
Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment


Abstract
:Simple Summary
Abstract
1. Introduction
2. Lipid Metabolism Is Regulated by SREBP and Interacts with AR Signaling in Prostate Cancer
3. Lipidomic Changes during PCa Progression and Castration Resistance
4. Lipidome as Biomarker in Prostate Cancer
5. Cholesterol Metabolism in PCa
6. Impact of Inhibition of the Cholesterol-Producing Mevalonate Pathway with Statins
7. Fatty Acid Metabolism in PCa
8. Changes in the Lipidome and Cholesterol Metabolism in Cancer-Associated Immune Cells
9. Pharmacological Interventions Targeting Lipid Metabolism Affect Immune Cells
10. Periprostatic Adipose Tissue
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, A.; Klein, E. Risk factors for prostate cancer. Nat. Rev. Urol. 2009, 6, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, B.; He, M.; Wang, Y.; Wang, Z.; Du, L. Prostate Cancer Incidence and Mortality: Global Status and Temporal Trends in 89 Countries From 2000 to 2019. Front. Public Health 2022, 10, 811044. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar]
- Swinnen, J.V.; Esquenet, M.; Goossens, K.; Heyns, W.; Verhoeven, G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res. 1997, 57, 1086–1090. [Google Scholar]
- Škara, L.; Huđek Turković, A.; Pezelj, I.; Vrtarić, A.; Sinčić, N.; Krušlin, B.; Ulamec, M. Prostate cancer-Focus on cholesterol. Cancers 2021, 13, 4696. [Google Scholar] [CrossRef]
- Galbraith, L.; Leung, H.Y.; Ahmad, I. Lipid pathway deregulation in advanced prostate cancer. Pharmacol. Res. 2018, 131, 177–184. [Google Scholar] [CrossRef]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar]
- Roberts, R.O.; Bergstralh, E.J.; Bass, S.E.; Lieber, M.M.; Jacobsen, S.J. Prostatitis as a Risk Factor for Prostate Cancer. Epidemiology 2004, 15, 93–99. [Google Scholar] [CrossRef]
- Javier-DesLoges, J.; McKay, R.R.; Swafford, A.D.; Sepich-Poore, G.D.; Knight, R.; Parsons, J.K. The microbiome and prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Vareki, S.M. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Reimers, M.A.; Knoche, E.M.; Pachynski, R.K. Immunotherapy and Immunotherapy Combinations in Metastatic Castration-Resistant Prostate Cancer. Cancers 2021, 13, 334. [Google Scholar] [CrossRef] [PubMed]
- Lounis, M.A.; Péant, B.; Leclerc-Desaulniers, K.; Ganguli, D.; Daneault, C.; Ruiz, M.; Zoubeidi, A.; Mes-Masson, A.M.; Saad, F. Modulation of de novo lipogenesis improves response to enzalutamide treatment in prostate cancer. Cancers 2020, 12, 3339. [Google Scholar] [CrossRef]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Eid, W.; Dauner, K.; Courtney, K.C.; Gagnon, A.; Parks, R.J.; Sorisky, A.; Zha, X. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. USA 2017, 114, 7999–8004. [Google Scholar] [CrossRef]
- Lawler, J.F., Jr.; Yin, M.; Diehl, A.M.; Roberts, E.; Chatterjee, S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J. Biol. Chem. 1998, 273, 5053–5059. [Google Scholar] [CrossRef]
- Natesan, V.; Kim, S.J. Lipid Metabolism, Disorders and Therapeutic Drugs—Review. Biomol. Ther. 2021, 29, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.R.; Kristiana, I.; Brown, A.J. Cholesterol homeostasis in two commonly used human prostate cancer cell-lines, LNCaP and PC-3. PLoS ONE 2009, 4, e8496. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.B.; Chung, L.W.; Huang, W.C. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6, 41018–41032. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640, Erratum in Proc. Natl. Acad. Sci. USA 2020, 117, 18893. [Google Scholar] [CrossRef]
