
Deep View of HCC Gene Expression Signatures and Their Comparison with Other Cancers


Abstract
:Simple Summary
Abstract

1. Introduction
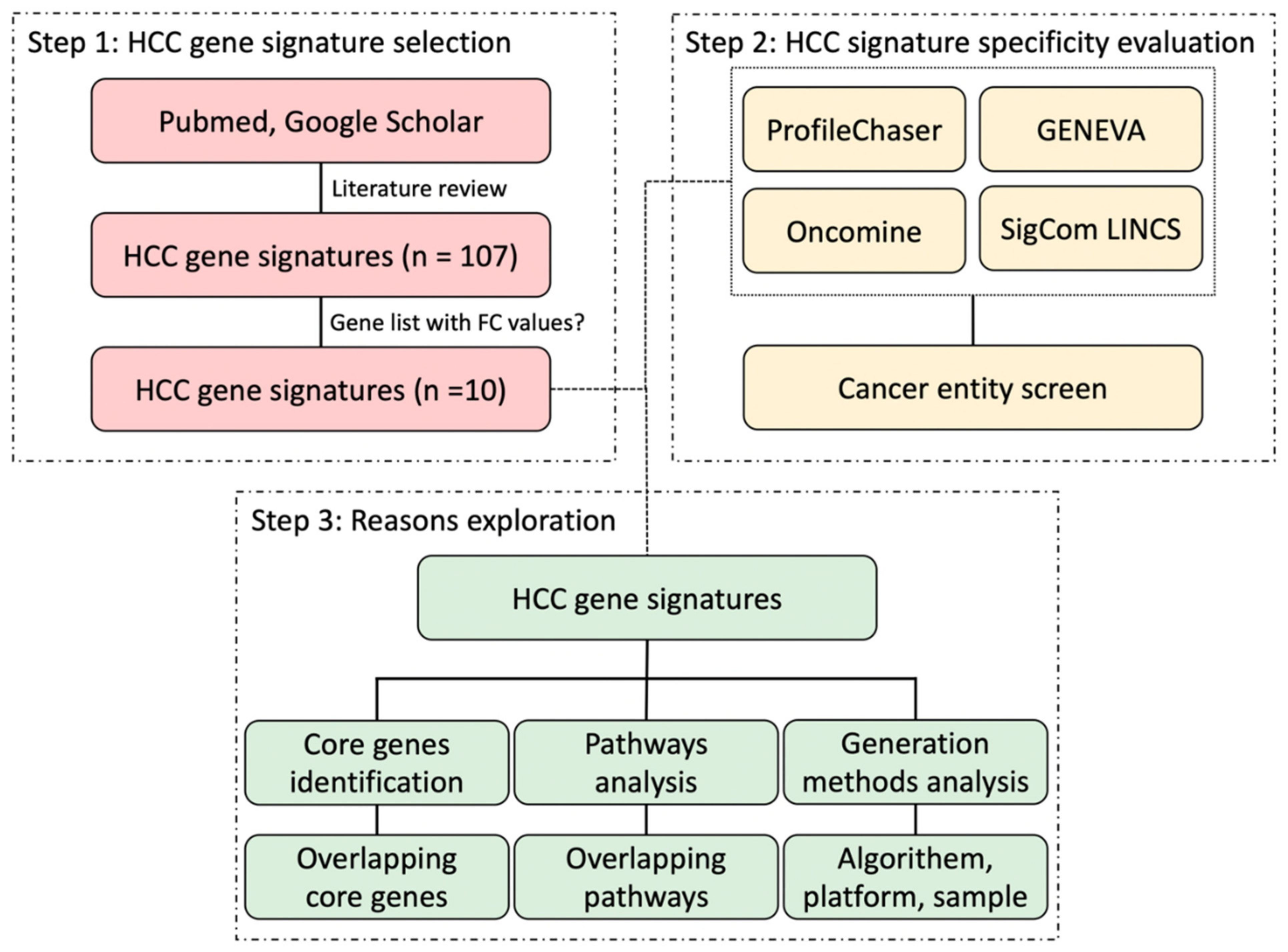
2. Material and Methods
2.1. Selection of HCC Gene Expression Signatures
2.2. ProfileChaser Analysis
2.3. Oncomine Concept Association Analysis
2.4. GENEVA Gene Signature Query
2.5. Sigcom LINCS Gene Signature Search
2.6. Core Genes and Pathways Identification
2.7. Signature Generation Methods
3. Results
3.1. Selected HCC Signatures Based on ProfileChaser
3.2. ProfileChaser and Oncomine Query Results of HCC Signatures
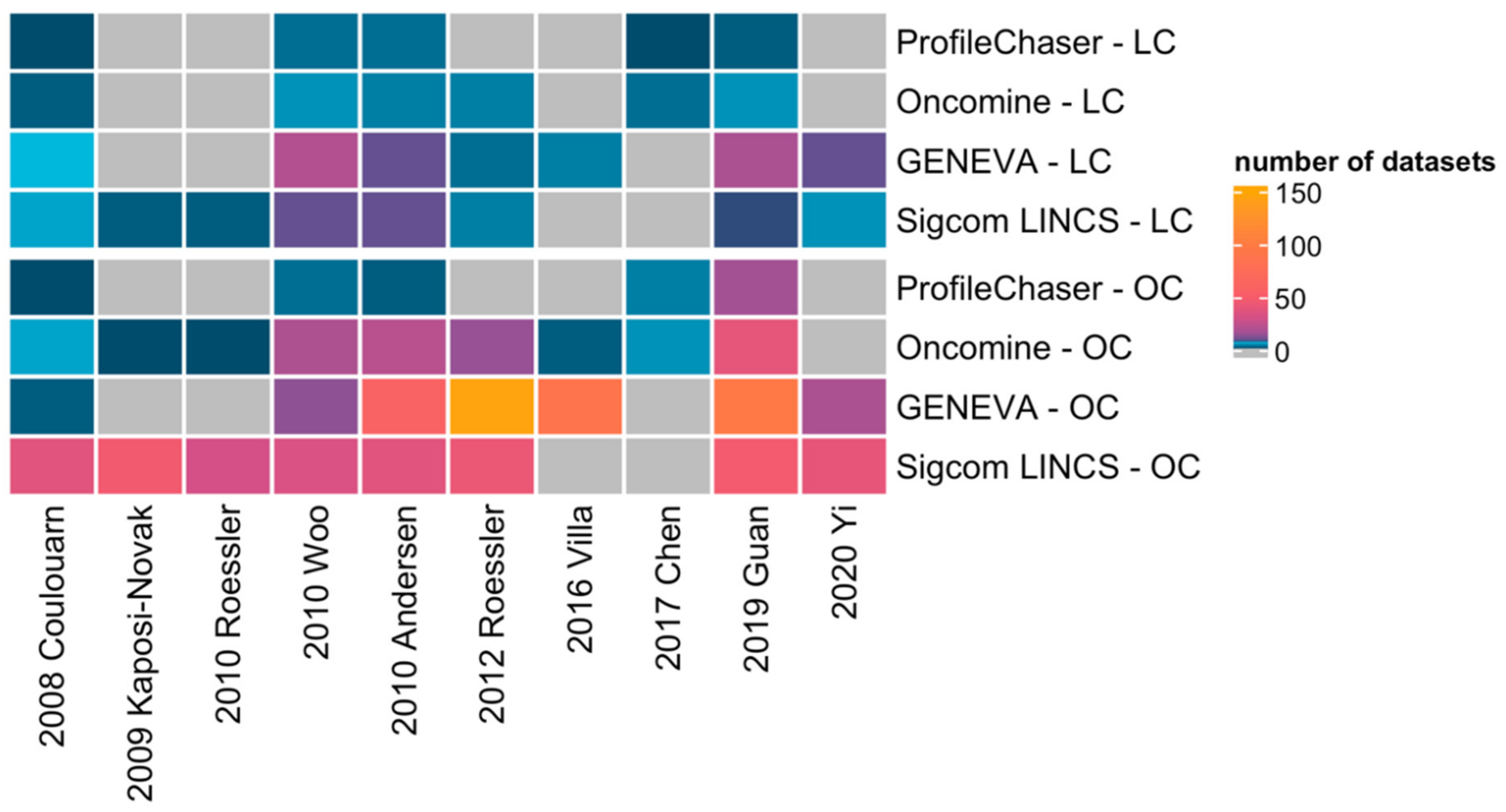
3.3. GENEVA and Sigcom LINCS Query Results of HCC Signatures
3.4. Similarities and Differences of Query Results between the Four Bio-Tools
3.5. Oncomine Results of Breast Cancer and Colorectal Cancer Gene Signatures
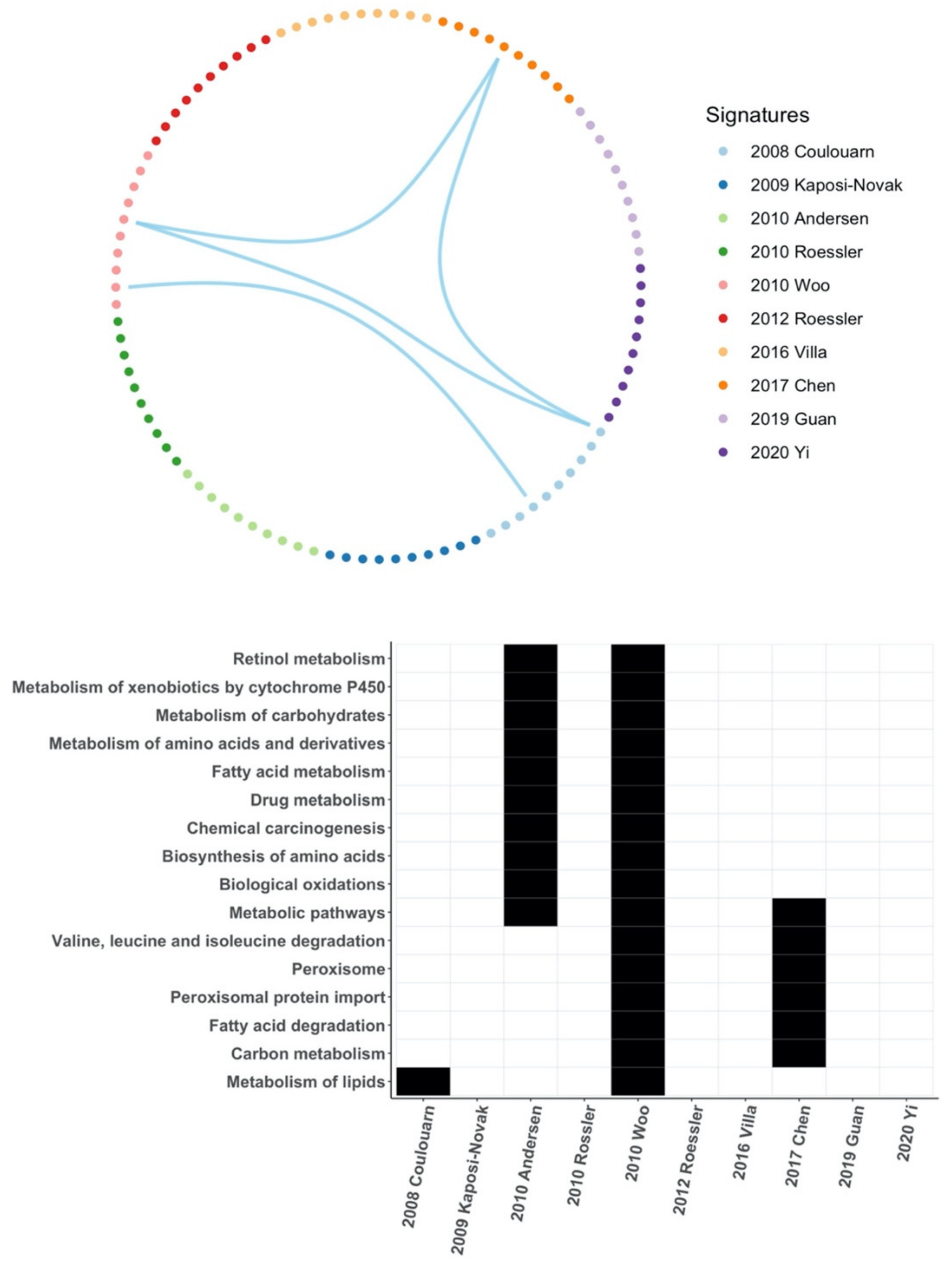
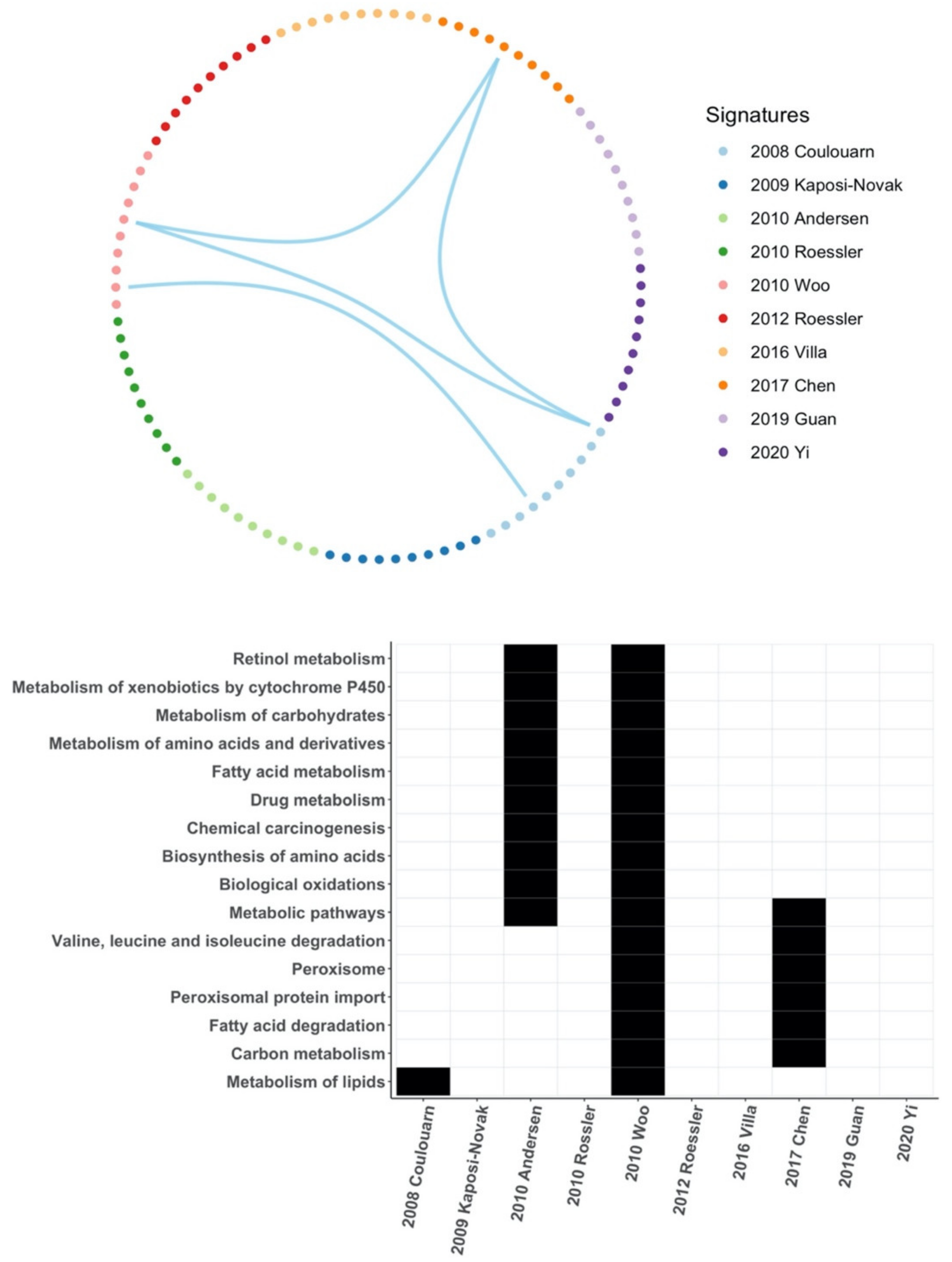
3.6. Common Core Genes and Common Pathways between HCC Gene Signatures
3.7. Signature Generation Differences
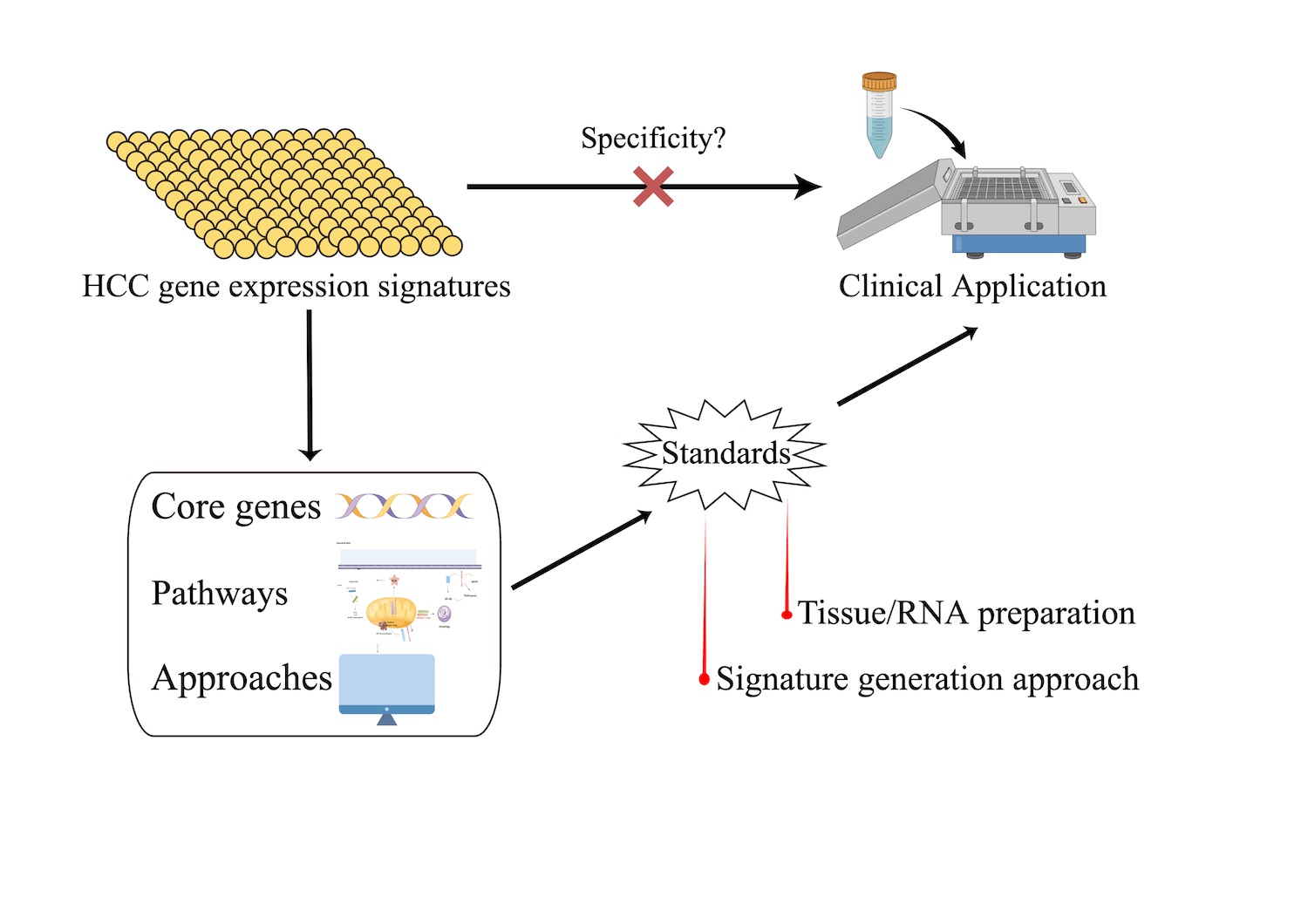
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of Hepatocellular Carcinoma: A Systematic Review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.-C.; Wang, M.-D.; Liu, S.-Y.; Ouyang, W.; Liang, L.; Pawlik, T.M.; Xu, Q.-R.; Huang, D.-S.; Shen, F.; Zhu, H.; et al. Early Diagnosis and Therapeutic Strategies for Hepatocellular Carcinoma: From Bench to Bedside. World J. Gastrointest. Oncol. 2021, 13, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Galle, P.R.; Teufel, A. Molecular Diagnosis and Therapy of Hepatocellular Carcinoma (HCC): An Emerging Field for Advanced Technologies. J. Hepatol. 2012, 56, 267–275. [Google Scholar] [CrossRef]
