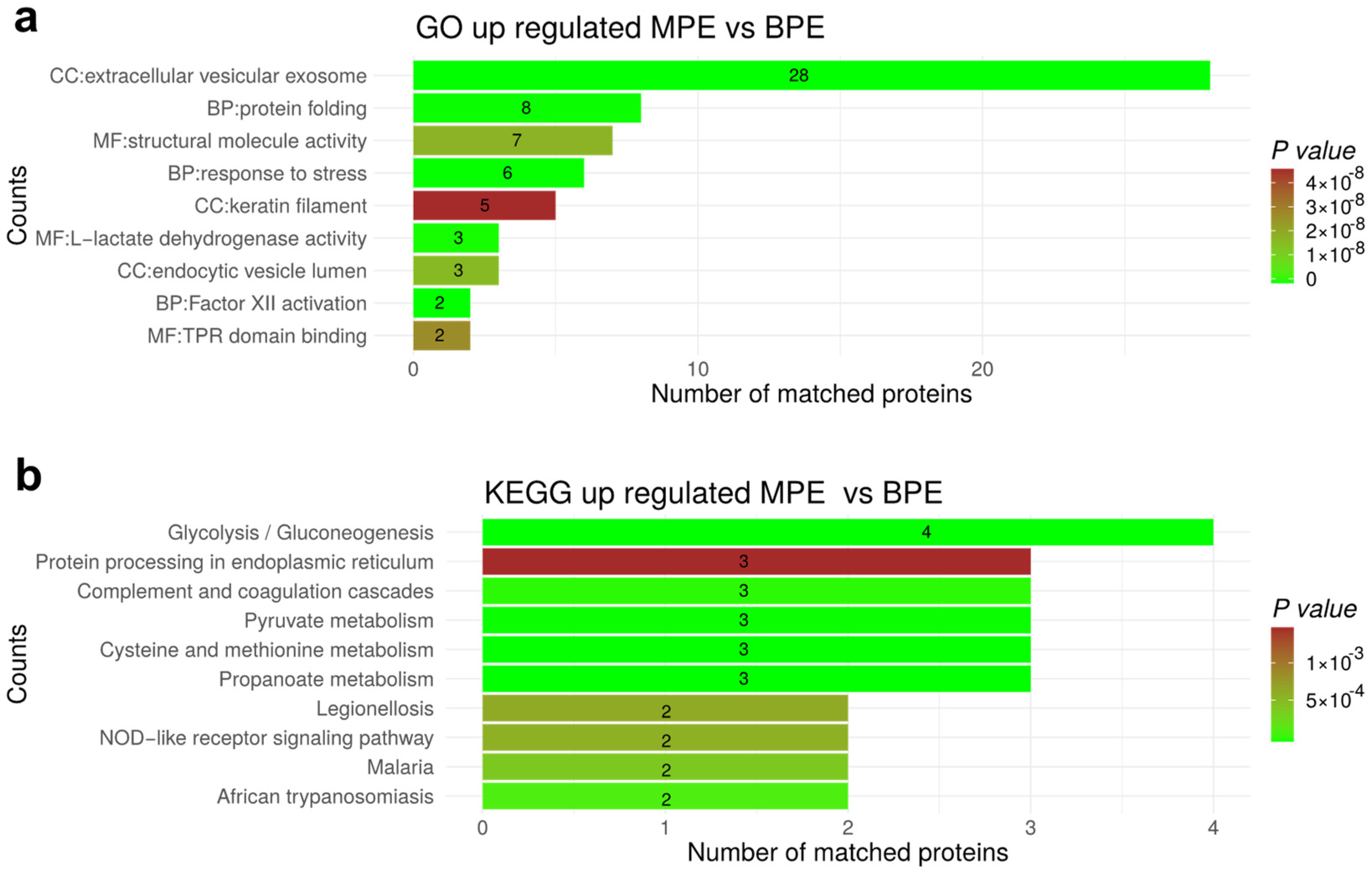
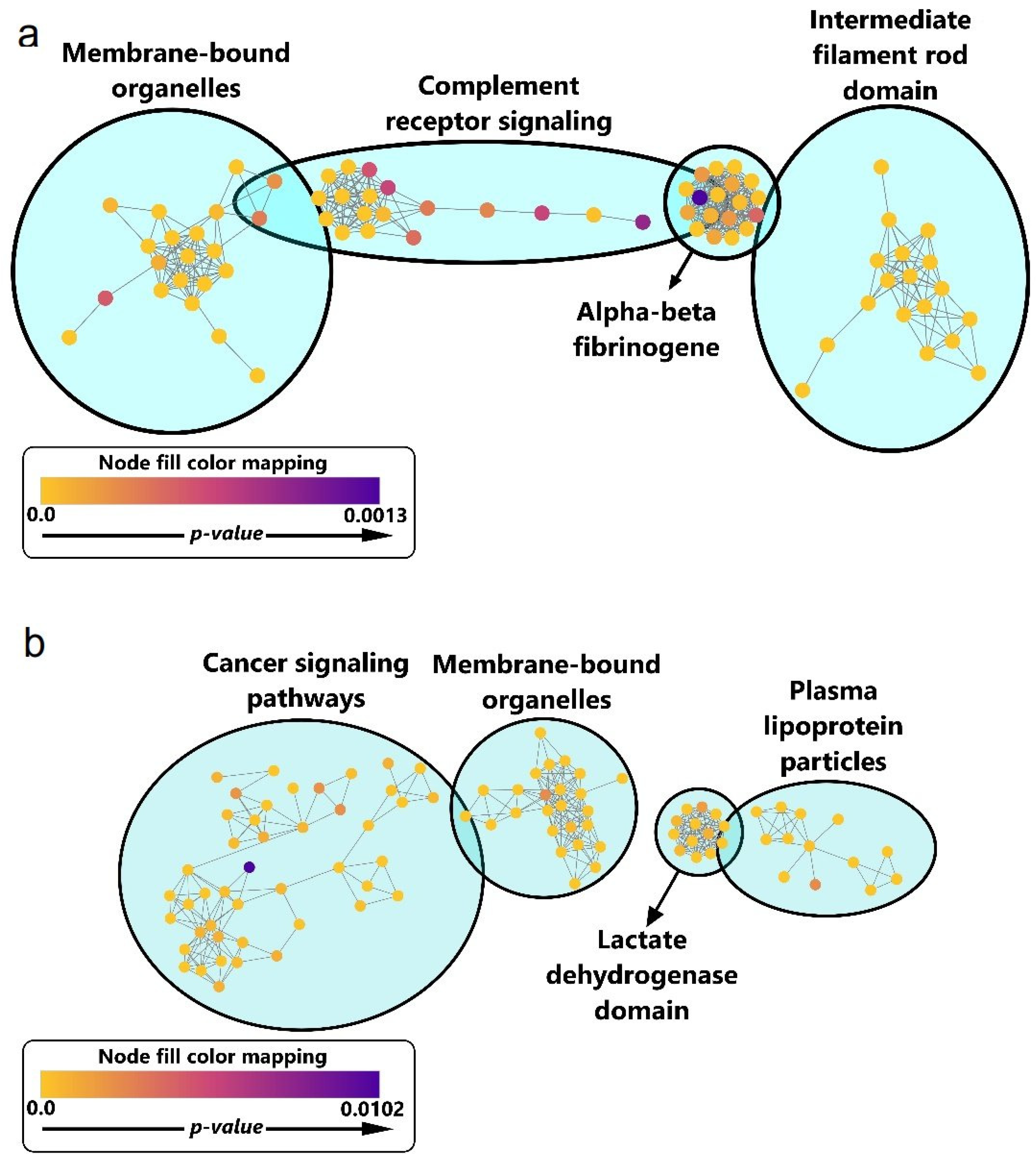
3.7. Functional Enrichment for Significantly Regulated Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A

References
- Sundaralingam, A.; Bedawi, E.O.; Rahman, N.M. Diagnostics in Pleural Disease. Diagnostics 2020, 10, 1046. [Google Scholar] [CrossRef]
- Jany, B.; Welte, T. Pleural Effusion in Adults—Etiology, Diagnosis, and Treatment. Dtsch. Arztebl. Int. 2019, 116, 377–386. [Google Scholar] [CrossRef]
- Baburaj, G.; Damerla, R.R.; Udupa, K.S.; Parida, P.; Munisamy, M.; Kolesar, J.; Rao, M. Liquid biopsy approaches for pleural effusion in lung cancer patients. Mol. Biol. Rep. 2020, 47, 8179–8187. [Google Scholar] [CrossRef] [PubMed]
- Principe, N.; Kidman, J.; Lake, R.A.; Lesterhuis, W.J.; Nowak, A.K.; McDonnell, A.M.; Chee, J. Malignant Pleural Effusions—A Window into Local Anti-Tumor T Cell Immunity? Front. Oncol. 2021, 11, 672747. [Google Scholar] [CrossRef] [PubMed]
- Kassirian, S.; Hinton, S.N.; Cuninghame, S.; Chaudhary, R.; Iansavitchene, A.; Amjadi, K.; Dhaliwal, I.; Zeman-Pocrnich, C.; Mitchell, M.A. Diagnostic sensitivity of pleural fluid cytology in malignant pleural effusions: Systematic review and meta-analysis. Thorax 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tian, S.; Li, Y.; Zhan, N.; Guo, Y.; Liu, Y.; Xu, J.; Ma, Y.; Zhang, S.; Song, S.; et al. Development and validation of a novel scoring system developed from a nomogram to identify malignant pleural effusion. eBioMedicine 2020, 58, 102924. [Google Scholar] [CrossRef]
- Meriggi, F. Malignant Pleural Effusion: Still a Long Way to Go. Rev. Recent Clin. Trials 2019, 14, 24–30. [Google Scholar] [CrossRef]
- Zhang, X.; Yi, F.-S.; Shi, H.-Z. Predicting Survival for Patients with Malignant Pleural Effusion: Development of the CONCH Prognostic Model. Cancer Manag. Res. 2021, 2021, 4699–4707. [Google Scholar] [CrossRef]
- Chen, K.-C.; Tsai, S.-W.; Zhang, X.; Zeng, C.; Yang, H.-Y. The investigation of the volatile metabolites of lung cancer from the microenvironment of malignant pleural effusion. Sci. Rep. 2021, 11, 13585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wan, B.; Wang, L.; Li, C.; Xu, Y.; Wang, X.; Liu, H.; Song, Y.; Lin, D.; Zhan, P.; et al. The diagnostic yield of closed needle pleural biopsy in exudative pleural effusion: A retrospective 10-year study. Ann. Transl. Med. 2020, 8, 491. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Han, Y.; Ma, Y.; Wu, Z.; Zhang, F.; Zheng, D.; Liu, X.; Tao, L.; Liang, Z.; Yang, Z.; Li, X.; et al. Histologic subtype classification of non-small cell lung cancer using PET/CT images. Eur. J. Pediatr. 2020, 48, 350–360. [Google Scholar] [CrossRef]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef]