- Segawa, T.; Nau, M.E.; Xu, L.L.; Chilukuri, R.N.; Makarem, M.; Zhang, W.; Petrovics, G.; Sesterhenn, I.A.; McLeod, D.G.; Moul, J.W.; et al. Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene 2002, 21, 8749–8758. [Google Scholar] [CrossRef]
- Burch, T.C.; Isaac, G.; Booher, C.L.; Rhim, J.S.; Rainville, P.; Langridge, J.; Baker, A.; Nyalwidhe, J.O. Comparative metabolomic and lipidomic analysis of phenotype stratified prostate cells. PLoS ONE 2015, 10, e0134206. [Google Scholar] [CrossRef]
- Ingram, L.M.; Finnerty, M.C.; Mansoura, M.; Chou, C.W.; Cummings, B.S. Identification of lipidomic profiles associated with drug-resistant prostate cancer cells. Lipids Health Dis. 2021, 20, 15. [Google Scholar] [CrossRef]
- Li, J.; Ren, S.; Piao, H.L.; Wang, F.; Yin, P.; Xu, C.; Lu, X.; Ye, G.; Shao, Y.; Yan, M.; et al. Integration of lipidomics and transcriptomics unravels aberrant lipid metabolism and defines cholesteryl oleate as potential biomarker of prostate cancer. Sci. Rep. 2016, 6, 20984. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Mah, C.Y.; Machiels, J.; Vincent, A.D.; Irani, S.; Mutuku, S.M.; Spotbeen, X.; Bagadi, M.; Waltregny, D.; Moldovan, M.; et al. Lipidomic profiling of clinical prostate cancer reveals targetable alterations in membrane lipid composition. Cancer Res. 2021, 81, 4981–4993. [Google Scholar] [CrossRef] [PubMed]
- Filella, X.; Alcover, J.; Zarco, M.A.; Beardo, P.; Molina, R.; Ballesta, A.M. Analysis of type T1 and T2 cytokines in patients with prostate cancer. Prostate 2000, 44, 271–274. [Google Scholar] [CrossRef]
- Lin, H.M.; Yeung, N.; Hastings, J.F.; Croucher, D.R.; Huynh, K.; Meikle, T.G.; Mellett, N.A.; Kwan, E.M.; Davis, I.D.; Tran, B.; et al. Relationship between circulating lipids and cytokines in metastatic castration-resistant prostate cancer. Cancers 2021, 13, 4964. [Google Scholar] [CrossRef]
- Zhou, X.; Mao, J.; Ai, J.; Deng, Y.; Roth, M.R.; Pound, C.; Henegar, J.; Welti, R.; Bigler, S.A. Identification of plasma lipid biomarkers for prostate cancer by lipidomics and bioinformatics. PLoS ONE 2012, 7, e48889. [Google Scholar] [CrossRef]
- Duscharla, D.; Bhumireddy, S.R.; Lakshetti, S.; Pospisil, H.; Murthy, P.V.; Walther, R.; Sripadi, P.; Ummanni, R. Prostate cancer associated lipid signatures in serum studied by ESI-tandem mass spectrometry as potential new biomarkers. PLoS ONE 2016, 11, e0150253. [Google Scholar] [CrossRef]
- Li, X.; Nakayama, K.; Goto, T.; Kimura, H.; Akamatsu, S.; Hayashi, Y.; Fujita, K.; Kobayashi, T.; Shimizu, K.; Nonomura, N.; et al. High level of phosphatidylcholines/lysophosphatidylcholine ratio in urine is associated with prostate cancer. Cancer Sci. 2021, 112, 4292–4302. [Google Scholar] [CrossRef]
- Lin, H.M.; Mahon, K.L.; Weir, J.M.; Mundra, P.A.; Spielman, C.; Briscoe, K.; Gurney, H.; Mallesara, G.; Marx, G.; Stockler, M.R.; et al. A distinct plasma lipid signature associated with poor prognosis in castration-resistant prostate cancer. Int. J. Cancer 2017, 141, 2112–2120. [Google Scholar] [CrossRef]