- Qian, Y.; Daza, J.; Itzel, T.; Betge, J.; Zhan, T.; Marmé, F.; Teufel, A. Prognostic Cancer Gene Expression Signatures: Current Status and Challenges. Cells 2021, 10, 648. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Chu, I.-S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and Prediction of Survival in Hepatocellular Carcinoma by Gene Expression Profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef]
- Nault, J.-C.; De Reyniès, A.; Villanueva, A.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Decaens, T.; Franco, D.; Imbeaud, S.; Rousseau, F.; et al. A Hepatocellular Carcinoma 5-Gene Score Associated with Survival of Patients after Liver Resection. Gastroenterology 2013, 145, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Park, E.S.; Cheon, J.H.; Kim, J.H.; Lee, J.-S.; Park, B.J.; Kim, W.; Park, S.C.; Chung, Y.J.; Kim, B.G.; et al. Gene Expression-Based Recurrence Prediction of Hepatitis B Virus-Related Human Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 2056–2064. [Google Scholar] [CrossRef]
- Roessler, S.; Jia, H.-L.; Budhu, A.; Forgues, M.; Ye, Q.-H.; Lee, J.-S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.-Y.; Qin, L.-X.; et al. A Unique Metastasis Gene Signature Enables Prediction of Tumor Relapse in Early-Stage Hepatocellular Carcinoma Patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Wang, F.; Feng, J.; Zhou, R.; Chang, Y.; Liu, J.; Zhao, Q. Co-Expression Network Analysis Identified Six Hub Genes in Association with Metastasis Risk and Prognosis in Hepatocellular Carcinoma. Oncotarget 2017, 8, 48948–48958. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Chen, R.; Morgan, A.A.; Dudley, J.T.; Mallelwar, R.; Butte, A.J. ProfileChaser: Searching Microarray Repositories Based on Genome-Wide Patterns of Differential Expression. Bioinformatics 2011, 27, 3317–3318. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Kaur, N.; Oskotsky, B.; Butte, A.J.; Hu, Z. Systematic Identification of ACE2 Expression Modulators Reveals Cardiomyopathy as a Risk Factor for Mortality in COVID-19 Patients. Genome Biol. 2022, 23, 15. [Google Scholar] [CrossRef]
- Evangelista, J.E.; Clarke, D.J.B.; Xie, Z.; Lachmann, A.; Jeon, M.; Chen, K.; Jagodnik, K.M.; Jenkins, S.L.; Kuleshov, M.V.; Wojciechowicz, M.L.; et al. SigCom LINCS: Data and Metadata Search Engine for a Million Gene Expression Signatures. Nucleic Acids Res. 2022, 50, W697–W709. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming Growth Factor-Beta Gene Expression Signature in Mouse Hepatocytes Predicts Clinical Outcome in Human Cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Coulouarn, C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central Role of C-Myc during Malignant Conversion in Human Hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef]
- Woo, H.G.; Lee, J.H.; Yoon, J.H.; Kim, C.Y.; Lee, H.S.; Jang, J.J.; Yi, N.J.; Suh, K.S.; Lee, K.U.; Park, E.S.; et al. Identification of a Cholangiocarcinoma-like Gene Expression Trait in Hepatocellular Carcinoma. Cancer Res. 2010, 70, 3034–3041. [Google Scholar] [CrossRef]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-Derived Hepatocellular Carcinoma Model in the Rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef]