- Jones, G.S.; Baldwin, D.R. Recent advances in the management of lung cancer. Clin. Med. 2018, 18, s41–s46. [Google Scholar] [CrossRef]
- Lai, Y.-H.; Chen, W.-N.; Hsu, T.-C.; Lin, C.; Tsao, Y.; Wu, S. Overall survival prediction of non-small cell lung cancer by integrating microarray and clinical data with deep learning. Sci. Rep. 2020, 10, 4679. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Z.; Qian, L.; Zeng, S.; Zhou, Y.; Chen, X.; Wei, J.; Gong, Z. Identification of CAV1 and DCN as potential predictive biomarkers for lung adenocarcinoma. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L630–L643. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, M.; Zhou, N.; Hou, H.; Li, T.; Yu, H.; Tan, Y.-D.; Zhang, X. Use tumor suppressor genes as biomarkers for diagnosis of non-small cell lung cancer. Sci. Rep. 2021, 11, 3596. [Google Scholar] [CrossRef] [PubMed]
- Zamay, T.N.; Zamay, G.S.; Kolovskaya, O.S.; Zukov, R.A.; Petrova, M.M.; Gargaun, A.; Berezovski, M.V.; Kichkailo, A.S. Current and Prospective Protein Biomarkers of Lung Cancer. Cancers 2017, 9, 155. [Google Scholar] [CrossRef]
- De Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e17. [Google Scholar] [CrossRef]
- Ortea, I.; Rodríguez-Ariza, A.; Chicano-Gálvez, E.; Vacas, M.A.; Gámez, B.J. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J. Proteom. 2016, 138, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, R. MS-Based Biomarker Discovery in Bronchoalveolar Lavage Fluid for Lung Cancer. Proteom. Clin. Appl. 2019, 14, e1900077. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.S.; Moraes, M.C.S.; Na, C.H.; Fierro-Monti, I.; Henriques, A.; Zahedi, S.; Bodo, C.; Tranfield, E.M.; Sousa, A.L.; Farinho, A.; et al. Is the Proteome of Bronchoalveolar Lavage Extracellular Vesicles a Marker of Advanced Lung Cancer? Cancers 2020, 12, 3450. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Cuco, C.M.; Lavareda, C.; Miguel, F.; Ventura, M.; Almeida, S.; Pinto, P.; De Abreu, T.T.; Rodrigues, L.; Seixas, S.; et al. Bronchoalveolar Lavage Proteomics in Patients with Suspected Lung Cancer. Sci. Rep. 2017, 7, srep42190. [Google Scholar] [CrossRef]
- Zhang, M.; Yan, L.; Lippi, G.; Hu, Z.-D. Pleural biomarkers in diagnostics of malignant pleural effusion: A narrative review. Transl. Lung Cancer Res. 2021, 10, 1557–1570. [Google Scholar] [CrossRef]
