
Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma
 , , ,
, , , 

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue
2.2. Isolation of Primary Liver Cells
2.2.1. Isolation of Primary Human Hepatocytes
2.2.2. Isolation of Primary Human Hepatoma Cells
2.3. Cell Culture and Sampling
2.4. Immunofluorescence Staining
2.5. RNA Isolation and Quantification
2.6. Western Blot Analyses
2.7. Functional Assays
2.7.1. Glucose Assay
2.7.2. Glycogen Assay
2.7.3. Pyruvate Assay
2.7.4. Lactate Assay
2.7.5. Lipid Determination (Oil Red O Assay)
2.7.6. Ketone Body Assay
2.7.7. Protein Determination (Bicinchoninic Acid Assay)
2.7.8. Quantification and Statistical Analyses
3. Results
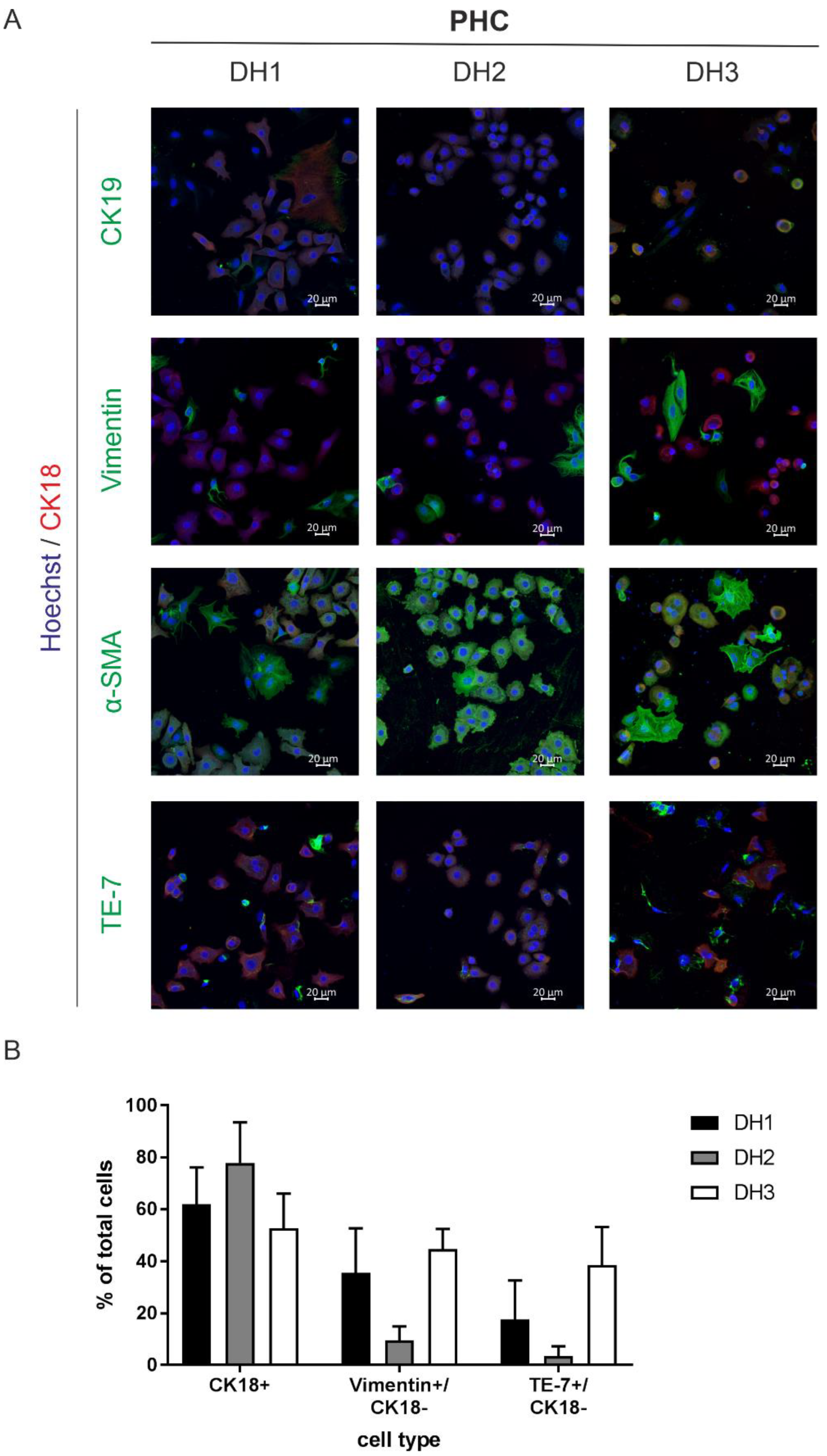
3.1. Characterization of the Isolated Primary Human Hepatoma Cells Revealed Mixed Populations with a High Yield of Cancer Cells


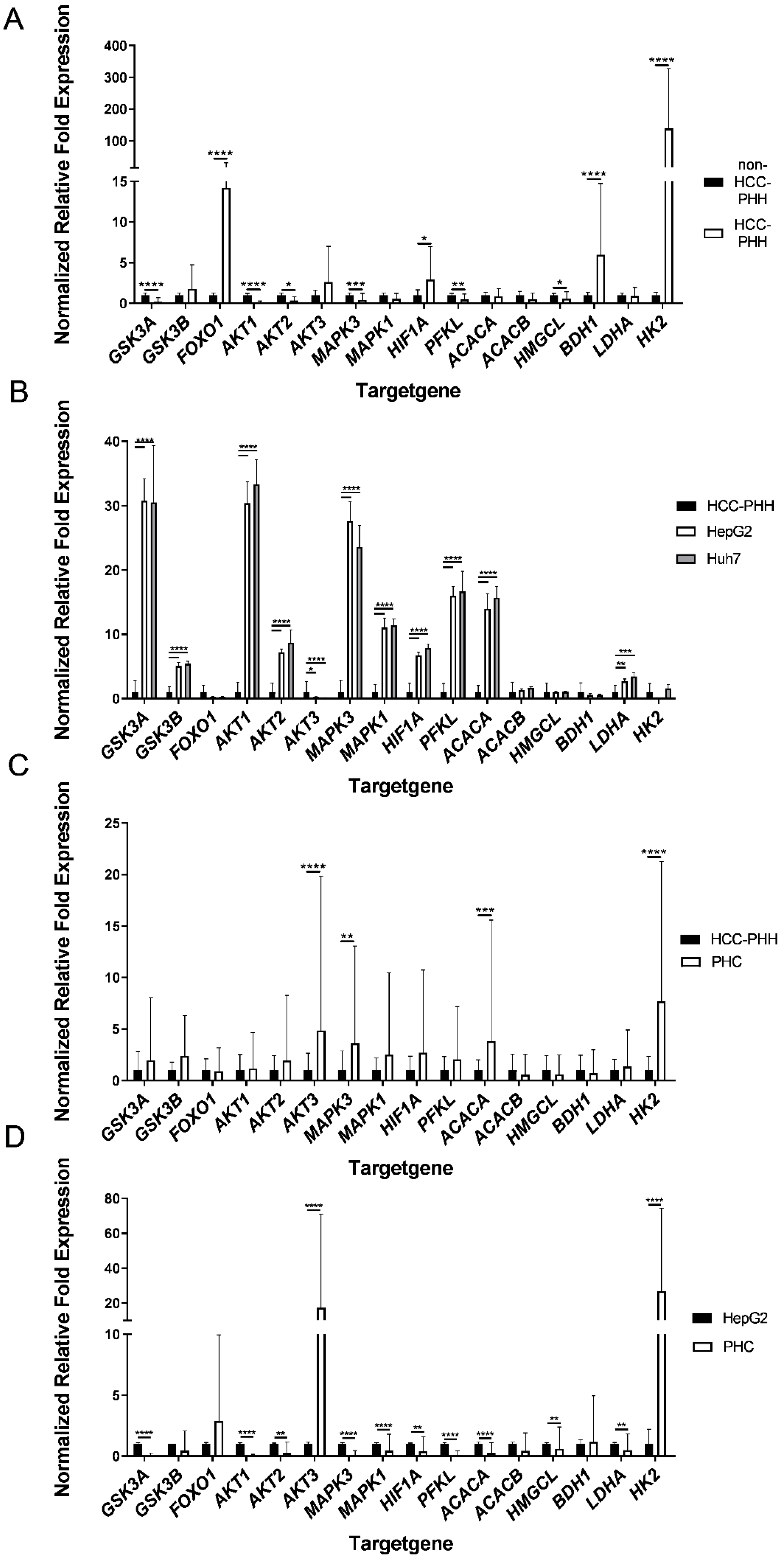
3.2. Primary Human Hepatoma Cells Showed a Metabolic Shift in Energy Metabolism Genes

3.3. Effects of Cell Types on the Expression Levels of Metabolic Proteins

3.4. Major Metabolic Changes Arise at a Premalignant Stage in the Diseased Liver

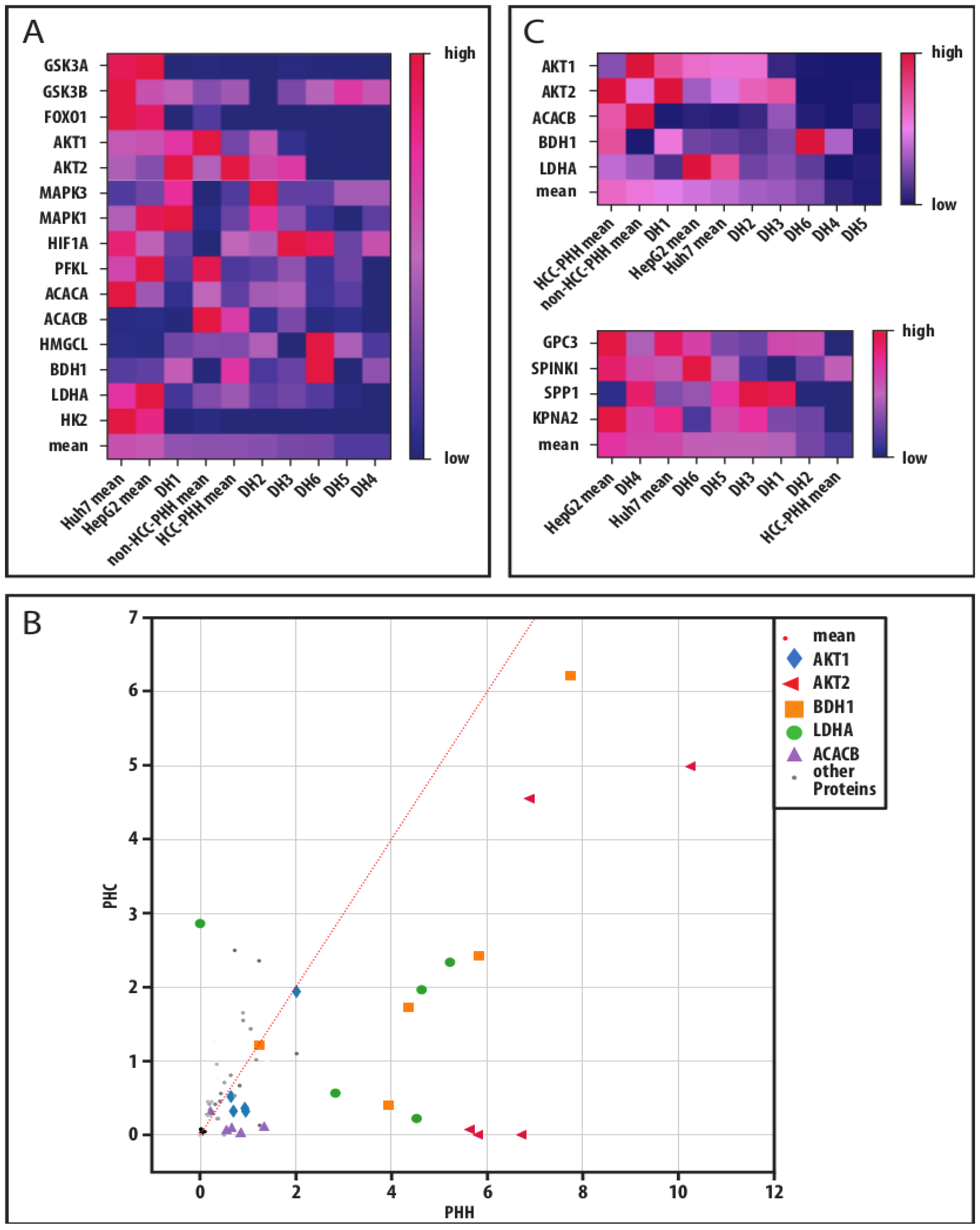
3.5. Global Data Analysis Shows Diverging Traits of Metabolic and Cancerous Dedifferentiation

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer; Global Cancer Observatory. Cancer Fact Sheets: Liver. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 25 August 2022).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xie, H.; Hu, M.; Huang, T.; Hu, Y.; Sang, N.; Zhao, Y. Recent progress in treatment of hepatocellular carcinoma. Am. J. Cancer Res. 2020, 10, 2993–3036. [Google Scholar] [PubMed]