- Roessler, S.; Long, E.L.; Budhu, A.; Chen, Y.; Zhao, X.; Ji, J.; Walker, R.; Jia, H.L.; Ye, Q.H.; Qin, L.X.; et al. Integrative Genomic Identification of Genes on 8p Associated with Hepatocellular Carcinoma Progression and Patient Survival. Gastroenterology 2012, 142, 957–966.e12. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Critelli, R.; Lei, B.; Marzocchi, G.; Camma, C.; Giannelli, G.; Pontisso, P.; Cabibbo, G.; Enea, M.; Colopi, S.; et al. Neoangiogenesis-Related Genes Are Hallmarks of Fast-Growing Hepatocellular Carcinomas and Worst Survival. Results from a Prospective Study. Gut 2016, 65, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Luo, Q.; Liang, N.; Liu, H. A Prognostic Prediction System for Hepatocellular Carcinoma Based on Gene Co-Expression Network. Exp. Ther. Med. 2019, 17, 4506–4516. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.; Tang, C.; Tao, Y.; Zhao, Z. Definition of a Novel Vascular Invasion-Associated Multi-Gene Signature for Predicting Survival in Patients with Hepatocellular Carcinoma. Oncol. Lett. 2020, 19, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Van ‘t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene Expression Profiling Predicts Clinical Outcome of Breast Cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Warf, M.B.; Rajamani, S.; Krappmann, K.; Doedt, J.; Cassiano, J.; Brown, K.; Reid, J.E.; Kronenwett, R.; Roa, B.B. Analytical Validation of a 12-Gene Molecular Test for the Prediction of Distant Recurrence in Breast Cancer. Future Sci. OA 2017, 3, FSO221. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Ma, X.J.; Salunga, R.; Dahiya, S.; Wang, W.; Carney, E.; Durbecq, V.; Harris, A.; Goss, P.; Sotiriou, C.; Erlander, M.; et al. A Five-Gene Molecular Grade Index and HOXB13:IL17BR Are Complementary Prognostic Factors in Early Stage Breast Cancer. Clin. Cancer Res. 2008, 14, 2601–2608. [Google Scholar] [CrossRef]
- Sgroi, D.C.; Carney, E.; Zarrella, E.; Steffel, L.; Binns, S.N.; Finkelstein, D.M.; Szymonifka, J.; Bhan, A.K.; Shepherd, L.E.; Zhang, Y.; et al. Prediction of Late Disease Recurrence and Extended Adjuvant Letrozole Benefit by the HOXB13/IL17BR Biomarker. JNCI J. Natl. Cancer Inst. 2013, 105, 1036–1042. [Google Scholar] [CrossRef]
- Clark-Langone, K.M.; Sangli, C.; Krishnakumar, J.; Watson, D. Translating Tumor Biology into Personalized Treatment Planning: Analytical Performance Characteristics of the Oncotype DX Colon Cancer Assay. BMC Cancer 2010, 10, 691. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Tabernero, J.; Rosenberg, R.; Jiang, Z.Q.; Moreno, V.; Bachleitner-Hofmann, T.; Lanza, G.; Stork-Sloots, L.; Maru, D.; Simon, I.; et al. Genomic Classifier ColoPrint Predicts Recurrence in Stage II Colorectal Cancer Patients More Accurately than Clinical Factors. Oncologist 2015, 20, 127–133. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Itzel, T.; Spang, R.; Maass, T.; Munker, S.; Roessler, S.; Ebert, M.P.; Schlitt, H.J.; Herr, W.; Evert, M.; Teufel, A. Random Gene Sets in Predicting Survival of Patients with Hepatocellular Carcinoma. J. Mol. Med. 2019, 97, 879–888. [Google Scholar] [CrossRef]
