
Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
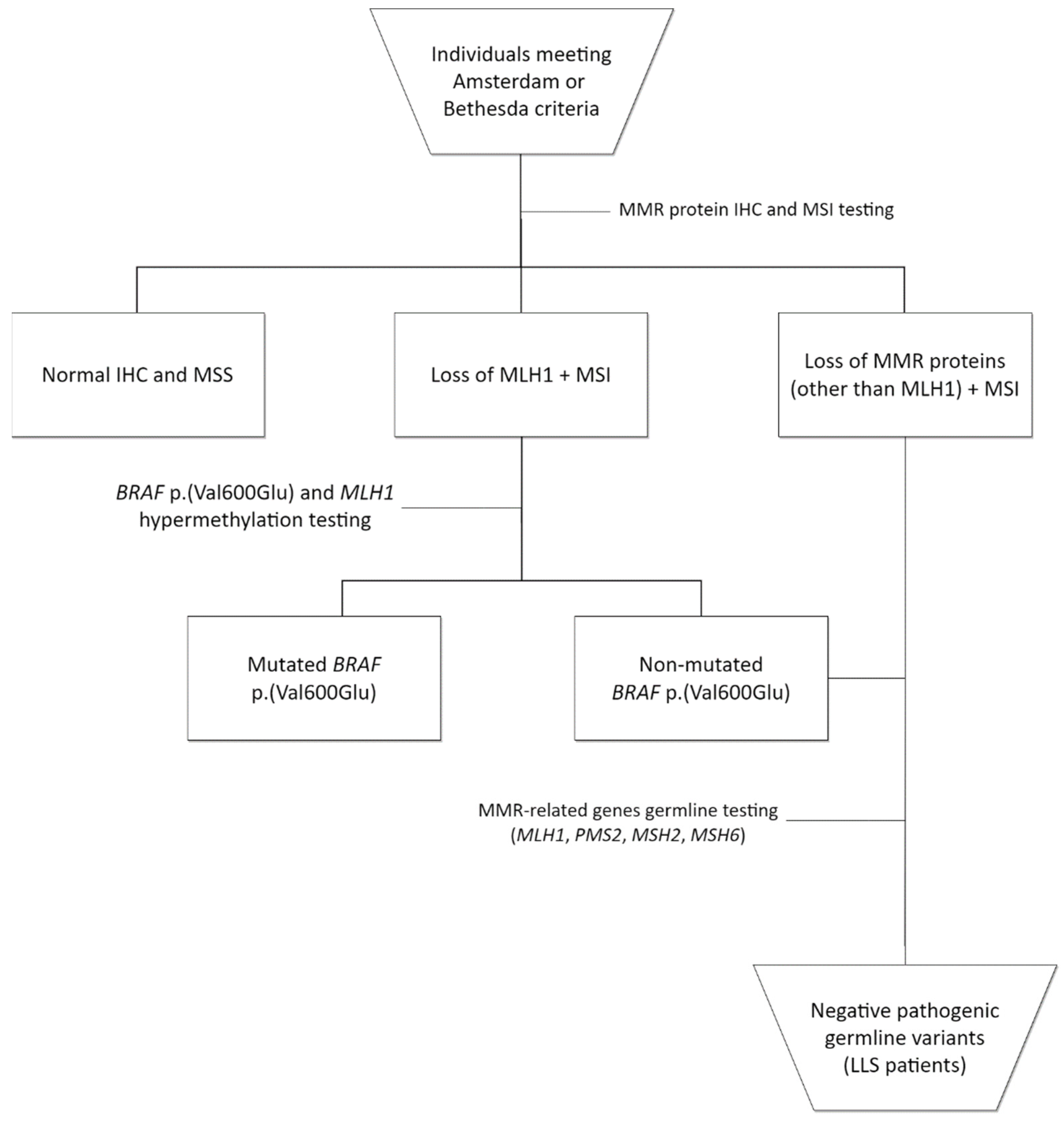
2.1. Patient Selection
2.2. DNA Isolation and Whole-Exome Sequencing
2.3. Sequence Quality Control, Alignment and Variant Calling
2.4. Variant Annotation and Classification
2.5. Statistical Analysis
3. Results
3.1. Patients
3.2. Germline Variants’ Profile
3.3. Germline Variants’ Classification
3.4. Variants in Patients with Loss of MLH1 or PMS2
3.5. Variants of Uncertain Significance (VUS)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Differentiating lynch-like from lynch syndrome. Gastroenterology 2014, 146, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.P.; Seppälä, T.T.; Peltomäki, P. Epidemiological, clinical and molecular characterization of lynch-like syndrome: A population-based study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef]
- Picó, M.D.; Castillejo, A.; Murcia, Ó.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Carrillo-Palau, M.; et al. Clinical and pathological characterization of lynch-like syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 368–374.e1. [Google Scholar] [CrossRef]
- Win, A.K.; Buchanan, D.D.; Rosty, C.; MacInnis, R.J.; Dowty, J.G.; Dite, G.S.; Giles, G.G.; Southey, M.C.; Young, J.P.; Clendenning, M.; et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2015, 64, 101–110. [Google Scholar] [CrossRef]
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in lynch-like syndrome. Fam. Cancer 2019, 18, 331–342. [Google Scholar] [CrossRef]
- de Paula, A.E.; de Galvão, H.C.R.; Bonatelli, M.; Sabato, C.; Fernandes, G.C.; Berardinelli, G.N.; Andrade, C.E.M.; Neto, M.C.; Romagnolo, L.G.C.; Campacci, N.; et al. Clinicopathological and molecular characterization of brazilian families at risk for lynch syndrome. Cancer Genet. 2021, 254–255, 82–91. [Google Scholar] [CrossRef]
- Castillejo, A.; Vargas, G.; Castillejo, M.I.; Navarro, M.; Barberá, V.M.; González, S.; Hernández-Illán, E.; Brunet, J.; Ramón, Y.; Cajal, T.; et al. Prevalence of germline MUTYH mutations among lynch-like syndrome patients. Eur. J. Cancer 2014, 50, 2241–2250. [Google Scholar] [CrossRef]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; De La Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.M.; Van Wezel, T.; Van Den Akker, B.E.; Ventayol Garcia, M.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gómez-García, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive mismatch repair gene panel identifies variants in patients with lynch-like syndrome. Mol. Genet. Genomic Med. 2019, 7, e850. [Google Scholar] [CrossRef] [PubMed]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset lynch-like syndrome. JCI Insight 2020, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Palmero, E.I.; Galvão, H.C.R.; Fernandes, G.C.; De Paula, A.E.; Oliveira, J.C.; Souza, C.P.; Andrade, C.E.; Romagnolo, L.G.C.; Volc, S.; Neto, M.C.; et al. Oncogenetics service and the brazilian public health system: The experience of a reference cancer hospital. Genet. Mol. Biol. 2016, 39, 168–177. [Google Scholar] [CrossRef]