- Lin, H.M.; Huynh, K.; Kohli, M.; Tan, W.; Azad, A.A.; Yeung, N.; Mahon, K.L.; Mak, B.; Sutherland, P.D.; Shepherd, A.; et al. Aberrations in circulating ceramide levels are associated with poor clinical outcomes across localised and metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 860–870. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, Y.; Jijiwa, M.; Nasu, M.; Ai, J.; Dai, S.; Jiang, B.; Zhang, J.; Huang, G.; Deng, Y. Identification of plasma lipid species as promising diagnostic markers for prostate cancer. BMC Med. Inform. Decis. Mak. 2020, 20 (Suppl. 9), 223. [Google Scholar] [CrossRef]
- Snider, A.J.; Seeds, M.C.; Johnstone, L.; Snider, J.M.; Hallmark, B.; Dutta, R.; Moraga Franco, C.; Parks, J.S.; Bensen, J.T.; Broeckling, C.D.; et al. Identification of plasma glycosphingolipids as potential biomarkers for prostate cancer (PCa) status. Biomolecules 2020, 10, 1393. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Solomon, K.R. Cholesterol and benign prostate disease. Differentiation 2011, 82, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtola, T.J.; Syvälä, H.; Pennanen, P.; Bläuer, M.; Solakivi, T.; Ylikomi, T.; Tammela, T.L. The importance of LDL and cholesterol metabolism for prostate epithelial cell growth. PLoS ONE 2012, 7, e39445. [Google Scholar] [CrossRef]
- Murtola, T.J.; Syvälä, H.; Pennanen, P.; Bläuer, M.; Solakivi, T.; Ylikomi, T.; Tammela, T.L. Comparative effects of high and low-dose simvastatin on prostate epithelial cells: The role of LDL. Eur. J. Pharmacol. 2011, 673, 96–100. [Google Scholar] [CrossRef]
- Hoque, A.; Chen, H.; Xu, X.C. Statin induces apoptosis and cell growth arrest in prostate cancer cells. Cancer Epidemiol. Biomark. Prev. 2008, 17, 88–94. [Google Scholar] [CrossRef]
- Murtola, T.J.; Pennanen, P.; Syvälä, H.; Bläuer, M.; Ylikomi, T.; Tammela, T.L. Effects of simvastatin, acetylsalicylic acid, and rosiglitazone on proliferation of normal and cancerous prostate epithelial cells at therapeutic concentrations. Prostate 2009, 69, 1017–1023. [Google Scholar] [CrossRef]
- Kochuparambil, S.T.; Al-Husein, B.; Goc, A.; Soliman, S.; Somanath, P.R. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J. Pharmacol. Exp. Ther. 2011, 336, 496–505. [Google Scholar] [CrossRef]
- Longo, J.; Mullen, P.J.; Yu, R.; van Leeuwen, J.E.; Masoomian, M.; Woon, D.T.S.; Wang, Y.; Chen, E.X.; Hamilton, R.J.; Sweet, J.M.; et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol. Metab. 2019, 25, 119–130. [Google Scholar] [CrossRef]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Zhang, Y.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal 2020, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Camacho, L.; Zabala-Letona, A.; Torrano, V.; Fernández-Ruiz, S.; Zamacola-Bascaran, K.; Arreal, L.; Valcárcel-Jiménez, L.; Martín-Martín, N.; Flores, J.M.; et al. Low-dose statin treatment increases prostate cancer aggressiveness. Oncotarget 2017, 9, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, S.; Su, S.; Yeon, A.; Lo, E.M.; Kim, S.; Mohler, J.L.; Freeman, M.R.; Kim, H.L. Cholesterol-lowering intervention decreases mTOR complex 2 signaling and enhances antitumor immunity. Clin. Cancer Res. 2022, 28, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Lin, S.C.; Lee, Y.C.; Yu, G.; Song, J.H.; Pan, J.; Titus, M.; Satcher, R.L.; Panaretakis, T.; Logothetis, C.; et al. Statins reduce castration-induced bone marrow adiposity and prostate cancer progression in bone. Oncogene 2021, 40, 4592–4603. [Google Scholar] [CrossRef]