- Giarnieri, E.; Bellipanni, G.; Macaluso, M.; Mancini, R.; Holstein, A.C.; Milanese, C.; Giovagnoli, M.R.; Giordano, A.; Russo, G. Review: Cell Dynamics in Malignant Pleural Effusions. J. Cell. Physiol. 2014, 230, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-J.; Chen, C.-D.; Wang, C.-L.; Wu, Y.-C.; Hsu, C.-W.; Lee, C.-W.; Huang, L.-H.; Yu, J.-S.; Chang, Y.-S.; Wu, C.-C.; et al. In-depth Proteomic Analysis of Six Types of Exudative Pleural Effusions for Nonsmall Cell Lung Cancer Biomarker Discovery. Mol. Cell. Proteom. 2015, 14, 917–932. [Google Scholar] [CrossRef]
- Yang, Z.; Song, Z.; Chen, Z.; Guo, Z.; Jin, H.; Ding, C.; Hong, Y.; Cai, Z. Metabolic and lipidomic characterization of malignant pleural effusion in human lung cancer. J. Pharm. Biomed. Anal. 2019, 180, 113069. [Google Scholar] [CrossRef]
- Lam, C.-W.; Law, C.-Y. Untargeted Mass Spectrometry-Based Metabolomic Profiling of Pleural Effusions: Fatty Acids as Novel Cancer Biomarkers for Malignant Pleural Effusions. J. Proteome Res. 2014, 13, 4040–4046. [Google Scholar] [CrossRef]
- Che, N.; Ma, Y.; Ruan, H.; Xu, L.; Wang, X.; Yang, X.; Liu, X. Integrated semi-targeted metabolomics analysis reveals distinct metabolic dysregulation in pleural effusion caused by tuberculosis and malignancy. Clin. Chim. Acta 2018, 477, 81–88. [Google Scholar] [CrossRef]
- Li, H.; Tang, Z.; Zhu, H.; Ge, H.; Cui, S.; Jiang, W. Proteomic study of benign and malignant pleural effusion. J. Cancer Res. Clin. Oncol. 2016, 142, 1191–1200. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, X.; Jia, H.; Huang, M.; Liu, F.; Wang, J.; Du, B.; Wei, R.; Sun, Q.; Xing, A.; et al. Label-Free Quantitative Proteomics Identifies Novel Biomarkers for Distinguishing Tuberculosis Pleural Effusion from Malignant Pleural Effusion. Proteom. Clin. Appl. 2019, 14, e1900001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, D.; Hu, Z.-D.; Huang, Y.-L. The diagnostic utility of pleural markers for tuberculosis pleural effusion. Ann. Transl. Med. 2020, 8, 607. [Google Scholar] [CrossRef]
- White, R.; Pulford, E.; Elliot, D.J.; Thurgood, L.A.; Klebe, S. Quantitative mass spectrometry to identify protein markers for diagnosis of malignant pleural mesothelioma. J. Proteom. 2018, 192, 374–382. [Google Scholar] [CrossRef]
- Lacerenza, S.; Ciregia, F.; Giusti, L.; Bonotti, A.; Greco, V.; Giannaccini, G.; D’Antongiovanni, V.; Fallahi, P.; Pieroni, L.; Cristaudo, A.; et al. Putative Biomarkers for Malignant Pleural Mesothelioma Suggested by Proteomic Analysis of Cell Secretome. Cancer Genom. Proteom. 2020, 17, 225–236. [Google Scholar] [CrossRef]
