
Prevalence of ARID1A Mutations in Cell-Free Circulating Tumor DNA in a Cohort of 71,301 Patients and Association with Driver Co-Alterations

 , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. Liquid Biopsy cfDNA NGS Assay
2.3. Statistical Analysis
3. Results
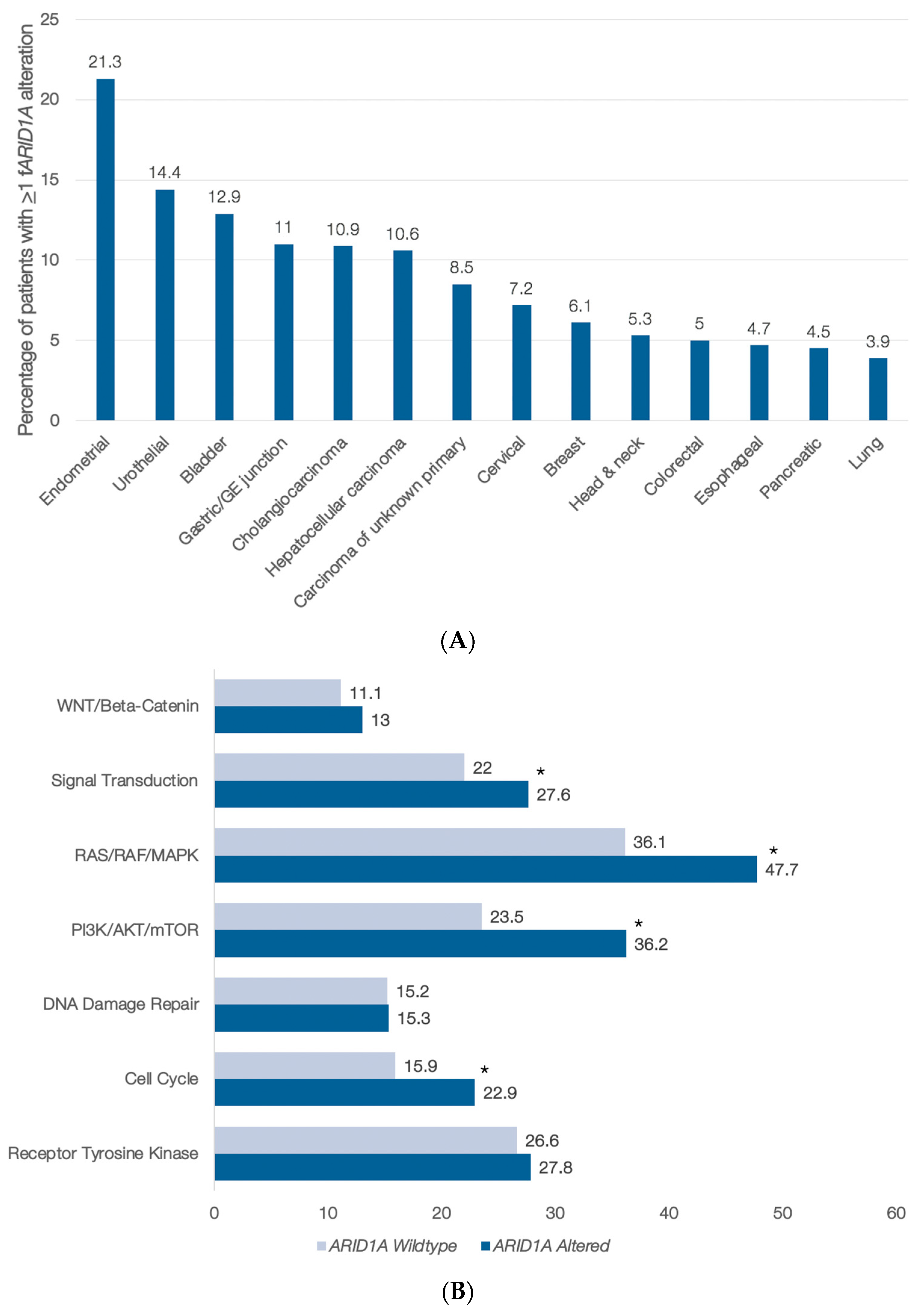
3.1. ARID1A Abnormalities in Blood-Derived cfDNA
3.2. cfDNA Genomic Alterations Co-Occurring with ARID1A Anomalies in the Pan-Cancer Setting
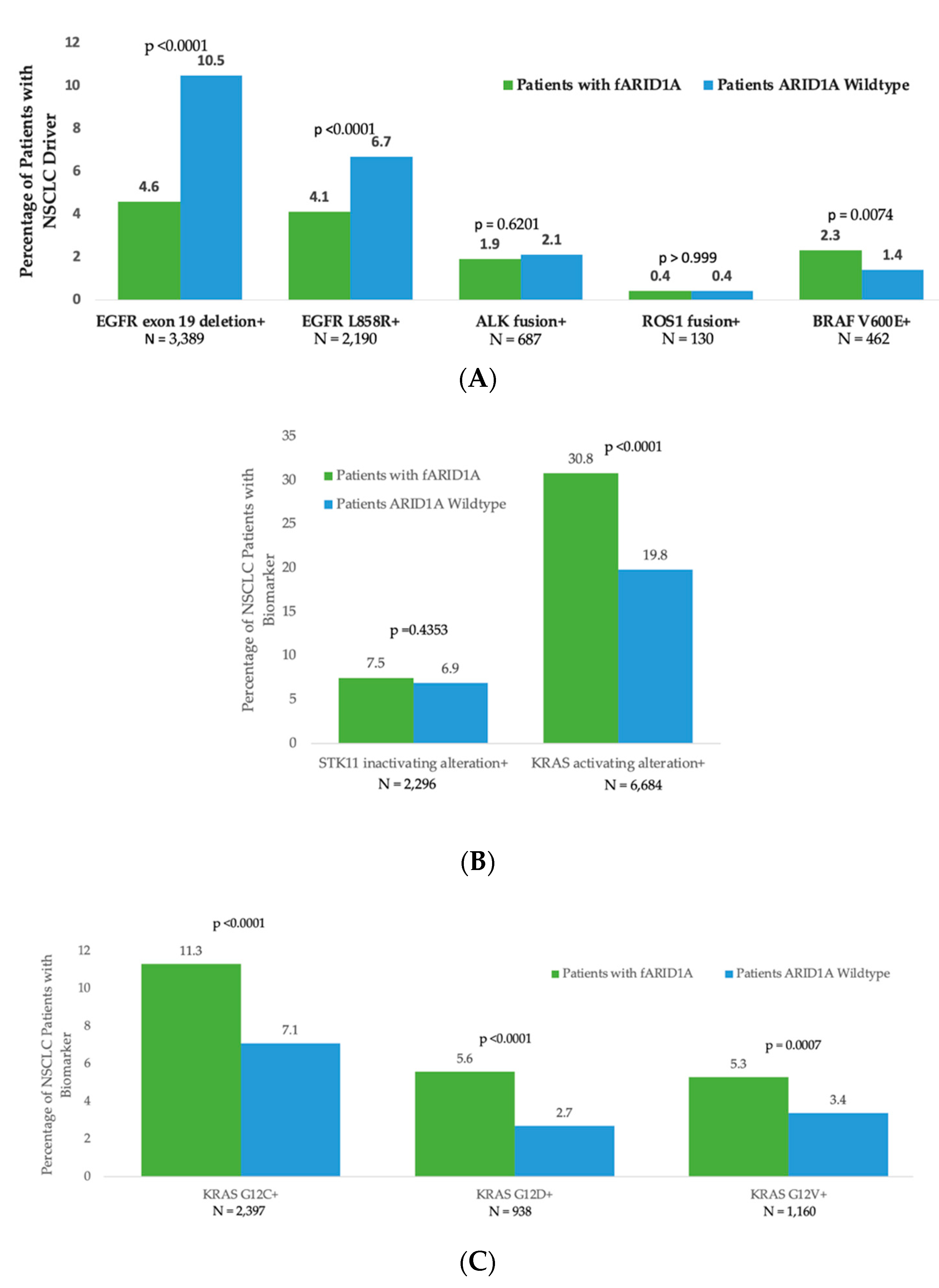
3.3. Segregation of ARID1A cfDNA Alterations with Other Oncogenic Drivers in Non-Small Cell Lung Cancer (NSCLC)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chen, X.; Su, C.; Ren, S.; Zhou, C. Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. J. Cancer 2020, 11, 776–780. [Google Scholar] [CrossRef]
- Pulice, J.L.; Kadoch, C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb. Symp. Quant. Biol. 2017, 81, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef]
- Rizvi, N.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.; Papadimitrakopoulou, V.; Heymach, J.; Scheuring, U.; Higgs, B.; et al. OA04.07 Mutations Associated with Sensitivity or Resistance to Immunotherapy in mNSCLC: Analysis from the MYSTIC Trial. J. Thorac. Oncol. 2019, 14, S217. [Google Scholar] [CrossRef]
- Johnson, R.M.; Qu, X.; Thomas, J.; Kschonsak, Y.; Huw, L.-Y.; Ou, F.-S.; Sokol, E.; Ihuegbu, N.; Zill, O.; Kabbarah, O.; et al. ARID1A mutations induce an EGFR-like gene expression signature and confer intrinsic and acquired resistance to cetuximab treatment in first line metastatic CRC. Cancer Res. 2020, 80 (Suppl. S16), LB-063. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef]