- Murtola, T.J.; Syvälä, H.; Tolonen, T.; Helminen, M.; Riikonen, J.; Koskimäki, J.; Pakarainen, T.; Kaipia, A.; Isotalo, T.; Kujala, P.; et al. Atorvastatin versus placebo for prostate cancer before radical prostatectomy-A randomized, double-blind, placebo-controlled clinical trial. Eur. Urol. 2018, 74, 697–701. [Google Scholar] [CrossRef]
- Knuuttila, E.; Riikonen, J.; Syvälä, H.; Auriola, S.; Murtola, T.J. Access and concentrations of atorvastatin in the prostate in men with prostate cancer. Prostate 2019, 79, 1412–1419. [Google Scholar] [CrossRef]
- Raittinen, P.V.H.; Syvälä, H.; Tammela, T.L.J.; Häkkinen, M.R.; Ilmonen, P.; Auriola, S.; Murtola, T.J. Atorvastatin induces adrenal androgen downshift in men with prostate cancer: A post Hoc analysis of a pilot adaptive randomised clinical trial. EBioMedicine 2021, 68, 103432. [Google Scholar] [CrossRef]
- Raittinen, P.; Niemistö, K.; Pennanen, E.; Syvälä, H.; Auriola, S.; Riikonen, J.; Lehtimäki, T.; Ilmonen, P.; Murtola, T. Circulatory and prostatic tissue lipidomic profiles shifts after high-dose atorvastatin use in men with prostate cancer. Sci. Rep. 2020, 10, 12016. [Google Scholar] [CrossRef]
- Enwald, M.; Lehtimäki, T.; Mishra, P.P.; Mononen, N.; Murtola, T.J.; Raitoharju, E. Human prostate tissue microRNAs and their predicted target pathways linked to prostate cancer risk factors. Cancers 2021, 13, 3537. [Google Scholar] [CrossRef]
- Jeong, I.G.; Lim, B.; Yun, S.C.; Lim, J.H.; Hong, J.H.; Kim, C.S. Adjuvant low-dose statin use after radical prostatectomy: The PRO-STAT randomized clinical trial. Clin. Cancer Res. 2021, 27, 5004–5011. [Google Scholar] [CrossRef]
- Siltari, A.; Riikonen, J.; Koskimäki, J.; Pakarainen, T.; Ettala, O.; Boström, P.; Seikkula, H.; Kotsar, A.; Tammela, T.; Helminen, M.; et al. Randomised double-blind phase 3 clinical study testing impact of atorvastatin on prostate cancer progression after initiation of androgen deprivation therapy: Study protocol. BMJ Open. 2022, 12, e050264. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, L.; Xie, T.; Li, J.; Zhai, T.S.; Xu, Y. Identification and validation of a prognostic signature for prostate cancer based on ferroptosis-related genes. Front. Oncol. 2021, 11, 623313. [Google Scholar] [CrossRef] [PubMed]
- Tousignant, K.D.; Rockstroh, A.; Poad, B.L.J.; Talebi, A.; Young, R.S.E.; Taherian Fard, A.; Gupta, R.; Zang, T.; Wang, C.; Lehman, M.L.; et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 2020, 8, 11. [Google Scholar] [CrossRef]
- Bordini, J.; Morisi, F.; Elia, A.R.; Santambrogio, P.; Pagani, A.; Cucchiara, V.; Ghia, P.; Bellone, M.; Briganti, A.; Camaschella, C.; et al. Iron induces cell death and strengthens the efficacy of antiandrogen therapy in prostate cancer models. Clin. Cancer Res. 2020, 26, 6387–6398. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, T.; Hu, C.; Xia, H.; Liu, W.; Chen, J.; Wu, S.; Jiang, Y.; Xu, Y.; Liu, W.; et al. Ferroptosis inducer erastin downregulates androgen receptor and its splice variants in castration resistant prostate cancer. Oncol. Rep. 2021, 45, 25. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Kumar, R.; Mendonca, J.; Owoyemi, O.; Boyapati, K.; Thomas, N.; Kanacharoen, S.; Coffey, M.; Topiwala, D.; Gomes, C.; Ozbek, B.; et al. Supraphysiologic testosterone induces ferroptosis and activates immune pathways through nucleophagy in prostate cancer. Cancer Res. 2021, 81, 5948–5962. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hsu, E.C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Tousignant, K.D.; Rockstroh, A.; Taherian Fard, A.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid uptake is an androgen-enhanced lipid supply pathway associated with prostate cancer disease progression and bone metastasis. Mol. Cancer Res. 2019, 17, 1166–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramhecha, Y.M.; Guérard, K.P.; Audet-Walsh, É.; Rouzbeh, S.; Kassem, O.; Pernet, E.; Scarlata, E.; Hamel, L.; Brimo, F.; Divangahi, M.; et al. Fatty acid oxidation enzyme Δ3, Δ2-enoyl-CoA isomerase 1 (ECI1) drives aggressive tumor phenotype and predicts poor clinical outcome in prostate cancer patients. Oncogene 2022, 41, 2798–2810. [Google Scholar] [CrossRef] [PubMed]
- Valentino, A.; Calarco, A.; Di Salle, A.; Finicelli, M.; Crispi, S.; Calogero, R.A.; Riccardo, F.; Sciarra, A.; Gentilucci, A.; Galderisi, U.; et al. Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: Molecular basis and pathophysiological consequences. Oncogene 2017, 36, 6030–6040. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Høiem, T.S.; Claes, B.S.R.; Balluff, B.; Martin-Lorenzo, M.; Richardsen, E.; Krossa, S.; Bertilsson, H.; Heeren, R.M.A.; Rye, M.B.; et al. Spatial differentiation of metabolism in prostate cancer tissue by MALDI-TOF MSI. Cancer Metab. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef]
- Zadra, G.; Loda, M. Metabolic vulnerabilities of prostate cancer: Diagnostic and therapeutic opportunities. Cold Spring Harb. Perspect. Med. 2018, 8, a030569. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife 2020, 9, e54166. [Google Scholar] [CrossRef] [PubMed]
- Blomme, A.; Ford, C.A.; Mui, E.; Patel, R.; Ntala, C.; Jamieson, L.E.; Planque, M.; McGregor, G.H.; Peixoto, P.; Hervouet, E.; et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat. Commun. 2020, 11, 2508. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A.; Wei, J.; Nguyen, T.M.; Shi, H.; Su, W.; Palacios, G.; Dhungana, Y.; Chapman, N.M.; Long, L.; Saravia, J.; et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8+ T cell-serived interferon-γ. Immunity 2019, 51, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jing, J.; Xu, H.; Xu, L.; Hu, H.; Tang, C.; Liu, S.; Wei, Q.; Duan, R.; Guo, J.; et al. N-cadherin inhibitor creates a microenvironment that protect TILs from immune checkpoints and Treg cells. J. Immunother Cancer 2021, 9, e002138. [Google Scholar] [CrossRef]
- Guan, X.; Polesso, F.; Wang, C.; Sehrawat, A.; Hawkins, R.M.; Murray, S.E.; Thomas, G.V.; Caruso, B.; Thompson, R.F.; Wood, M.A.; et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 2022, 606, 791–796. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019, 30, 143–156. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Huang, C.; Lu, Y.; Xue, G.; Guo, X.; Wang, A.; Yang, M.; Qian, J.; Dong, C.; et al. Cholesterol negatively regulates IL-9-producing CD8+ T cell differentiation and antitumor activity. J. Exp. Med. 2018, 215, 1555–1569. [Google Scholar] [CrossRef]