- Cerciello, F.; Choi, M.; Sinicropi-Yao, S.L.; Lomeo, K.; Amann, J.M.; Felley-Bosco, E.; Stahel, R.A.; Robinson, B.W.; Creaney, J.; Pass, H.I.; et al. Verification of a Blood-Based Targeted Proteomics Signature for Malignant Pleural Mesothelioma. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1973–1982. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Huang, X.; Shen, Y.; Shen, J.; Ying, K. Differential proteome profiling of pleural effusions from lung cancer and benign inflammatory disease patients. Biochim. Biophys. Acta BBA Proteins Proteom. 2012, 1824, 692–700. [Google Scholar] [CrossRef]
- Choi, H.; Ko, Y.; Lee, C.Y. Pro-cathepsin D as a diagnostic marker in differentiating malignant from benign pleural effusion: A retrospective cohort study. BMC Cancer 2020, 20, 825. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.S.; Baeta, H.; Henriques, A.F.A.; Ejtehadifar, M.; Tranfield, E.M.; Sousa, A.L.; Farinho, A.; Silva, B.C.; Cabeçadas, J.; Gameiro, P.; et al. Proteomic Landscape of Extracellular Vesicles for Diffuse Large B-Cell Lymphoma Subtyping. Int. J. Mol. Sci. 2021, 22, 11004. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Ribeiro, H.; Voabil, P.; Penque, D.; Jensen, O.N.; Molina, H.; Matthiesen, R. Global Mass Spectrometry and Transcriptomics Array Based Drug Profiling Provides Novel Insight into Glucosamine Induced Endoplasmic Reticulum Stress. Mol. Cell. Proteom. 2014, 13, 3294–3307. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Matthiesen, R.; Prieto, G.; Amorim, A.; Aloria, K.; Fullaondo, A.; Carvalho, A.S.; Arizmendi, J.M. SIR: Deterministic protein inference from peptides assigned to MS data. J. Proteom. 2012, 75, 4176–4183. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Müller, J.B.; Buchholtz, M.L.; Bader, J.M.; Teupser, D.; et al. Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Kosinski, M.; Biecek, P. RTCGA: The Cancer Genome Atlas Data Integration. R Package Version 1.26.0. 2022. Available online: https://www.bioconductor.org/packages/devel/bioc/manuals/RTCGA/man/RTCGA.pdf (accessed on 15 July 2022).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. caret: Classification and Regression Training. R Package Version 6.0-88. 2021. Available online: https://cran.r-project.org/web/packages/caret/caret.pdf (accessed on 15 July 2022).
- Klein, J.P.; Van Houwelingen, H.C.; Ibrahim, J.G.; Scheike, T.H. Handbook of Survival Analysis; Chapman and Hall: London, UK, 2016. [Google Scholar]
- Liu, K.; Kang, M.; Li, J.; Qin, W.; Wang, R. Prognostic value of the mRNA expression of members of the HSP90 family in non-small cell lung cancer. Exp. Ther. Med. 2019, 17, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Cleven, A.H.; Balluff, B.; de Graaff, M.; Kostine, M.; Briaire-de Bruijn, I.; McDonnell, L.A.; Bovee, J.V. High nuclear expression of proteasome activator complex subunit 1 predicts poor survival in soft tissue leiomyosarcomas. Clin. Sarcoma Res. 2016, 6, 17. [Google Scholar] [CrossRef]
