
Investigation of the Optimal Prime Boost Spacing Regimen for a Cancer Therapeutic Vaccine Targeting Human Papillomavirus



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Cell Lines
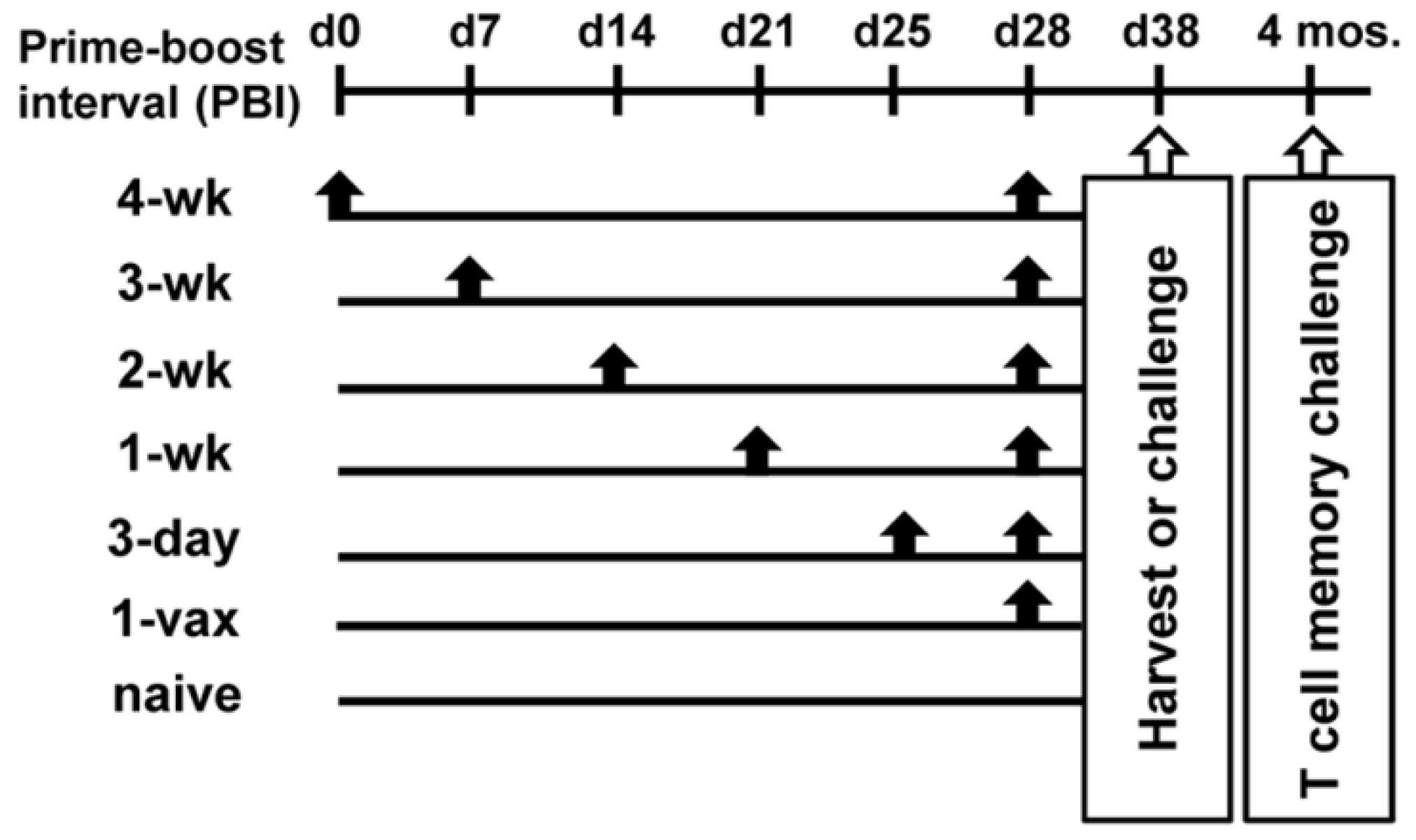
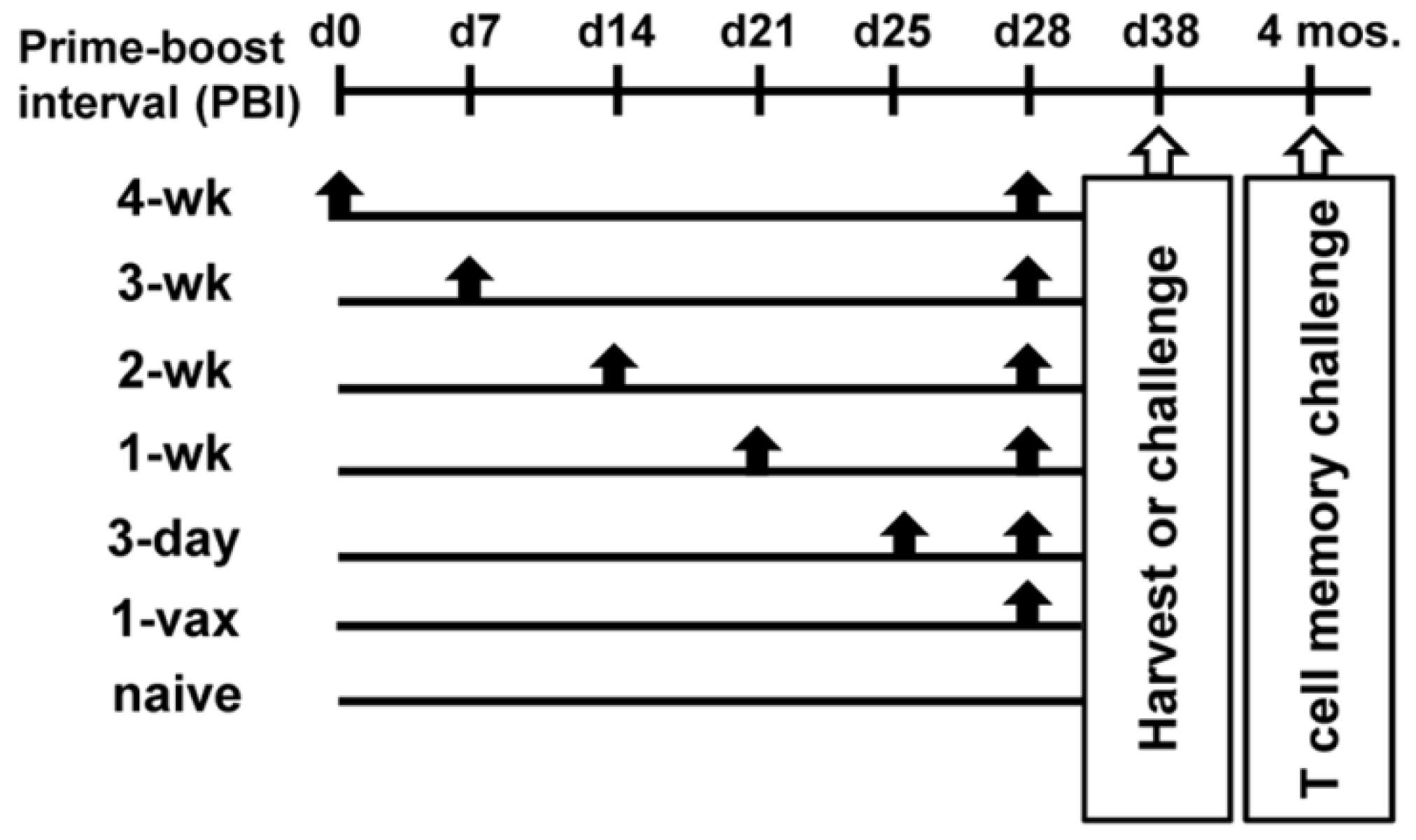
2.2. Vaccine Regimens
2.3. Peptides
2.4. Enzyme-Linked Immunospot Assay (ELISpot)
2.5. MHC Tetramer Analysis
2.6. In Vivo Cytoxicity Assay
2.7. In Vivo Tumor Studies
2.8. Statistical Analysis
3. Results
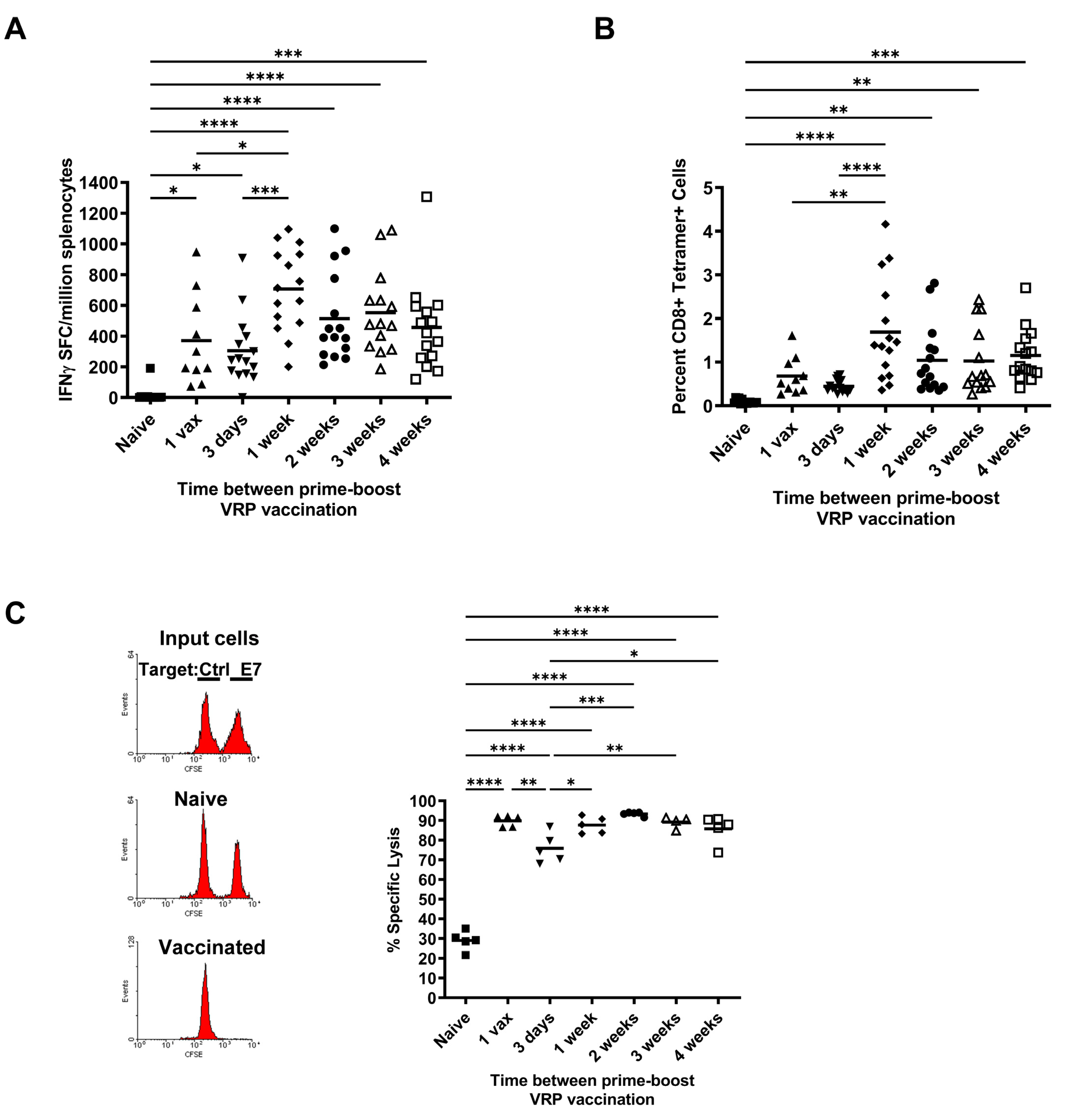
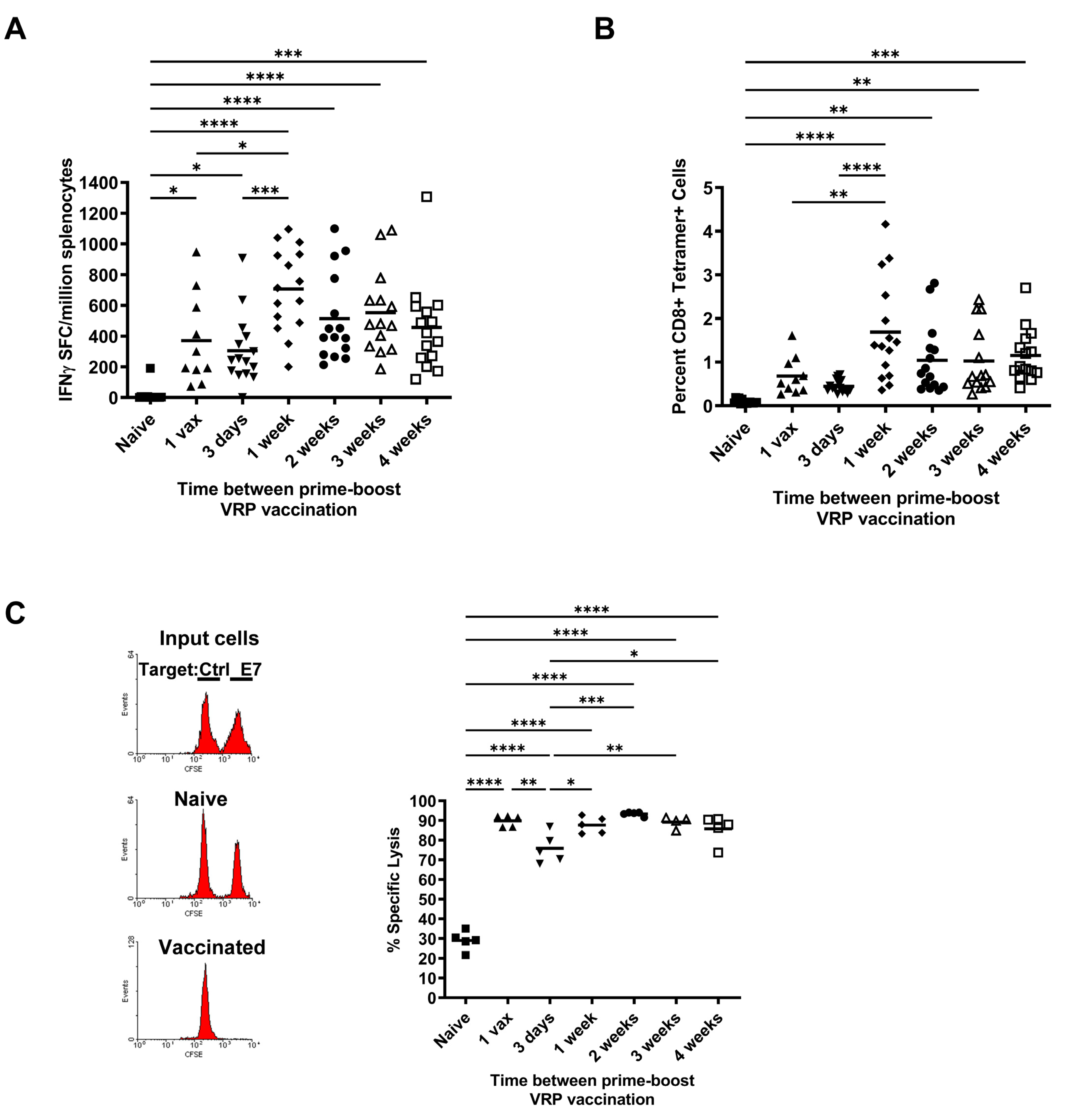
3.1. Effect of Increasing Prime-Boost Intervals on Magnitude of Induced Effector HPV Specific CD8+ T Cell Population
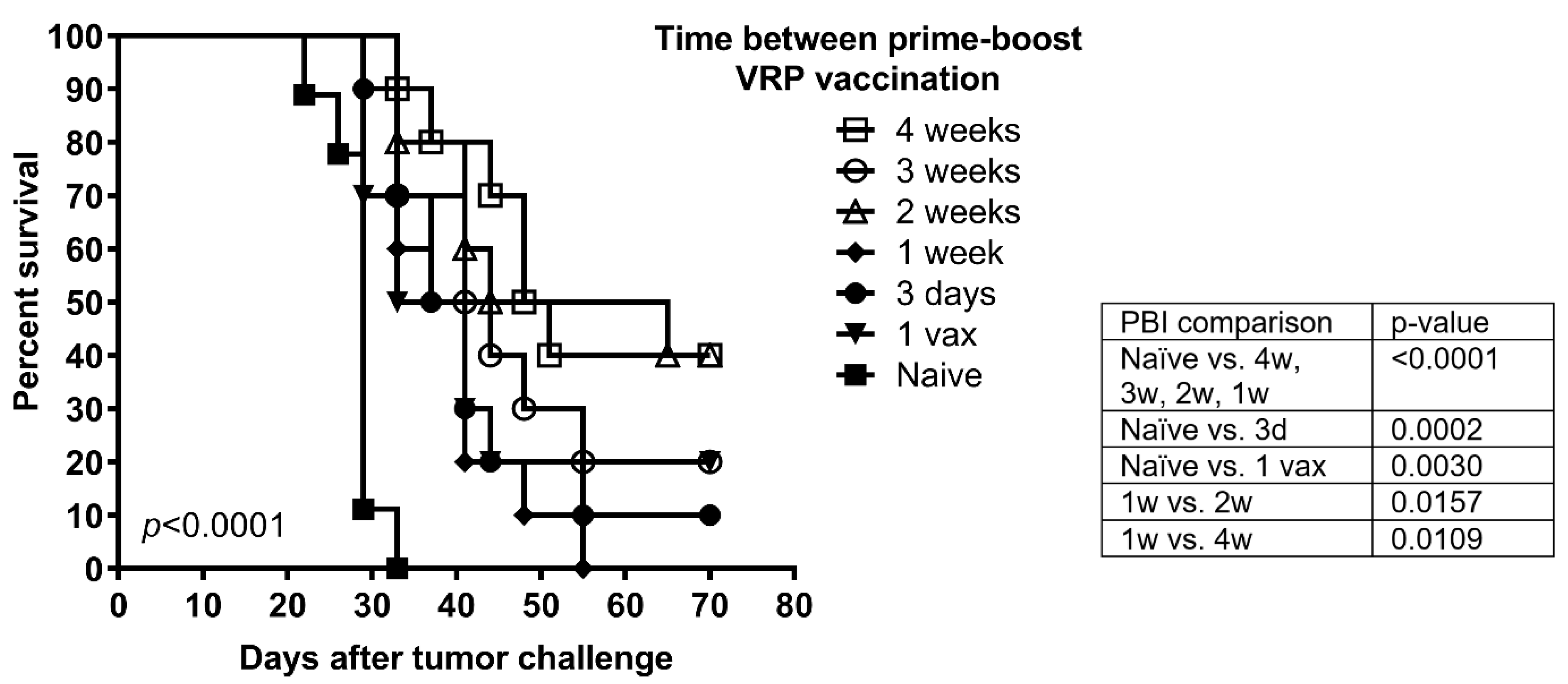
3.2. Vaccination Protects against Tumor Challenge Independent of Prime-Boost Interval in a Prophylactic HPV16 Tumor Model
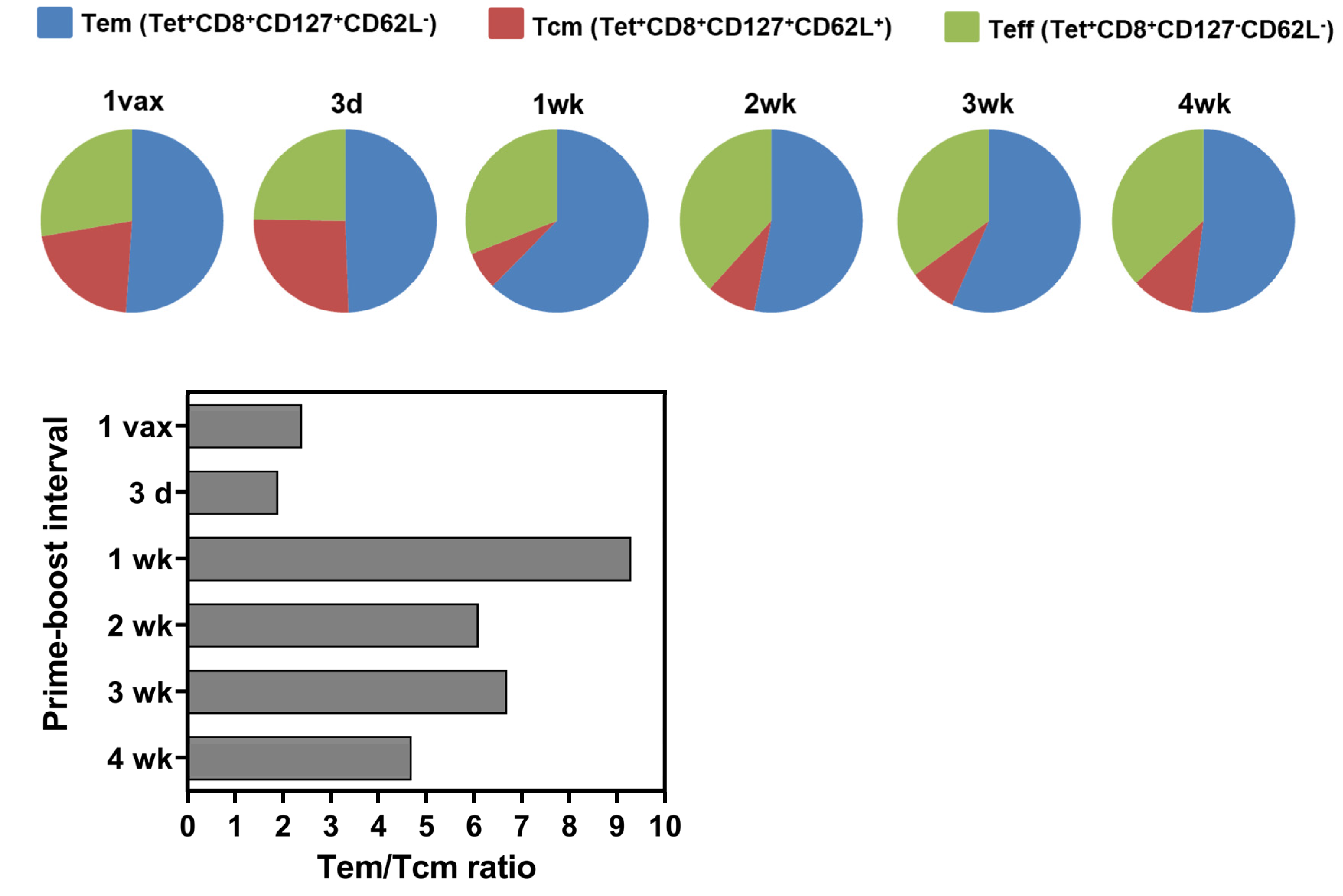
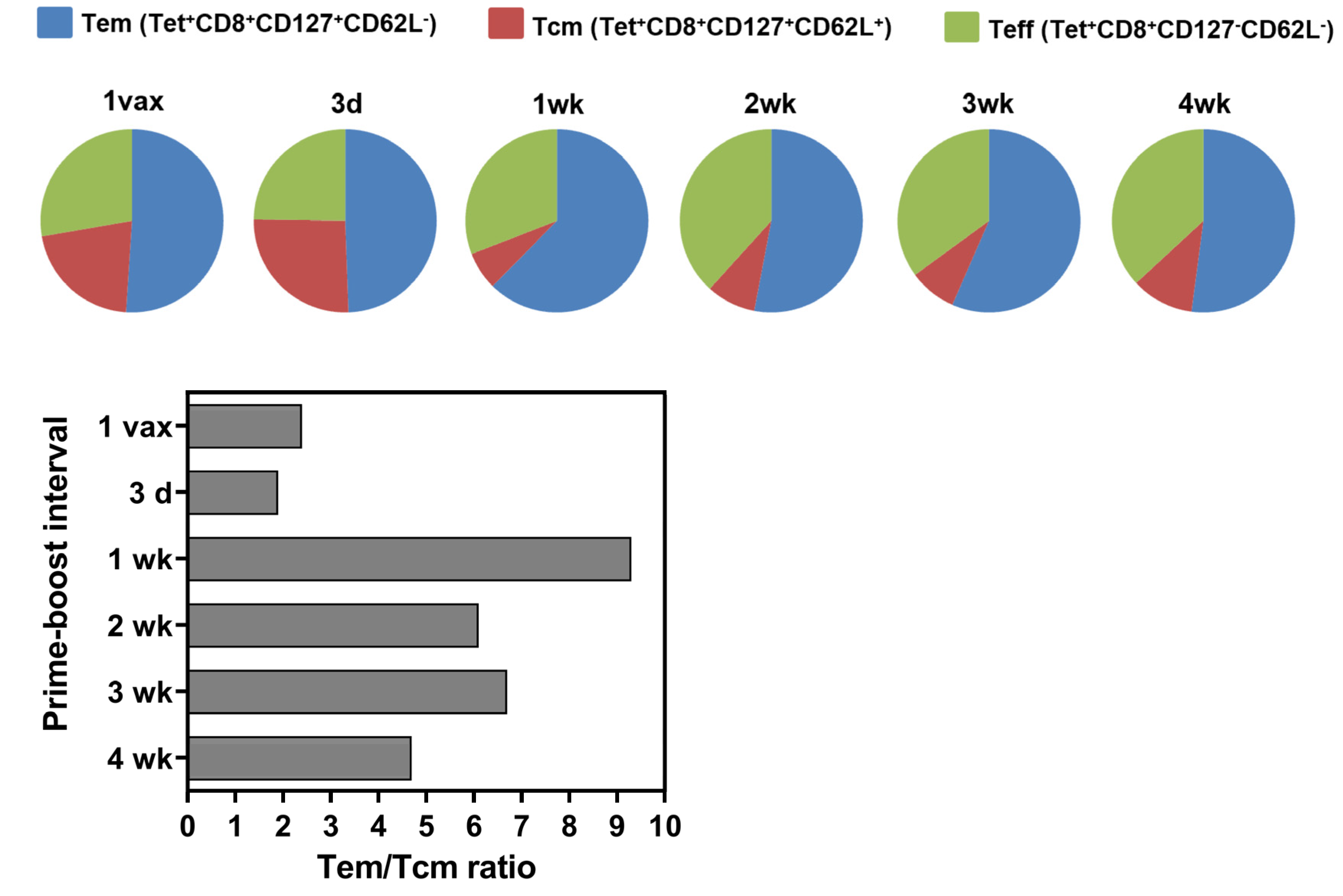
3.3. Longer Prime-Boost Intervals Lead to Enhanced Memory Recall Response to Tumor and Differential Induction of Memory T Cell Phenotypes