- Adashek, J.J.; Janku, F.; Kurzrock, R. Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers 2021, 13, 3600. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.; Kato, S.M.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A Mutations in Cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Okamoto, A.; Hollis, R.L.; Gourley, C.; Herrington, C.S. Clear cell carcinoma of the ovary: A clinical and molecular perspective. Int. J. Gynecol. Cancer 2021, 31, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Toumpeki, C.; Liberis, A.; Tsirkas, I.; Tsirka, T.; Kalagasidou, S.; Inagamova, L.; Anthoulaki, X.; Tsatsaris, G.; Kontomanolis, E.N. The Role of ARID1A in Endometrial Cancer and the Molecular Pathways Associated with Pathogenesis and Cancer Progression. In Vivo 2019, 33, 659–667. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef]
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted therapy in advanced non-small cell lung cancer: Current advances and future trends. J. Hematol. Oncol. 2021, 14, 108–115. [Google Scholar] [CrossRef]
- Fountzilas, E.; Kurzrock, R.; Hiep Vo, H.; Tsimberidou, A.M. Wedding of Molecular Alterations and Immune Checkpoint Blockade: Genomics as a Matchmaker. J. Natl. Cancer Inst. 2021, 113, 1634–1647. [Google Scholar] [CrossRef]
- Torralvo, J.; Friedlaender, A.; Achard, V.; Addeo, A. The Activity of Immune Checkpoint Inhibition in KRAS Mutated Non-small Cell Lung Cancer: A Single Centre Experience. Cancer Genom. Proteom. 2019, 16, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.-M.; Conejo-Garcia, J.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kim, K.H.; Lim, H.J.; Boichard, A.; Nikanjam, M.; Weihe, E.; Kuo, D.J.; Eskander, R.N.; Goodman, A.; Galanina, N.; et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 2020, 11, 4965. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Pathway | Genes Included |
|---|---|