- Jiao, Y.; Du, P. Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 2016, 4, 320–330. [Google Scholar] [CrossRef]
- Xu, B.; Wang, Y.; Wang, Z.; Zhou, J.; Zhou, S.; Guan, J. An effective approach to detecting both small and large complexes from protein-protein interaction networks. BMC Bioinform. 2017, 18, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.; Yu, I.-J.; Seo, D.; Kang, U.; Sael, L. PS-MCL: Parallel shotgun coarsened Markov clustering of protein interaction networks. BMC Bioinform. 2019, 20, 381. [Google Scholar] [CrossRef]
- Brohée, S.; Van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinform. 2006, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Robak, A.; Kistowski, M.; Wojtas, G.; Perzanowska, A.; Targowski, T.; Michalak, A.; Krasowski, G.; Dadlez, M.; Domański, D. Diagnosing pleural effusions using mass spectrometry-based multiplexed targeted proteomics quantitating mid- to high-abundance markers of cancer, infection/inflammation and tuberculosis. Sci. Rep. 2022, 12, 3054. [Google Scholar] [CrossRef]
- Chen, C.-D.; Wang, C.-L.; Yu, C.-J.; Chien, K.-Y.; Chen, Y.-T.; Chen, M.-C.; Chang, Y.-S.; Wu, C.-C.; Yu, J.-S. Targeted Proteomics Pipeline Reveals Potential Biomarkers for the Diagnosis of Metastatic Lung Cancer in Pleural Effusion. J. Proteome Res. 2014, 13, 2818–2829. [Google Scholar] [CrossRef] [PubMed]
- Cappellesso, R.; Millioni, R.; Arrigoni, G.; Simonato, F.; Caroccia, B.; Iori, E.; Guzzardo, V.; Ventura, L.; Tessari, P.; Fassina, A. Lumican Is Overexpressed in Lung Adenocarcinoma Pleural Effusions. PLoS ONE 2015, 10, e0126458. [Google Scholar] [CrossRef]
- Shi, J.; Li, P.; Zhou, L.; Qi, S.; Wang, B.; Li, D.; Duan, L.; Chen, W.X.; Xia, J.; Zou, L.; et al. Potential biomarkers for antidiastole of tuberculous and malignant pleural effusion by proteome analysis. Biomark. Med. 2019, 13, 123–133. [Google Scholar] [CrossRef]
- Li, Y.; Lian, H.; Jia, Q.; Wan, Y. Proteome screening of pleural effusions identifies IL1A as a diagnostic biomarker for non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2015, 457, 177–182. [Google Scholar] [CrossRef]
- Sheng, S.-H.; Zhu, H.-L. Proteomic analysis of pleural effusion from lung adenocarcinoma patients by shotgun strategy. Clin. Transl. Oncol. 2014, 16, 153–157. [Google Scholar] [CrossRef]