- de Garcia, F.A.O.; de Andrade, E.S.; de Campos Reis Galvão, H.; da Silva Sábato, C.; Campacci, N.; de Paula, A.E.; Evangelista, A.F.; Santana, I.V.V.; Melendez, M.E.; Reis, R.M.; et al. New insights on familial colorectal cancer type X syndrome. Sci. Rep. 2022, 12, 1–11. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Picard Toolkit, 2019. Broad Institute, Repository. Available online: http://broadinstitute.github.io/picard/ (accessed on 12 April 2022).
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 1–22. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease-gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The genecards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinforma. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Spatz, M.A. Genetics home reference. J. Med. Libr. Assoc. 2004, 92, 282–283. [Google Scholar]
- Das, R.; Ghosh, S.K. Genetic variants of the DNA repair genes from exome aggregation consortium (EXAC) database: Significance in cancer. DNA Repair 2017, 52, 92–102. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A Joint Consensus Recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870. [Google Scholar] [CrossRef]
- Bertier, G.; Hétu, M.; Joly, Y. Unsolved challenges of clinical whole-exome sequencing: A systematic literature review of end-users’ views Donna Dickenson, Sandra Soo-Jin Lee, and Michael Morrison. BMC Med. Genom. 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Jurado, C.; Garre, P.; Vila, M.; Lozano, J.J.; Pristoupilova, A.; Beltrán, S.; Abulí, A.; Muñoz, J.; Balaguer, F.; Ocaña, T.; et al. New genes emerging for colorectal cancer predisposition. World J. Gastroenterol. 2014, 20, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Trujillano, D.; Bertoli-Avella, A.M.; Kumar Kandaswamy, K.; Weiss, M.E.; Köster, J.; Marais, A.; Paknia, O.; Schröder, R.; Garcia-Aznar, J.M.; Werber, M.; et al. Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 2017, 25, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017, 3, 464. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Brosens, L.A.A.; Offerhaus, G.J.A.; Giardiello, F.M.; de Leng, W.W.J.; Montgomery, E.A. Pathology and genetics of hereditary colorectal cancer. Pathology 2018, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Kim, H.; Cleary, S.; Cupples, C.; Gallinger, S.; Bristow, R. Characterization of Mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology 2008, 135, 499–507. [Google Scholar] [CrossRef]
- Jones, N.; Vogt, S.; Nielsen, M.; Christian, D.; Wark, P.A.; Eccles, D.; Edwards, E.; Evans, D.G.; Maher, E.R.; Vasen, H.F.; et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology 2009, 137, 489–494.e1. [Google Scholar] [CrossRef]
- Win, A.K.; Cleary, S.P.; Dowty, J.G.; Baron, J.A.; Young, J.P.; Buchanan, D.D.; Southey, M.C.; Burnett, T.; Parfrey, P.S.; Green, R.C.; et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int. J. Cancer 2011, 129, 2256–2262. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; Madhavan, M.V.; Mirchandani, K.D.; McCaffrey, R.M.; Vinciguerra, P.; D’Andrea, A.D. DNA polymerase poln participates in cross-link repair and homologous recombination. Mol. Cell. Biol. 2010, 30, 1088–1096. [Google Scholar] [CrossRef]