- Fontana, E.; Eason, K.; Cervantes, A.; Salazar, R.; Sadanandam, A. Context Matters—Consensus Molecular Subtypes of Colorectal Cancer as Biomarkers for Clinical Trials. Ann. Oncol. 2019, 30, 520–527. [Google Scholar] [CrossRef]
- Manjang, K.; Tripathi, S.; Yli-Harja, O.; Dehmer, M.; Glazko, G.; Emmert-Streib, F. Prognostic Gene Expression Signatures of Breast Cancer Are Lacking a Sensible Biological Meaning. Sci. Rep. 2021, 11, 156. [Google Scholar] [CrossRef]
- Polenkowski, M.; Burbano de Lara, S.; Allister, A.B.; Nguyen, T.N.Q.; Tamura, T.; Tran, D.D.H. Identification of Novel Micropeptides Derived from Hepatocellular Carcinoma-Specific Long Noncoding RNA. Int. J. Mol. Sci. 2022, 23, 58. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 Catalog of Human Long Noncoding RNAs: Analysis of Their Gene Structure, Evolution, and Expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L.; Gu, J.; Zhang, H.; Yuan, J.; Lian, Q.; Lv, G.; Wang, S.; Wu, Y.; Yang, Y.-C.T.; et al. Recurrently Deregulated LncRNAs in Hepatocellular Carcinoma. Nat. Commun. 2017, 8, 14421. [Google Scholar] [CrossRef]
- Brazma, A.; Hingamp, P.; Quackenbush, J.; Sherlock, G.; Spellman, P.; Stoeckert, C.; Aach, J.; Ansorge, W.; Ball, C.A.; Causton, H.C.; et al. Minimum Information about a Microarray Experiment (MIAME)-toward Standards for Microarray Data. Nat. Genet. 2001, 29, 365–371. [Google Scholar] [CrossRef] [PubMed]
- FGED Society. MINSEQE. Available online: https://www.fged.org/projects/minseqe (accessed on 19 June 2022).
- FGED Society. MIAME. Available online: https://www.fged.org/projects/miame (accessed on 19 June 2022).