- Lei, K.; Kurum, A.; Kaynak, M.; Bonati, L.; Han, Y.; Cencen, V.; Gao, M.; Xie, Y.Q.; Guo, Y.; Hannebelle, M.T.M.; et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat. Biomed. Eng. 2021, 5, 1411–1425. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Schmidt, N.M.; Wing, P.A.C.; Diniz, M.O.; Pallett, L.J.; Swadling, L.; Harris, J.M.; Burton, A.R.; Jeffery-Smith, A.; Zakeri, N.; Amin, O.E.; et al. Targeting human Acyl-CoA: Cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat. Commun. 2021, 12, 2814. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ding, H.; Jia, Y.; Shi, M.; Guo, D.; Yang, P.; Wang, Y.; Liu, F.; Zhang, Y.; Zhu, Z. Associations of genetically proxied inhibition of HMG-CoA reductase, NPC1L1, and PCSK9 with breast cancer and prostate cancer. Breast Cancer Res. 2022, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- De Angulo, A.; Travis, P.; Galvan, G.C.; Jolly, C.; de Graffenried, L. Obesity-modified CD4+ T-cells promote an epithelial-mesenchymal transition phenotype in prostate cancer cells. Nutr. Cancer 2022, 74, 650–659. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Dominguez-Viqueira, W.; Liu, M.; Awasthi, S.; Abraham-Miranda, J.; Keske, A.; Steiner, K.K.; Noel, L.; Serna, A.N.; Dhillon, J.; et al. Macrophage-derived cholesterol contributes to therapeutic resistance in prostate cancer. Cancer Res. 2021, 81, 5477–5490. [Google Scholar] [CrossRef]
- Sag, D.; Cekic, C.; Wu, R.; Linden, J.; Hedrick, C.C. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat. Commun. 2015, 6, 6354. [Google Scholar] [CrossRef]
- Lee, M.K.; Moore, X.L.; Fu, Y.; Al-Sharea, A.; Dragoljevic, D.; Fernandez-Rojo, M.A.; Parton, R.; Sviridov, D.; Murphy, A.J.; Chin-Dusting, J.P. High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br. J. Pharmacol. 2016, 173, 741–751. [Google Scholar] [CrossRef]
- Masetti, M.; Carriero, R.; Portale, F.; Marelli, G.; Morina, N.; Pandini, M.; Iovino, M.; Partini, B.; Erreni, M.; Ponzetta, A.; et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 2022, 219, e20210564. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-fat diet-induced inflammation accelerates prostate cancer Ggowth via IL6 signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef]
- Gevariya, N.; Besançon, M.; Robitaille, K.; Picard, V.; Diabaté, L.; Alesawi, A.; Julien, P.; Fradet, Y.; Bergeron, A.; Fradet, V. Omega-3 fatty acids decrease prostate cancer progression associated with an anti-tumor immune response in eugonadal and castrated mice. Prostate 2019, 79, 9–20. [Google Scholar] [CrossRef]
- Allott, E.H.; Howard, L.E.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freedland, S.J. Statin use, serum lipids, and prostate inflammation in men with a negative prostate biopsy: Results from the REDUCE Trial. Cancer Prev. Res. 2017, 10, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Lucia, M.S.; Thompson, I.M., Jr.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856. [Google Scholar] [CrossRef]
- Murtola, T.J.; Gurel, B.; Umbehr, M.; Lucia, M.S.; Thompson, I.M., Jr.; Goodman, P.J.; Kristal, A.R.; Parnes, H.L.; Lippman, S.M.; Sutcliffe, S.; et al. Inflammation in benign prostate tissue and prostate cancer in the finasteride arm of the Prostate Cancer Prevention Trial. Cancer Epidemiol. Biomark. Prev. 2016, 25, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, L.M.; Kulac, I.; Gumuskaya, B.; Valle, J.A.B.D.; Benedetti, I.; Pan, F.; Liu, J.O.; Marrone, M.T.; Arnold, K.B.; Goodman, P.J.; et al. Use of aspirin and statins in relation to inflammation in benign prostate tissue in the placebo arm of the Prostate Cancer Prevention Trial. Cancer Prev. Res. 2020, 13, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Kauerova, S.; Bartuskova, H.; Muffova, B.; Janousek, L.; Fronek, J.; Petras, M.; Poledne, R.; Kralova