- Grant, R.C.; Denroche, R.E.; Borgida, A.; Virtanen, C.; Cook, N.; Smith, A.L.; Connor, A.A.; Wilson, J.M.; Peterson, G.; Roberts, N.J.; et al. Exome-wide association study of pancreatic cancer risk. Gastroenterology 2018, 154, 719–722.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wei, Y.; Pan, J.; Jin, S.; Gu, W.; Gan, H.; Zhu, Y.; Ye, D.W. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int. J. Cancer 2021, 148, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Dicks, E.; Song, H.; Ramus, S.J.; Van Oudenhove, E.; Tyrer, J.P.; Intermaggio, M.P.; Kar, S.; Harrington, P.; Bowtell, D.D.; Cicek, M.S.; et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017, 8, 50930–50940. [Google Scholar] [CrossRef] [PubMed]
- Tarafa, G.; Villanueva, A.; Farré, L.; Rodríguez, J.; Musulén, E.; Reyes, G.; Seminago, R.; Olmedo, E.; Paules, A.B.; Peinado, M.A.; et al. DCC and SMAD4 Alterations in human colorectal and pancreatic tumor dissemination. Oncogene 2000, 19, 546–555. [Google Scholar] [CrossRef]
- Polvi, A.; Linnankivi, T.; Kivelä, T.; Herva, R.; Keating, J.P.; Mäkitie, O.; Pareyson, D.; Vainionpää, L.; Lahtinen, J.; Hovatta, I.; et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am. J. Hum. Genet. 2012, 90, 540–549. [Google Scholar] [CrossRef]
- Anderson, B.H.; Kasher, P.R.; Mayer, J.; Szynkiewicz, M.; Jenkinson, E.M.; Bhaskar, S.S.; Urquhart, J.E.; Daly, S.B.; Dickerson, J.E.; O’Sullivan, J.; et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause coats plus. Nat. Genet. 2012, 44, 338–342. [Google Scholar] [CrossRef]
- Shen, W.; Kerr, C.M.; Przychozen, B.; Mahfouz, R.Z.; LaFramboise, T.; Nagata, Y.; Hanna, R.; Radivoyevitch, T.; Nazha, A.; Sekeres, M.A.; et al. Impact of germline CTC1 alterations on telomere length in acquired bone marrow failure. Br. J. Haematol. 2019, 185, 935–939. [Google Scholar] [CrossRef]
- Kim, B.; Yun, W.; Lee, S.T.; Choi, J.R.; Yoo, K.H.; Koo, H.H.; Jung, C.W.; Kim, S.H. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci. Rep. 2020, 1–7. [Google Scholar] [CrossRef]
- Liao, H.F.; Lee, H.H.; Chang, Y.S.; Lin, C.L.; Liu, T.Y.; Chen, Y.C.; Yen, J.C.; Lee, Y.T.; Lin, C.Y.; Wu, S.H.; et al. Down-regulated and commonly mutated ALPK1 in lung and colorectal cancers. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Jandrig, B.; Seitz, S.; Hinzmann, B.; Arnold, W.; Micheel, B.; Koelble, K.; Siebert, R.; Schwartz, A.; Ruecker, K.; Schlag, P.M.; et al. ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene 2004, 23, 9295–9302. [Google Scholar] [CrossRef]
- Capaccio, D.; Ciccodicola, A.; Sabatino, L.; Casamassimi, A.; Pancione, M.; Fucci, A.; Febbraro, A.; Merlino, A.; Graziano, G.; Colantuoni, V. A novel germline mutation in peroxisome proliferator-activated receptor γ gene associated with large intestine polyp formation and dyslipidemia. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 572–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.M.; Zhou, X.P.; Kurose, K.; Gao, X.; Latif, F.; Kroll, T.; Sugano, K.; Cannistra, S.A.; Clinton, S.K.; Maher, E.R.; et al. Opposite association of two PPARG variants with cancer: Overrepresentation of H449H in endometrial carcinoma cases and underrepresentation of P12A in renal cell carcinoma cases. Hum. Genet. 2001, 109, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Mitui, M.; Campbell, C.; Coutinho, G.; Sun, X.; Lai, C.H.; Thorstenson, Y.; Castellvi-Bel, S.; Fernandez, L.; Monros, E.; Carvalho, B.T.C.; et al. Independent mutational events are rare in the ATM Gene: Haplotype prescreening enhances mutation detection rate. Hum. Mutat. 2003, 22, 43–50. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef]