- Yu, C.-J.; Wang, C.-L.; Wang, C.-I.; Chen, C.-D.; Dan, Y.-M.; Wu, C.-C.; Wu, Y.-C.; Lee, I.-N.; Tsai, Y.-H.; Chang, Y.-S.; et al. Comprehensive Proteome Analysis of Malignant Pleural Effusion for Lung Cancer Biomarker Discovery by Using Multidimensional Protein Identification Technology. J. Proteome Res. 2011, 10, 4671–4682. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Shapiro, G.I. Heat Shock Protein 90 Inhibition in Lung Cancer. J. Thorac. Oncol. 2008, 3, S152–S159. [Google Scholar] [CrossRef]
- Cong, L.; Maishi, N.; Annan, D.A.; Young, M.F.; Morimoto, H.; Morimoto, M.; Nam, J.-M.; Hida, Y.; Hida, K. Inhibition of stromal biglycan promotes normalization of the tumor microenvironment and enhances chemotherapeutic efficacy. Breast Cancer Res. BCR 2021, 23, 51. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.K.; Kumar, D.; Naik, M.; Dixit, M. The expression profile and prognostic significance of eukaryotic translation elongation factors in different cancers. PLoS ONE 2018, 13, e0191377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, P.; Liu, M.; Zhang, X.; Si, Q.; Yang, T.; Ye, H.; Song, C.; Shi, J.; Wang, K.; et al. Identification of tumor-associated antigens of lung cancer: SEREX combined with bioinformatics analysis. J. Immunol. Methods 2021, 492, 112991. [Google Scholar] [CrossRef] [PubMed]
- Guinde, J.; Frankel, D.; Perrin, S.; Delecourt, V.; Lévy, N.; Barlesi, F.; Astoul, P.; Roll, P.; Kaspi, E. Lamins in Lung Cancer: Biomarkers and Key Factors for Disease Progression through miR-9 Regulation? Cells 2018, 7, 78. [Google Scholar] [CrossRef]
- Jackson, D.; Craven, R.A.; Hutson, R.C.; Graze, I.; Lueth, P.; Tonge, R.P.; Hartley, J.L.; Nickson, J.A.; Rayner, S.J.; Johnston, C.; et al. Proteomic Profiling Identifies Afamin as a Potential Biomarker for Ovarian Cancer. Clin. Cancer Res. 2007, 13, 7370–7379. [Google Scholar] [CrossRef]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/α-albumin. eLife 2016, 5, e11621. [Google Scholar] [CrossRef] [PubMed]
- Dieplinger, H.; Dieplinger, B. Afamin—A pleiotropic glycoprotein involved in various disease states. Clin. Chim. Acta 2015, 446, 105–110. [Google Scholar] [CrossRef]
- Dong, H.; Zou, M.; Bhatia, A.; Jayaprakash, P.; Hofman, F.; Ying, Q.; Chen, M.; Woodley, D.T.; Li, W. Breast Cancer MDA-MB-231 Cells Use Secreted Heat Shock Protein-90alpha (Hsp90α) to Survive a Hostile Hypoxic Environment. Sci. Rep. 2016, 6, 20605. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperon 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, X.; Jin, J.; He, Y. The Expression Patterns and Prognostic Value of the Proteasome Activator Subunit Gene Family in Gastric Cancer Based on Integrated Analysis. Front. Cell Dev. Biol. 2021, 9, 663001. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; Craze, M.L.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. The multifunctional solute carrier 3A2 (SLC3A2) confers a poor prognosis in the highly proliferative breast cancer subtypes. Br. J. Cancer 2018, 118, 1115–1122. [Google Scholar] [CrossRef]