- Opitz, L.; Salinas-Riester, G.; Grade, M.; Jung, K.; Jo, P.; Emons, G.; Ghadimi, B.M.; Beissbarth, T.; Gaedcke, J. Impact of RNA Degradation on Gene Expression Profiling. BMC Med. Genom. 2010, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Ibberson, D.; Benes, V.; Muckenthaler, M.U.; Castoldi, M. RNA Degradation Compromises the Reliability of MicroRNA Expression Profiling. BMC Biotechnol. 2009, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Jun, J.; Park, M.; Park, S.K.; Choi, W.; Park, K.; Jang, K.-T.; Lee, M. Biobanking of Fresh-Frozen Cancer Tissue: RNA Is Stable Independent of Tissue Type with Less Than 1 Hour of Cold Ischemia. Biopreserv. Biobank. 2018, 16, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Florell, S.R.; Coffin, C.M.; Holden, J.A.; Zimmermann, J.W.; Gerwels, J.W.; Summers, B.K.; Jones, D.A.; Leachman, S.A. Preservation of RNA for Functional Genomic Studies: A Multidisciplinary Tumor Bank Protocol. Mod. Pathol. 2001, 14, 116–128. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signature | Clinical Outcome | No. Genes | Comparison |
|---|---|---|---|
| 2008, Coulouarn [16] | Survival | 249 | early vs. late |
| 2009, Kaposi-Novak [17] | Malignant Transformation | 85 | Dysplastic nodules vs. cirrhotic (regenerative) nodules |
| 2010, Roessler [10] | Metastasis, Recurrence | 161 | metastasis vs. metastasis-free |
| 2010, Woo [18] | Survival | 625 | cholangiocarcinoma-like HCC vs. other HCCs |
| 2010, Andersen [19] | Survival | 110 | poor prognosis vs. better prognosis |
| 2012, Roessler [20] | Survival | 10 | good survival vs. poor survival |
| 2016, Villa [21] | Growth, Survival | 5 | fast vs. slow growing tumors |
| 2017, Chen [11] | Metastasis | 6 | HCC tumor tissue vs. non-tumor tissues |
| 2019, Guan [22] | Survival | 55 | good prognostic group vs. poor prognostic group |
| 2020, Yi [23] | Survival | 14 | with vs. without vascular invasion |
| 2008, Coulouarn [16] | 2009, Kaposi-Novak [17] | 2010, Roessler [10] | 2010, Woo [18] | 2010, Andersen [19] | 2012, Roessler [20] | 2016, Villa [21] | 2017, Chen [11] | 2019, Guan [22] | 2020, Yi [23] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | On | Pro | |
| Liver | 2 | 1 | 5 | 3 | 4 | 3 | 4 | 3 | 1 | 5 | 2 | |||||||||
| Lung | 1 | 8 | 2 | |||||||||||||||||
| Colorectal | 1 | 1 | 4 | 1 | 3 | 3 | 1 | |||||||||||||
| Breast | 1 | 1 | 4 | 1 | 2 | 10 | 7 | |||||||||||||
| Kidney | 1 | 3 | 1 | 4 | 1 | 2 | 4 | 4 | 1 | |||||||||||
| Lymphoma | 2 | 3 | 1 | |||||||||||||||||
| Leukemia | 1 | 1 | 1 | |||||||||||||||||
| Sarcoma | 1 | 2 | 1 | 4 | 4 | 1 | ||||||||||||||
| Glioma | 1 | |||||||||||||||||||
| Esophageal | 2 | 1 | 3 | |||||||||||||||||
| Cervical | 1 | 1 | ||||||||||||||||||
| Gastric | 2 | 1 | 3 | |||||||||||||||||
| Head and Neck | 1 | 1 | 1 | |||||||||||||||||
| Ovarian | 1 | 1 | 1 | |||||||||||||||||
| Melanoma | 1 | 1 | ||||||||||||||||||
| Prostate | 1 | 1 | 2 | |||||||||||||||||
| Pancreatic | 1 | 1 | ||||||||||||||||||
| Bladder | 1 | 1 | 3 | |||||||||||||||||
| Other Cancer | 1 | 2 | 1 | 1 | 2 | |||||||||||||||
| 2008, Coulouarn [16] | 2009, Kaposi-Novak [17] | 2010, Roessler [10] | 2010, Woo [18] | 2010, Andersen [19] | 2012, Roessler [20] | 2016, Villa [21] | 2017, Chen [11] | 2019, Guan [22] | 2020, Yi [23] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | GE | Sig | |
| Liver | 7 | 6 | 2 | 2 | 20 | 9 | 9 | 9 | 3 | 4 | 4 | 18 | 8 | 9 | 5 | |||||
| Lung | 2 | 1 | 1 | 2 | 3 | 7 | 3 | 7 | 2 | 3 | 6 | 2 | 3 | 2 | ||||||
| Colorectal | 5 | 2 | 1 | 3 | 4 | 3 | 6 | 3 | 3 | 5 | 3 | 2 | 4 | |||||||
| Breast | 4 | 7 | 7 | 1 | 5 | 8 | 9 | 17 | 11 | 10 | 18 | 8 | 5 | 4 | ||||||
| Kidney | 2 | 1 | 3 | 1 | 2 | 5 | 4 | 1 | 3 | 1 | 2 | |||||||||
| Lymphoma | 1 | 1 | 4 | 6 | 1 | 1 | 1 | 1 | ||||||||||||
| Leukemia | 2 | 5 | 1 | 8 | 1 | 21 | 3 | 6 | 9 | 4 | 2 | 2 | ||||||||
| Sarcoma | 1 | 1 | 2 | 2 | 4 | 16 | 3 | 4 | 5 | 1 | 2 | |||||||||
| Glioma | 1 | 1 | 2 | 2 | 7 | 4 | 4 | 2 | 3 | |||||||||||
| Esophageal | 1 | 1 | 2 | 1 | 1 | 1 | ||||||||||||||
| Cervical | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 3 | ||||||||||||