- De Matteis, S.; Ragusa, A.; Marisi, G.; de Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant metabolism in hepatocellular carcinoma provides diagnostic and therapeutic opportunities. Oxid. Med. Cell. Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [PubMed]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.-M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Cui, Z.; Chen, P.; Han, H.; Peng, J.; Leng, X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med. Oncol. 2012, 29, 909–914. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Tenen, D.G.; Chai, L.; Tan, J.L. Metabolic alterations and vulnerabilities in hepatocellular carcinoma. Gastroenterol. Rep. 2021, 9, 1–13. [Google Scholar] [CrossRef]
- Fraczek, J.; Bolleyn, J.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Primary hepatocyte cultures for pharmaco-toxicological studies: At the busy crossroad of various anti-dedifferentiation strategies. Arch. Toxicol. 2013, 87, 577–610. [Google Scholar] [CrossRef]
- Shi, J.; Wang, X.; Lyu, L.; Jiang, H.; Zhu, H.-J. Comparison of protein expression between human livers and the hepatic cell lines HepG2, Hep3B, and Huh7 using SWATH and MRM-HR proteomics: Focusing on drug-metabolizing enzymes. Drug Metab. Pharmacokinet. 2018, 33, 133–140. [Google Scholar] [CrossRef]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef]
- Zhuo, J.-Y.; Lu, D.; Tan, W.-Y.; Zheng, S.-S.; Shen, Y.-Q.; Xu, X. CK19-positive hepatocellular carcinoma is a characteristic subtype. J. Cancer 2020, 11, 5069–5077. [Google Scholar] [CrossRef] [PubMed]
- Castro-Muñozledo, F.; Meza-Aguilar, D.G.; Domínguez-Castillo, R.; Hernández-Zequinely, V.; Sánchez-Guzmán, E. Vimentin as a marker of early differentiating, highly motile corneal epithelial cells. J. Cell. Physiol. 2017, 232, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Scearce, R.M.; Lobach, D.F.; Hensley, L.L. Phenotypic characterization and ontogeny of mesodermal-derived and endocrine epithelial components of the human thymic microenvironment. J. Exp. Med. 1984, 159, 1149–1168. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef]
- Hass, H.; Jobst, J.; Scheurlen, M.; Vogel, U.; Nehls, O. Gene expression analysis for evaluation of potential biomarkers in hepatocellular carcinoma. Anticancer. Res. 2015, 35, 2021–2028. [Google Scholar]
- Huang, P.; Zhang, L.; Gao, Y.; He, Z.; Yao, D.; Wu, Z.; Cen, J.; Chen, X.; Liu, C.; Hu, Y.; et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 2014, 14, 370–384. [Google Scholar] [CrossRef]
- Hamilton, S.R. Pathology and Genetics of Tumours of the Digestive System: Reflects the Views of a Working Group that Convened for an Editorial and Consensus Conference in Lyon, France, 6–9 November 1999; IARC Press: Lyon, France, 2000; ISBN 9283224108. [Google Scholar]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for isolation of primary human hepatocytes and corresponding major populations of non-parenchymal liver cells. J. Vis. Exp. 2016, 109, e53069. [Google Scholar] [CrossRef]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nüssler, A.K.; Seehofer, D.; Damm, G. Featured article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef]