- Zhou, X.; Curbo, S.; Li, F.; Krishnan, S.; Karlsson, A. Inhibition of glutamate oxaloacetate transaminase 1 in cancer cell lines results in altered metabolism with increased dependency of glucose. BMC Cancer 2018, 18, 559. [Google Scholar] [CrossRef]
- Whitney, D.H.; Elashoff, M.R.; Porta-Smith, K.; Gower, A.C.; Vachani, A.; Ferguson, J.S.; Silvestri, G.A.; Brody, J.S.; Lenburg, M.E.; Spira, A. Derivation of a bronchial genomic classifier for lung cancer in a prospective study of patients undergoing diagnostic bronchoscopy. BMC Med. Genom. 2015, 8, 18. [Google Scholar] [CrossRef]
- Li, L.; Wei, Y.; To, C.; Zhu, C.Q.; Tong, J.; Pham, N.-A.; Taylor, P.; Ignatchenko, V.; Ignatchenko, A.; Zhang, W.; et al. Integrated Omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nat. Commun. 2014, 5, 5469. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.A.; Tanner, N.T.; Kearney, P.; Vachani, A.; Massion, P.P.; Porter, A.; Springmeyer, S.C.; Fang, K.C.; Midthun, D.; Mazzone, P.J.; et al. Assessment of Plasma Proteomics Biomarker’s Ability to Distinguish Benign from Malignant Lung Nodules: Results of the PANOPTIC (Pulmonary Nodule Plasma Proteomic Classifier) Trial. Chest 2018, 154, 491–500. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Malignant (MPE) | Suspected Malignant | Non-Malignant (BPE) | p |
|---|---|---|---|---|
| Observations | ||||
| N = 97 | 35 | 5 | 57 | |
| Age (Years) | ||||
| Mean (SD) | 67 (15) | 77 (9.5) | 75 (15) | 0.024 |
| valid (missing) | 35 (0) | 5 (0) | 57 (0) | |
| Gender | ||||
| F | 51% (18) | 60% (3) | 42% (24) | 0.56 |
| M | 49% (17) | 40% (2) | 58% (33) | |
| Pleural fluid | ||||
| Exudate | 97% (34) | 100% (5) | 53% (30) | <0.001 |
| Transudate | 2.9% (1) | 47% (27) | ||
| Pleural LDH units | ||||
| Mean (SD) | 372 (325) | 237 (108) | 266 (580) | <0.001 |
| valid (missing) | 34 (1) | 5 (0) | 57 (0) | |
| Pleural proteins mg/dL | ||||
| Mean (SD) | 4.5 (0.97) | 4.2 (0.3) | 3.2 (1.4) | <0.001 |
| valid (missing) | 34 (1) | 5 (0) | 57 (0) | |
| Ethnicity | ||||
| Black | 17% (6) | 0% (0) | 1.8% (1) | 0.018 |
| Caucasian | 83% (29) | 100% (5) | 98% (56) | |
| Smoking status | ||||
| Ex-smoker | 31% (11) | 40% (2) | 39% (22) | 0.47 |
| Non-smoker | 57% (20) | 20% (1) | 53% (30) | |
| Smoker | 11% (4) | 20% (1) | 5.3% (3) | |
| missing | 0% (0) | 20% (1) | 3.5% (2) | |
| Cytology | ||||
| Negative | 29% (10) | 100% (5) | 98% (56) | <0.001 |
| Positive | 54% (19) | 0% (0) | 0% (0) | |
| missing | 17% (6) | 0% (0) | 1.8% (1) | |