| Gastric | 2 | 1 | 1 | 2 | 1 | 2 | 1 | 3 | 1 | 1 | ||||||||||
| Head and Neck | 3 | 4 | 5 | 4 | 1 | 2 | 10 | 3 | 3 | 5 | 4 | |||||||||
| Ovarian | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | ||||||||||
| Melanoma | 1 | 2 | 3 | 2 | 1 | 5 | 3 | 8 | 7 | 1 | ||||||||||
| Prostate | 2 | 5 | 1 | 3 | 6 | 1 | 7 | 2 | 7 | 19 | 1 | |||||||||
| Pancreatic | 3 | 1 | 1 | 1 | 4 | 7 | 3 | 1 | 1 | |||||||||||
| Bladder | 2 | 2 | 1 | |||||||||||||||||
| Other Cancer | 8 | 13 | 3 | 5 | 8 | 9 | 6 | 30 | 9 | 27 | 15 | 12 | 4 | 7 | ||||||
| Oncotype DX Breast | MammaPrint | Endopredict | Prosigna/PAM50 | Breast Cancer Index | Oncotype DX Colon | ColoPrint | |
|---|---|---|---|---|---|---|---|
| Bladder | 2 | 2 | 3 | 3 | |||
| Brain and CNS | 4 | 4 | |||||
| Breast | 6 | 12 | 20 | 6 | 4 | ||
| Cervical | 2 | 3 | 3 | 1 | |||
| Colorectal | 9 | 15 | 6 | 4 | |||
| Esophageal | 2 | 3 | 2 | 1 | |||
| Gastric | 2 | 5 | 6 | 3 | 1 | ||
| Head and Neck | 4 | 8 | 12 | ||||
| Kidney | 2 | ||||||
| Leukemia | 1 | 2 | 3 | 2 | |||
| Liver | 3 | 4 | 3 | ||||
| Lung | 7 | 8 | 17 | 9 | |||
| Lymphoma | 2 | 4 | 1 | ||||
| Melanoma | 2 | ||||||
| Other cancer | 1 | 4 | 7 | 3 | |||
| Ovarian | 2 | 3 | 6 | 1 | |||
| Pancreatic | 1 | 2 | 2 | ||||
| Prostate | 2 | ||||||
| Sarcoma | 3 | 9 | 11 | 6 |
| 2008, Coulouarn [16] | 2009, Kaposi-Novak [17] | 2010, Roessler [10] | 2010, Woo [18] | 2010, Andersen [19] | 2012, Roessler [20] | 2016, Villa [21] | 2017, Chen [11] | 2019, Guan [22] | 2020, Yi [23] |
|---|---|---|---|---|---|---|---|---|---|
| SQLE | NUBPL | RAD50 | FGA | RPS3A | SH2D4A | ESM1 | AGXT | STIL | |
| HMGCS1 | CIAO1 | FEN1 | C8A | RPS18 | CCDC25 | DLL4 | DAO | RAD51AP1 | |
| SREBF2 | ISCA2 | RPA2 | CPB2 | RPL27A | SORBS3 | ANGPT2 | EHHADH | CDC20 | |
| MSMO1 | RFC5 | F11 | RPL3 | PROSC | ABAT | CEP55 | |||
| CYP51A1 | GTF2H1 | SERPINA10 | RPS25 | ALDH6A1 | POLE2 | ||||
| IDI1 | CHEK1 | FETUB | RPS12 | SPC24 | |||||
| FDPS | GTF2H4 | HRG | RPS17 | CCNB1 | |||||
| DHCR24 | F13B | RPS14 | KIF20A | ||||||
| LDLR | FTCD | RPL13A | CDCA3 | ||||||
| DHCR7 | SPP2 | RPL9 | CDT1 | ||||||
| RPL35A | |||||||||
| CCT2 | |||||||||
| RPL10A |
| Signature | Algorithm for Signature Generation | Platform | Origin of Samples |
|---|---|---|---|
| 2008, Coulouarn [16] | Differential gene expression | Custom NCI array | America, Asia, Europe |
| 2009, Kaposi-Novak [17] | Differential gene expression | Custom NCI array | Europe |
| 2010, Roessler [10] | Cox regression | Affymetrix HG-U133A, Custom NCI array | America, Asia, Europe |
| 2010, Woo [18] | Differential gene expression | Affymetrix HG-U133A | Asia |
| 2010, Andersen [19] | External signature | Illumina humanRef-8 | America, Asia, Europe |
| 2012, Roessler [20] | Unsupervised hierarchical clustering | Affymetrix HG-U133A, Agilent-014698 Human Genome CGH Microarray 105A | America, Asia |
| .2015, Villa [21] | Cox regression | Agilent-014850 Whole Human Genome Microarray 4 × 44K G4112F | Europe |
| 2017, Chen [10] | gene co-expression network analysis | Affymetrix HG-U133A, Affymetrix HG-U133_Plus_2, IlluminiaHiseq | America, Asia, Europe |
| 2019, Guan [22] | Cox regression | Illumina HumanHT-12 V4.0 | Asia |
| 2020, Yi [23] | Cox regression | Illumina Hiseq | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; Itzel, T.; Ebert, M.; Teufel, A. Deep View of HCC Gene Expression Signatures and Their Comparison with Other Cancers. Cancers 2022, 14, 4322. https://doi.org/10.3390/cancers14174322
Qian Y, Itzel T, Ebert M, Teufel A. Deep View of HCC Gene Expression Signatures and Their Comparison with Other Cancers. Cancers. 2022; 14(17):4322. https://doi.org/10.3390/cancers14174322
Chicago/Turabian StyleQian, Yuquan, Timo Itzel, Matthias Ebert, and Andreas Teufel. 2022. "Deep View of HCC Gene Expression Signatures and Their Comparison with Other Cancers" Cancers 14, no. 17: 4322. https://doi.org/10.3390/cancers14174322
APA StyleQian, Y., Itzel, T., Ebert, M., & Teufel, A. (2022). Deep View of HCC Gene Expression Signatures and Their Comparison with Other Cancers. Cancers, 14(17), 4322. https://doi.org/10.3390/cancers14174322