Lesna, I. Statins directly influence the polarization of adipose tissue macrophages: A role in chronic inflammation. Biomedicines 2021, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Choi, J.E.; Tien, J.C.; Simko, S.A.; Rajendiran, T.; Vo, J.N.; Delekta, A.D.; Wang, L.; Xiao, L.; Hodge, N.B.; et al. Autophagy inhibition by targeting PIKfyve potentiates response to immune checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 978–993. [Google Scholar] [CrossRef]
- Guth, A.; Monk, E.; Agarwal, R.; Bergman, B.C.; Zemski-Berry, K.A.; Minic, A.; Jordan, K.; Schlaepfer, I.R. Targeting fat oxidation in mouse prostate cancer decreases tumor growth and stimulates anti-cancer immunity. Int. J. Mol. Sci. 2020, 21, 9660. [Google Scholar] [CrossRef]
- Estève, D.; Roumiguié, M.; Manceau, C.; Milhas, D.; Muller, C. Periprostatic adipose tissue: A heavy player in prostate cancer progression. Curr. Opin. Endocr. Metab. Res. 2020, 10, 29–35. [Google Scholar] [CrossRef]
- Kapoor, J.; Namdarian, B.; Pedersen, J.; Hovens, C.; Moon, D.; Peters, J.; Costello, A.J.; Ruljancich, P.; Corcoran, N.M. Extraprostatic extension into periprostatic fat is a more important determinant of prostate cancer recurrence than an invasive phenotype. J. Urol. 2013, 190, 2061–2066. [Google Scholar] [CrossRef]
- Hoda, M.R.; Theil, G.; Mohammed, N.; Fischer, K.; Fornara, P. The adipocyte-derived hormone leptin has proliferative actions on androgen-resistant prostate cancer cells linking obesity to advanced stages of prostate cancer. J. Oncol. 2012, 2012, 280386. [Google Scholar] [CrossRef]
- Onuma, M.; Bub, J.D.; Rummel, T.L.; Iwamoto, Y. Prostate cancer cell-adipocyte interaction: Leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J. Biol. Chem. 2003, 278, 42660–42667. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, L.J.; Yang, Z.G.; Zhang, G.M.; Huo, R.C. Influence of adipocytokines in periprostatic adipose tissue on prostate cancer aggressiveness. Cytokine 2016, 85, 148–156. [Google Scholar] [CrossRef]
- Ribeiro, R.J.; Monteiro, C.P.; Cunha, V.F.; Azevedo, A.S.; Oliveira, M.J.; Monteiro, R.; Fraga, A.M.; Príncipe, P.; Lobato, C.; Lobo, F.; et al. Tumor cell-educated periprostatic adipose tissue acquires an aggressive cancer-promoting secretory profile. Cell Physiol. Biochem. 2012, 29, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Dahran, N.; Szewczyk-Bieda, M.; Vinnicombe, S.; Fleming, S.; Nabi, G. Periprostatic fat adipokine expression is correlated with prostate cancer aggressiveness in men undergoing radical prostatectomy for clinically localized disease. BJU Int. 2019, 123, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
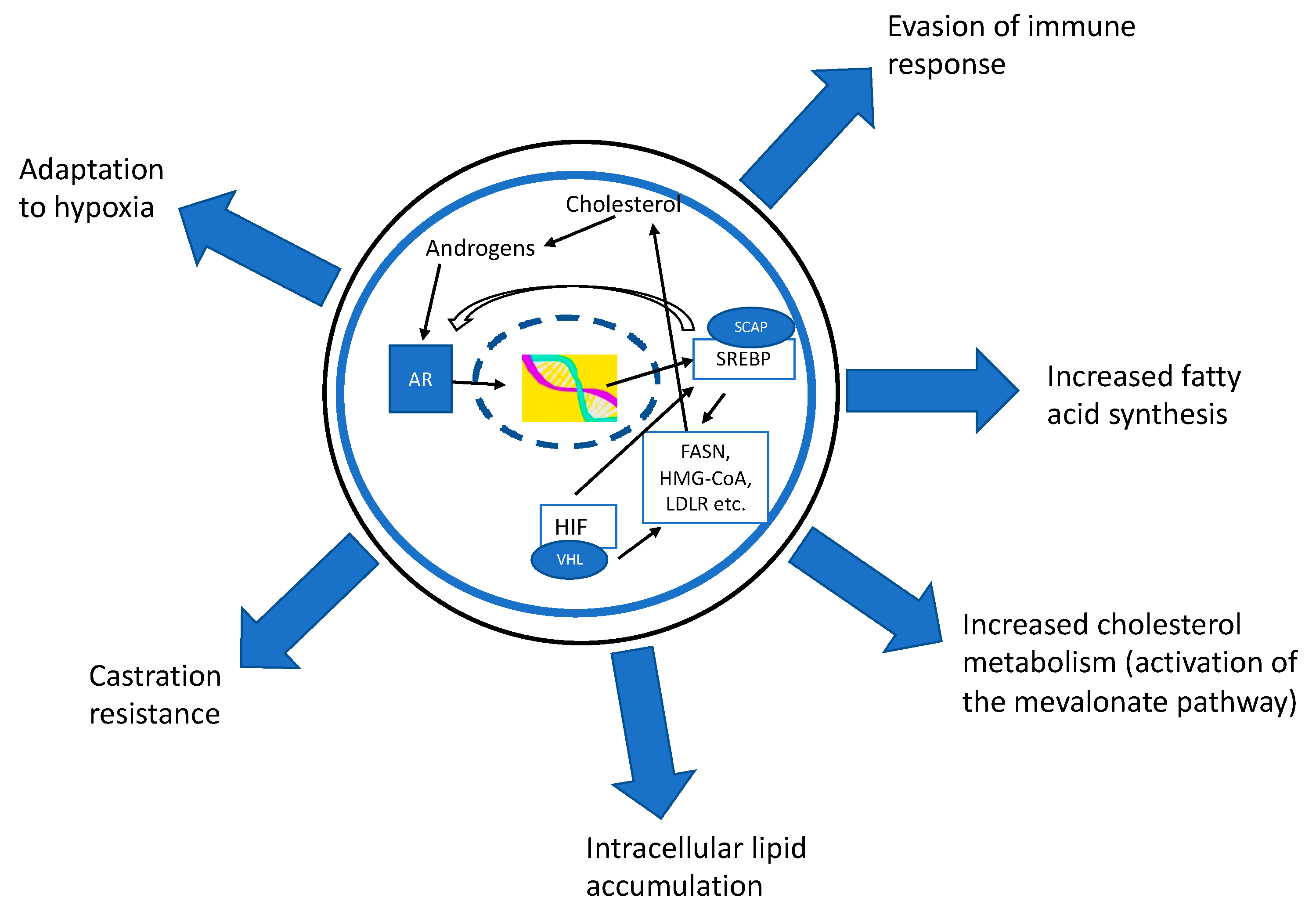

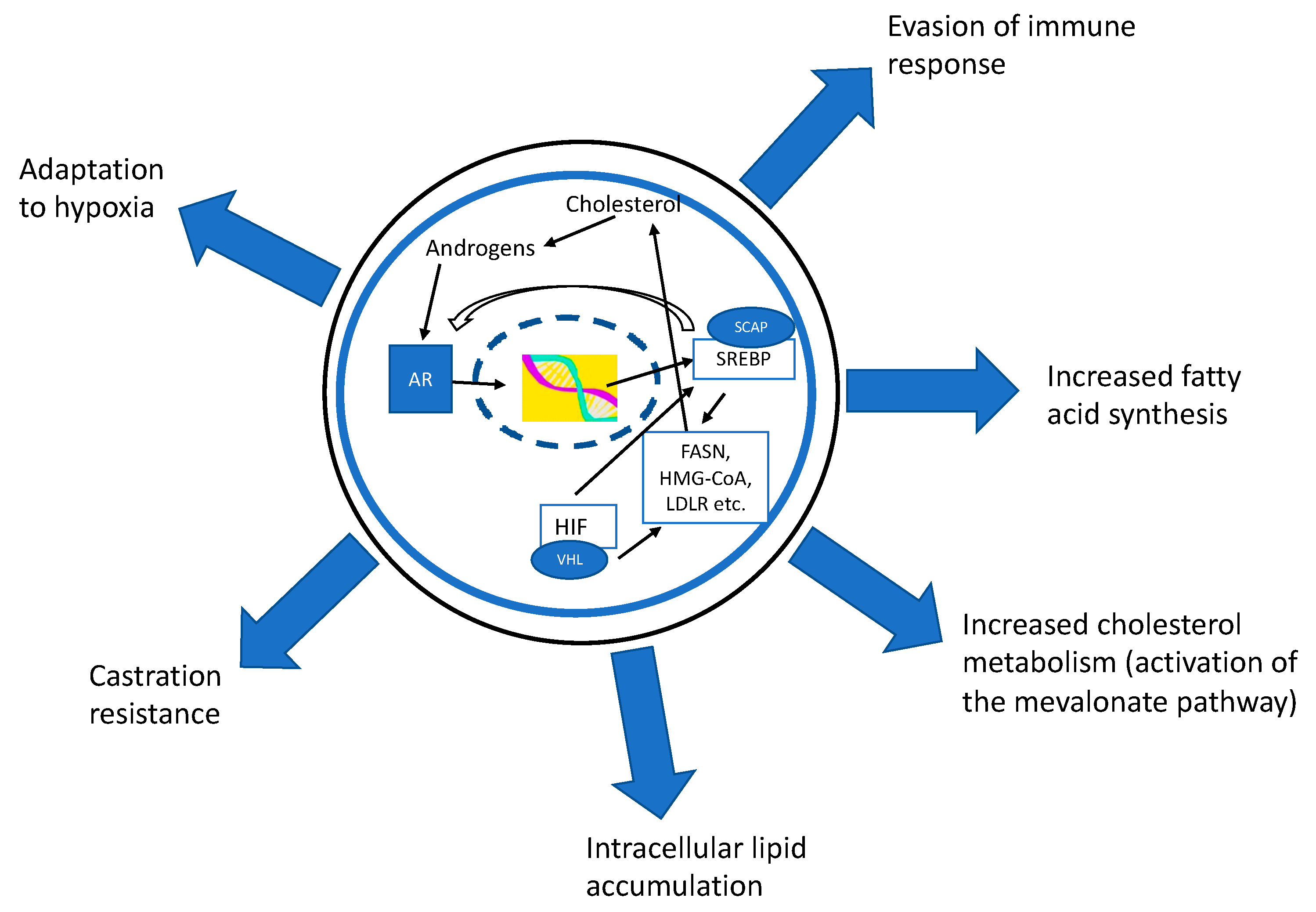{kind=link}
| Specific Cell Type | Specific Cancer Type (If Known) | Impact of Lipid Metabolism on Immune Cells | Reference |
|---|---|---|---|
| CD8+ T-cells | Cholesterol content in tumor microenvironment increased expression of immune checkpoints leading to T-cell exhaustion. Cholesterol depletion restore the activity | Ma et al., 2019 [87] | |
| Anticancer Tc9 cells (CD8+ T-cells) | Cholesterol depletion increased antitumor activity | Ma et al., 2018 [88] | |
| CD8+ T-cells | Cholesterol in cancer cell’s plasma memrane decrease CD8+ cytotoxicity | Lei et al., 2021 [89] | |