| Status § | ||||
| Alive | 29% (10) | 20% (1) | 100% (57) | <0.001 |
| Dead | 71% (25) | 80% (4) | 0% (0) |
| Protein | p | Log-Rank | Protein | p | Log-Rank |
|---|---|---|---|---|---|
| IGLV9_49 | 0.00014266 | 2.87 × 10−5 | ACTC1 | 0.0048079 | 0.00023111 |
| PSME1 | 0.00037219 | 1.68 × 10−6 | ACTA2 | 0.00482817 | 0.00023881 |
| HSP90AA1 | 0.0005116 | 0.00030706 | SAA2 | 0.00527138 | 0.00336226 |
| POTEKP | 0.0005359 | 0.00012375 | ACTA1 | 0.00547701 | 0.00029288 |
| SERPINA3 | 0.00132541 | 0.00110944 | LPA | 0.006096 | 0.0044481 |
| VTN | 0.00148688 | 0.00096543 | DSP | 0.0064211 | 0.0015112 |
| HSP90AB1 | 0.00165501 | 0.00114973 | ITIH2 | 0.00652889 | 0.00572679 |
| HSP90AB2P | 0.00246227 | 0.00172132 | ITIH4 | 0.00694899 | 0.00580105 |
| HSP90AB3P | 0.00248381 | 0.00175161 | POTEE | 0.00887962 | 0.00111277 |
| SFTPD | 0.00356403 | 0.00212968 | POTEI | 0.01041618 | 0.00164127 |
| HPX | 0.0036552 | 0.00398468 | POTEF | 0.01052107 | 0.00150148 |
| ACTG1 | 0.00367013 | 0.00028455 | SAA4 | 0.01084473 | 0.00922537 |
| SAA2_SAA4 | 0.00397487 | 0.0029843 | CPB2 | 0.01106905 | 0.0005958 |
| HSP90AA5P | 0.00400309 | 0.00297193 | SAA1 | 0.0124629 | 0.00516249 |
| ACTB | 0.00401629 | 0.00031496 | AKR1B10 | 0.01503984 | 0.00520114 |
| H0YJW9 | 0.00436481 | 0.00344019 | IGHV5_51 | 0.01558887 | 0.00675904 |
| ACTG2 | 0.00448447 | 0.00020827 | POTEJ | 0.02431411 | 0.00718341 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahedi, S.; Carvalho, A.S.; Ejtehadifar, M.; Beck, H.C.; Rei, N.; Luis, A.; Borralho, P.; Bugalho, A.; Matthiesen, R. Assessment of a Large-Scale Unbiased Malignant Pleural Effusion Proteomics Study of a Real-Life Cohort. Cancers 2022, 14, 4366. https://doi.org/10.3390/cancers14184366
Zahedi S, Carvalho AS, Ejtehadifar M, Beck HC, Rei N, Luis A, Borralho P, Bugalho A, Matthiesen R. Assessment of a Large-Scale Unbiased Malignant Pleural Effusion Proteomics Study of a Real-Life Cohort. Cancers. 2022; 14(18):4366. https://doi.org/10.3390/cancers14184366
Chicago/Turabian StyleZahedi, Sara, Ana Sofia Carvalho, Mostafa Ejtehadifar, Hans C. Beck, Nádia Rei, Ana Luis, Paula Borralho, António Bugalho, and Rune Matthiesen. 2022. "Assessment of a Large-Scale Unbiased Malignant Pleural Effusion Proteomics Study of a Real-Life Cohort" Cancers 14, no. 18: 4366. https://doi.org/10.3390/cancers14184366
APA StyleZahedi, S., Carvalho, A. S., Ejtehadifar, M., Beck, H. C., Rei, N., Luis, A., Borralho, P., Bugalho, A., & Matthiesen, R. (2022). Assessment of a Large-Scale Unbiased Malignant Pleural Effusion Proteomics Study of a Real-Life Cohort. Cancers, 14(18), 4366. https://doi.org/10.3390/cancers14184366