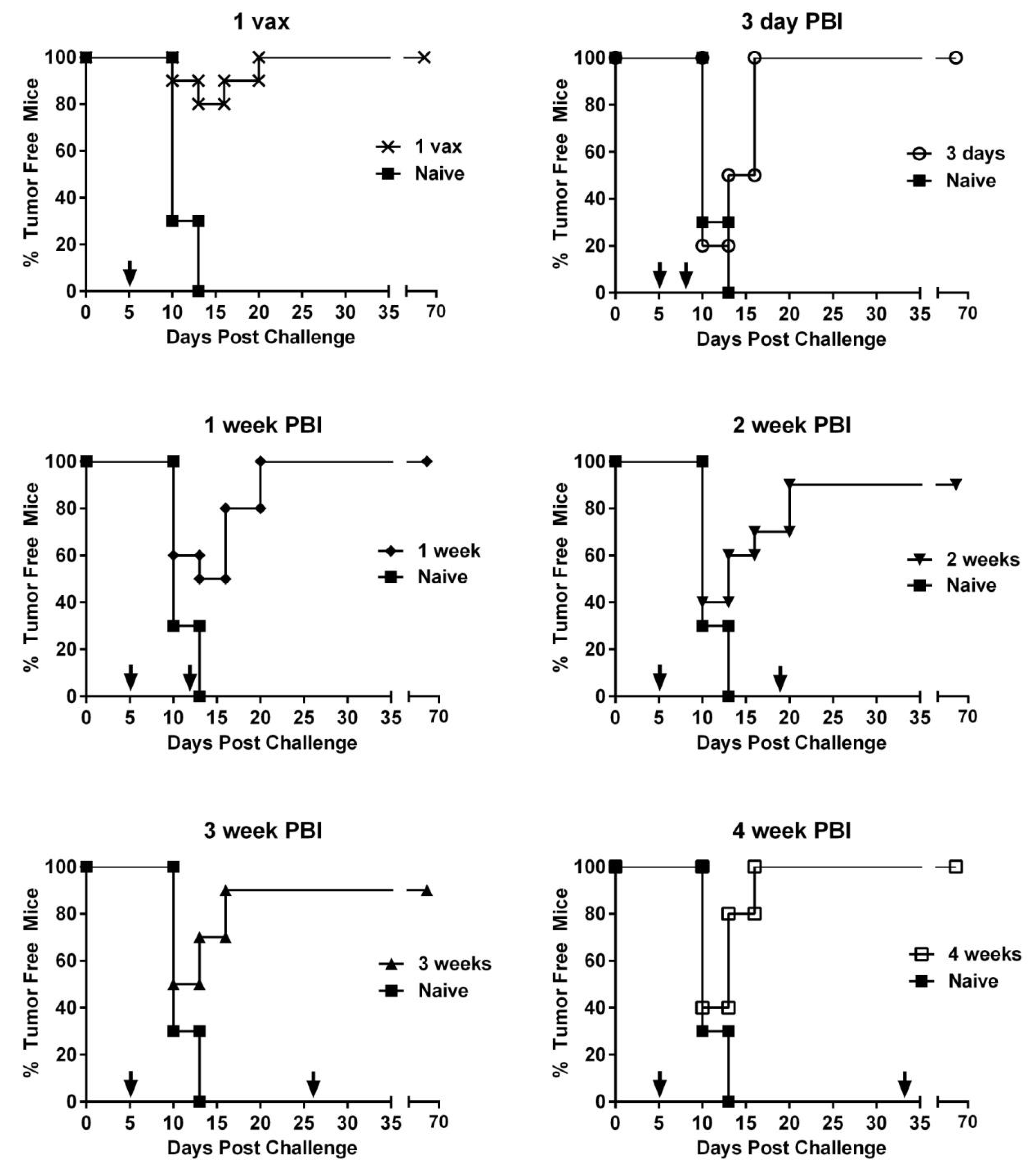
3.4. Effect of PBI Regimens on Anti-Tumor Efficacy Using VRP-Based Vaccines in a Therapeutic Tumor Setting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perret, R.; Ronchese, F. Memory T cells in cancer immunotherapy: Which CD8 T-cell population provides the best protection against tumours? Tissue Antigens 2008, 72, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, Y.; Wherry, E.J. Memory T-Cell Heterogeneity and Terminology. Cold Spring Harb. Perspect. Biol. 2021, 13, a037929. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef]
- Estcourt, M.J.; Letourneau, S.; McMichael, A.J.; Hanke, T. Vaccine route, dose and type of delivery vector determine patterns of primary CD8+ T cell responses. Eur. J. Immunol. 2005, 35, 2532–2540. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Messingham, K.A.; Jabbari, A.; Haring, J.S.; Harty, J.T. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 2005, 11, 748–756. [Google Scholar] [CrossRef]