| CD8+ T-cells | melanoma (in a mouse model) | Inhibition of acetyl-CoA cholesterol acyltransferase increased proliferation of CD8+ T-cells | Yang et al., 2016 [90] |
| CD8+ T-cells | Liver carcinoma cells | Inhibition on cholesterol acyltransferase increased the amount of CD8+ T-cells and restorored CD8+ exhausted T-cell activity | Schmidt et al., 2021 [91] |
| T-cells | breast cancer, colon cancer, and melanoma | Inhibition of protein convertase subtilisin/kexin 9 decreased tumor growth and mortality in mice by reducing cholesterol metabolism and increasing T-cell infiltration in the tumors | Liu et al., 2020 [92] |
| Treg cells | melanoma (in a mouse model) | Lipid associated metabolic pathways by SREBP activity were enriched in tumors’ Treg cells. Inhibition of SREBP activity in Treg cells decreased tumor growth | Lim et al., 2021 [83] |
| T cells and tumor-associated macrophages | Treg cells were able to activate tumor associated macrophages by modulating interferon γ secretion in CD8+ T-cells | Liu et al., 2019 [84] | |
| CD4+ T-cells | Prostate cancer | Condition median from obesity-modified CD4+ T cells (decreased expression of IFNγ and IL-2 and increased expression of IL-6) increased the expression of epithelial-mesenchymal transition markers and showed a higher invasive and migratory capacity | De Angulo et al., 2022 [94] |
| Macrophages | Prostate cancer | Macrophages were associated with cholesterol transport and androgen synthesis in prostate cancer cells | El-Kenawi et al., 2021 [95] |
| Macrophages’ polarization | In ABCG1 (transporter which efflux cholesterol from the cells) knockdown mouse strain, macrophage polarization switch from tumor-promoting M2 to anti-tumor-promoting M1 phenotype | Sag et al., 2015 [96] | |
| Macrophages’ polarization | Inhibition of ABCG1 in human macrophages switch polarization from tumor-promoting M2 to anti-tumor-promoting M1 phenotype. Stimulation with HDL also downregulated polarization to M1 phenotype | Lee et al., 2016 [97] | |
| Macrophages | Prostate cancer | Cancer cell derived IL-1β enhanced expression of scavenger receptor, marco, on subset of macrophages. This was associated with prostate cancer progression and shorter disease-free survival. Marco was shown to regulate accumulation of lipids into the macrophages | Masetti et al., 2022 [98] |
| Myeloid-derived suppressor cells and macrophages | Prostate cancer | In mice fed with high-fat diet, number of myeloid-derived suppressor cells and ratio of M2/M1 macrophages were increased | Hayashi et al., 2018 [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siltari, A.; Syvälä, H.; Lou, Y.-R.; Gao, Y.; Murtola, T.J. Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment. Cancers 2022, 14, 4293. https://doi.org/10.3390/cancers14174293
Siltari A, Syvälä H, Lou Y-R, Gao Y, Murtola TJ. Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment. Cancers. 2022; 14(17):4293. https://doi.org/10.3390/cancers14174293
Chicago/Turabian StyleSiltari, Aino, Heimo Syvälä, Yan-Ru Lou, Yuan Gao, and Teemu J. Murtola. 2022. "Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment" Cancers 14, no. 17: 4293. https://doi.org/10.3390/cancers14174293
APA StyleSiltari, A., Syvälä, H., Lou, Y.-R., Gao, Y., & Murtola, T. J. (2022). Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment. Cancers, 14(17), 4293. https://doi.org/10.3390/cancers14174293