| Receptor Tyrosine Kinase | EGFR, FGFR1, FGFR2, KIT, MET, MPL, and PDGFRA |
| Cell Cycle | CCNE1, CDH1, CDK4, CDK6, CDKN2A, FBXW7, and RB1 |
| DNA Damage | ATM, BRCA1, BRCA2, CCND1, and MLH1 |
| PI3K/AKT/mTOR | AKT1, MTOR, PIK3CA, PTEN, STK11, and TSC1 |
| RAS/RAF/MAPK | ARAF, BRAF, ERBB2, GNA11, HRAS, KRAS, MAP2K1, MAP2K2, MAPK1, MAPK3, NF1, NRAS, RAF1, and RIT1 |
| Signal Transduction | ALK, AR, DDR, ESR1, GATA3, GNAS, GNAQ, MYC, NOTCH1, NTRK1, NTRK3, PTPN11, RET, RHOA, ROS1, and SMAD4 |
| -Catenin | APC and CTNNB1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurzrock, R.; Aggarwal, C.; Weipert, C.; Kiedrowski, L.; Riess, J.; Lenz, H.-J.; Gandara, D. Prevalence of ARID1A Mutations in Cell-Free Circulating Tumor DNA in a Cohort of 71,301 Patients and Association with Driver Co-Alterations. Cancers 2022, 14, 4281. https://doi.org/10.3390/cancers14174281
Kurzrock R, Aggarwal C, Weipert C, Kiedrowski L, Riess J, Lenz H-J, Gandara D. Prevalence of ARID1A Mutations in Cell-Free Circulating Tumor DNA in a Cohort of 71,301 Patients and Association with Driver Co-Alterations. Cancers. 2022; 14(17):4281. https://doi.org/10.3390/cancers14174281
Chicago/Turabian StyleKurzrock, Razelle, Charu Aggarwal, Caroline Weipert, Lesli Kiedrowski, Jonathan Riess, Heinz-Josef Lenz, and David Gandara. 2022. "Prevalence of ARID1A Mutations in Cell-Free Circulating Tumor DNA in a Cohort of 71,301 Patients and Association with Driver Co-Alterations" Cancers 14, no. 17: 4281. https://doi.org/10.3390/cancers14174281
APA StyleKurzrock, R., Aggarwal, C., Weipert, C., Kiedrowski, L., Riess, J., Lenz, H.-J., & Gandara, D. (2022). Prevalence of ARID1A Mutations in Cell-Free Circulating Tumor DNA in a Cohort of 71,301 Patients and Association with Driver Co-Alterations. Cancers, 14(17), 4281. https://doi.org/10.3390/cancers14174281