- Wherry, E.J.; Barber, D.L.; Kaech, S.M.; Blattman, J.N.; Ahmed, R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 16004–16009. [Google Scholar] [CrossRef]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From vaccines to memory and back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef]
- Serrano, B.; Brotons, M.; Bosch, F.X.; Bruni, L. Epidemiology and burden of HPV-related disease. Best Pr. Res Clin. Obs. Gynaecol. 2018, 47, 14–26. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar]
- Smalley Rumfield, C.; Roller, N.; Pellom, S.T.; Schlom, J.; Jochems, C. Therapeutic Vaccines for HPV-Associated Malignancies. Immunotargets Ther. 2020, 9, 167–200. [Google Scholar] [CrossRef]
- von Knebel Doeberitz, M.; Rittmuller, C.; Aengeneyndt, F.; Jansen-Durr, P.; Spitkovsky, D. Reversible repression of papillomavirus oncogene expression in cervical carcinoma cells: Consequences for the phenotype and E6-p53 and E7-pRB interactions. J. Virol. 1994, 68, 2811–2821. [Google Scholar] [CrossRef]
- Feltkamp, M.C.; Smits, H.L.; Vierboom, M.P.; Minnaar, R.P.; de Jongh, B.M.; Drijfhout, J.W.; ter Schegget, J.; Melief, C.J.; Kast, W.M. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol. 1993, 23, 2242–2249. [Google Scholar] [CrossRef]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar]
- Eiben, G.L.; Velders, M.P.; Schreiber, H.; Cassetti, M.C.; Pullen, J.K.; Smith, L.R.; Kast, W.M. Establishment of an HLA-A*0201 human papillomavirus type 16 tumor model to determine the efficacy of vaccination strategies in HLA-A*0201 transgenic mice. Cancer Res. 2002, 62, 5792–5799. [Google Scholar]
- Velders, M.P.; McElhiney, S.; Cassetti, M.C.; Eiben, G.L.; Higgins, T.; Kovacs, G.R.; Elmishad, A.G.; Kast, W.M.; Smith, L.R. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001, 61, 7861–7867. [Google Scholar]
- Smith, K.A.; Meisenburg, B.L.; Tam, V.L.; Pagarigan, R.R.; Wong, R.; Joea, D.K.; Lantzy, L.; Carrillo, M.A.; Gross, T.M.; Malyankar, U.M.; et al. Lymph node-targeted immunotherapy mediates potent immunity resulting in regression of isolated or metastatic human papillomavirus-transformed tumors. Clin. Cancer Res. 2009, 15, 6167–6176. [Google Scholar] [CrossRef]