- Damm, G.; Schicht, G.; Zimmermann, A.; Rennert, C.; Fischer, N.; Kießig, M.; Wagner, T.; Kegel, V.; Seehofer, D. Effect of glucose and insulin supplementation on the isolation of primary human hepatocytes. EXCLI J. 2019, 18, 1071–1091. [Google Scholar] [CrossRef]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006, 1, 2753–2758. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose metabolism on tumor plasticity, diagnosis, and treatment. Front. Oncol. 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef]
- Pilling, J.; Garside, H.; Ainscow, E. Development of a quantitative 96-well method to image glycogen storage in primary rat hepatocytes. Mol. Cell. Biochem. 2010, 341, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; de Fabiani, E.; Crestani, M. Ketogenic diet: A new light shining on old but gold biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Vidalain, P.-O.; Jacquemin, C.; Aublin-Gex, A.; Olmstead, K.; Panthu, B.; Rautureau, G.J.P.; André, P.; Nyczka, P.; Hütt, M.-T.; et al. A hexokinase isoenzyme switch in human liver cancer cells promotes lipogenesis and enhances innate immunity. Commun. Biol. 2021, 4, 217. [Google Scholar] [CrossRef]
- Ozaki, I.; Yamamoto, K.; Mizuta, T.; Kajihara, S.; Fukushima, N.; Setoguchi, Y.; Morito, F.; Sakai, T. Differential expression of laminin receptors in human hepatocellular carcinoma. Gut 1998, 43, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qiao, X.; Qiao, S.; Yu, L. Targeting integrins in hepatocellular carcinoma. Expert Opin. Ther. Targets 2011, 15, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.; Pereira, J.; Paredes, J. The crosstalk between cell adhesion and cancer metabolism. Int. J. Mol. Sci. 2019, 20, 1933. [Google Scholar] [CrossRef]
- Wang, J.; Hao, F.; Fei, X.; Chen, Y. SPP1 functions as an enhancer of cell growth in hepatocellular carcinoma targeted by miR-181c. Am. J. Transl. Res. 2019, 11, 6924–6937. [Google Scholar]
- Gao, C.-L.; Wang, G.-W.; Yang, G.-Q.; Yang, H.; Zhuang, L. Karyopherin subunit-α 2 expression accelerates cell cycle progression by upregulating CCNB2 and CDK1 in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 2815–2820. [Google Scholar] [CrossRef]
- Shirakawa, H.; Kuronuma, T.; Nishimura, Y.; Hasebe, T.; Nakano, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; et al. Glypican-3 is a useful diagnostic marker for a component of hepatocellular carcinoma in human liver cancer. Int. J. Oncol. 2009, 34, 649–656. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ying, H.Y.; Gong, C.J.; Feng, Y.; Jing, D.D.; Lu, L.G. Serine protease inhibitor Kazal type 1 (SPINK1) downregulates E-cadherin and induces EMT of hepatoma cells to promote hepatocellular carcinoma metastasis via the MEK/ERK signaling pathway. J. Dig. Dis. 2017, 18, 349–358. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 25 August 2022).