- Young, E.L.; Thompson, B.A.; Neklason, D.W.; Firpo, M.A.; Werner, T.; Bell, R.; Berger, J.; Fraser, A.; Gammon, A.; Koptiuch, C.; et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer 2018, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Characteristics (n = 20) | n | (%) |
|---|---|---|
| Gender | ||
| Female | 12 | (60) |
| Male | 8 | (40) |
| Survival status | ||
| Followed-up | 19 | (95) |
| Deceased | 1 | (5) |
| Mean age of first diagnosed tumor (SD) | 48.2 (7.7) | |
| First diagnosed tumor | ||
| Colorectal cancer | 15 | (75) |
| Endometrium | 2 | (10) |
| Ovary | 2 | (10) |
| Stomach | 1 | (5) |
| Mean age of second diagnosed tumor (SD) | 59.2 (7) | |
| Second diagnosed tumor | ||
| Colorectal cancer | 1 | (5) |
| Endometrium | 2 | (10) |
| Breast | 1 | (5) |
| Non-melanoma skin | 1 | (5) |
| Clinical criteria | ||
| Amsterdam | 5 | (25) |
| Bethesda | 8 | (40) |
| Revised Bethesda | 7 | (35) |
| ID Case | Gene | Pathogenic Variant | REVEL | AF | (Class) 1 | Tumor Site | Age 2 | Criteria Fulfilled |
|---|---|---|---|---|---|---|---|---|
| 1194 | PPARG | NM_015869.5:c.1230C > A p.(Ser410Arg) | 0.767 | 1.60 × 10−5 | (IV) | ovary | 44 | Bethesda * |
| 142 | MUTYH | NM_001128425.2:c.1187G > A p. (Gly396Asp) | 0.954 | 3.00 × 10−3 | (V) | colorectal | 39 | Bethesda |
| 1728 | POLN | NC_000004.11(NM_181808.2):c.1375-2A > G splicing variant | - | 4.07 × 10−6 | (V) | colorectal | 57 | Bethesda * |
| 313 | CTC1 | NM_025099.6:c.19C > T p. (Gln7Ter) | - | 1.68 × 10−5 | (V) | colorectal | 48 | Bethesda * |
| 573 | ALPK1 | NM_001102406.2:c.3428_3431del p. (Asn1143ThrfsTer5) | - | - | (IV) | stomach colorectal | 44 49 | Bethesda |
| 635 | DCC | NM_005215.4:c.1861G > A p. (Val621Met) | 0.303 | 2.00 × 10−4 | (IV) | colorectal non-melanoma skin | 50 56 | Bethesda * |
| 837 | ATM | NC_000011.9(NM_000051.3):c.3993 + 1G > A splicing variant | - | 1.60 × 10−5 | (V) | endometrium breast | 53 58 | Bethesda * |
| ST18 | NM_014682.2:c.2093del p. (Lys698SerfsTer24) | - | - | (IV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, W.; de Andrade, E.S.; Garcia, F.A.d.O.; Campacci, N.; Sábato, C.d.S.; Melendez, M.E.; Reis, R.M.; Galvão, H.d.C.R.; Palmero, E.I. Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome. Cancers 2022, 14, 4233. https://doi.org/10.3390/cancers14174233
dos Santos W, de Andrade ES, Garcia FAdO, Campacci N, Sábato CdS, Melendez ME, Reis RM, Galvão HdCR, Palmero EI. Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome. Cancers. 2022; 14(17):4233. https://doi.org/10.3390/cancers14174233
Chicago/Turabian Styledos Santos, Wellington, Edilene Santos de Andrade, Felipe Antonio de Oliveira Garcia, Natália Campacci, Cristina da Silva Sábato, Matias Eliseo Melendez, Rui Manuel Reis, Henrique de Campos Reis Galvão, and Edenir Inez Palmero. 2022. "Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome" Cancers 14, no. 17: 4233. https://doi.org/10.3390/cancers14174233
APA Styledos Santos, W., de Andrade, E. S., Garcia, F. A. d. O., Campacci, N., Sábato, C. d. S., Melendez, M. E., Reis, R. M., Galvão, H. d. C. R., & Palmero, E. I. (2022). Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome. Cancers, 14(17), 4233. https://doi.org/10.3390/cancers14174233