- Cassetti, M.C.; McElhiney, S.P.; Shahabi, V.; Pullen, J.K.; Le Poole, I.C.; Eiben, G.L.; Smith, L.R.; Kast, W.M. Antitumor efficacy of Venezuelan equine encephalitis virus replicon particles encoding mutated HPV16 E6 and E7 genes. Vaccine 2004, 22, 520–527. [Google Scholar] [CrossRef]
- Garcia-Hernandez Mde, L.; Gray, A.; Hubby, B.; Klinger, O.J.; Kast, W.M. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008, 68, 861–869. [Google Scholar] [CrossRef]
- Feltkamp, M.C.; Vreugdenhil, G.R.; Vierboom, M.P.; Ras, E.; van der Burg, S.H.; ter Schegget, J.; Melief, C.J.; Kast, W.M. Cytotoxic T lymphocytes raised against a subdominant epitope offered as a synthetic peptide eradicate human papillomavirus type 16-induced tumors. Eur. J. Immunol. 1995, 25, 2638–2642. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Krasovsky, J.; Steinman, R.M.; Bhardwaj, N. Mature dendritic cells boost functionally superior CD8(+) T-cell in humans without foreign helper epitopes. J. Clin. Investig. 2000, 105, R9–R14. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Arias, C.; Arias, C.F.; Zhang, M.; Herrero, M.A.; Acosta, F.J.; Tsuji, M. Modeling the effect of boost timing in murine irradiated sporozoite prime-boost vaccines. PLoS ONE 2018, 13, e0190940. [Google Scholar] [CrossRef] [Green Version]
- Pettini, E.; Pastore, G.; Fiorino, F.; Medaglini, D.; Ciabattini, A. Short or Long Interval between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity? Vaccines 2021, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Sallusto, F. Understanding the generation and function of memory T cell subsets. Curr. Opin. Immunol. 2005, 17, 326–332. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Oxenius, A. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J. Immunol. 2005, 175, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef]
- Ricupito, A.; Grioni, M.; Calcinotto, A.; Hess Michelini, R.; Longhi, R.; Mondino, A.; Bellone, M. Booster vaccinations against cancer are critical in prophylactic but detrimental in therapeutic settings. Cancer Res. 2013, 73, 3545–3554. [Google Scholar] [CrossRef]
- Valmori, D.; Scheibenbogen, C.; Dutoit, V.; Nagorsen, D.; Asemissen, A.M.; Rubio-Godoy, V.; Rimoldi, D.; Guillaume, P.; Romero, P.; Schadendorf, D.; et al. Circulating Tumor-reactive CD8(+) T cells in melanoma patients contain a CD45RA(+)CCR7(-) effector subset exerting ex vivo tumor-specific cytolytic activity. Cancer Res. 2002, 62, 1743–1750. [Google Scholar]
- Kanodia, S.; Da Silva, D.M.; Kast, W.M. Recent advances in strategies for immunotherapy of human papillomavirus-induced lesions. Int. J. Cancer 2008, 122, 247–259. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prime-Boost Regimen | Tumor-Free Mice after 60 Days | Significance 2 |
|---|---|---|
| PBI of 4 weeks | 10/10 | p < 0.0001 |
| PBI of 3 weeks | 10/10 | |
| PBI of 2 weeks | 10/10 | |
| PBI of 1 week | 10/10 | |
| PBI of 3 days | 10/10 | |
| 1 vaccination | 10/10 | |
| Naive | 0/10 | Reference |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Silva, D.M.; Martinez, E.A.; Bogaert, L.; Kast, W.M. Investigation of the Optimal Prime Boost Spacing Regimen for a Cancer Therapeutic Vaccine Targeting Human Papillomavirus. Cancers 2022, 14, 4339. https://doi.org/10.3390/cancers14174339
Da Silva DM, Martinez EA, Bogaert L, Kast WM. Investigation of the Optimal Prime Boost Spacing Regimen for a Cancer Therapeutic Vaccine Targeting Human Papillomavirus. Cancers. 2022; 14(17):4339. https://doi.org/10.3390/cancers14174339
Chicago/Turabian StyleDa Silva, Diane M., Emma A. Martinez, Lies Bogaert, and W. Martin Kast. 2022. "Investigation of the Optimal Prime Boost Spacing Regimen for a Cancer Therapeutic Vaccine Targeting Human Papillomavirus" Cancers 14, no. 17: 4339. https://doi.org/10.3390/cancers14174339
APA StyleDa Silva, D. M., Martinez, E. A., Bogaert, L., & Kast, W. M. (2022). Investigation of the Optimal Prime Boost Spacing Regimen for a Cancer Therapeutic Vaccine Targeting Human Papillomavirus. Cancers, 14(17), 4339. https://doi.org/10.3390/cancers14174339