- Ma, L.; Wang, L.; Khatib, S.A.; Chang, C.-W.; Heinrich, S.; Dominguez, D.A.; Forgues, M.; Candia, J.; Hernandez, M.O.; Kelly, M.; et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, W.; Athavale, D.; Ge, X.; Desert, R.; Das, S.; Han, H.; Nieto, N. Osteopontin takes center stage in chronic liver disease. Hepatology 2021, 73, 1594–1608. [Google Scholar] [CrossRef]
- Mossenta, M.; Busato, D.; Bo, M.D.; Toffoli, G. Glucose metabolism and oxidative stress in hepatocellular carcinoma: Role and possible implications in novel therapeutic strategies. Cancers 2020, 12, 1668. [Google Scholar] [CrossRef]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-regulation of key glycolysis proteins in cancer development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Chai, F.; Li, Y.; Liu, K.; Li, Q.; Sun, H. Caveolin enhances hepatocellular carcinoma cell metabolism, migration, and invasion in vitro via a hexokinase 2-dependent mechanism. J. Cell. Physiol. 2019, 234, 1937–1946. [Google Scholar] [CrossRef]
- Meng, P.; Zhang, Y.-F.; Zhang, W.; Chen, X.; Xu, T.; Hu, S.; Liang, X.; Feng, M.; Yang, X.; Ho, M. Identification of the atypical cadherin FAT1 as a novel glypican-3 interacting protein in liver cancer cells. Sci. Rep. 2021, 11, 40. [Google Scholar] [CrossRef]
- Yao, G.; Yin, J.; Wang, Q.; Dong, R.; Lu, J. Glypican-3 enhances reprogramming of glucose metabolism in liver cancer cells. BioMed Res. Int. 2019, 2019, 2560650. [Google Scholar] [CrossRef]
- Zhang, W.W.; Churchill, S.; Lindahl, R.; Churchill, P. Regulation of D-beta-hydroxybutyrate dehydrogenase in rat hepatoma cell lines. Cancer Res. 1989, 49, 2433–2437. [Google Scholar]
- Huang, D.; Li, T.; Wang, L.; Zhang, L.; Yan, R.; Li, K.; Xing, S.; Wu, G.; Hu, L.; Jia, W.; et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016, 26, 1112–1130. [Google Scholar] [CrossRef]
- Yang, H.; Zheng, W.; Shuai, X.; Chang, R.-M.; Yu, L.; Fang, F.; Yang, L.-Y. MicroRNA-424 inhibits Akt3/E2F3 axis and tumor growth in hepatocellular carcinoma. Oncotarget 2015, 6, 27736–27750. [Google Scholar] [CrossRef]
- Wang, L.; Huang, D.; Jiang, Z.; Luo, Y.; Norris, C.; Zhang, M.; Tian, X.; Tang, Y. Akt3 is responsible for the survival and proliferation of embryonic stem cells. Biol. Open 2017, 6, 850–861. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Mauriz, J.L.; García-Palomo, A.; González-Gallego, J. FoxO proteins: Regulation and molecular targets in liver cancer. Curr. Med. Chem. 2014, 21, 1231–1246. [Google Scholar] [CrossRef]
- Cervello, M.; Augello, G.; Cusimano, A.; Emma, M.R.; Balasus, D.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Pivotal roles of glycogen synthase-3 in hepatocellular carcinoma. Adv. Biol. Regul. 2017, 65, 59–76. [Google Scholar] [CrossRef]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.; Loizzi, D.; Serviddio, G. Lipid metabolism in development and progression of hepatocellular carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Wuensch, T.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhütter, H.-G. Metabolic heterogeneity of human hepatocellular carcinoma: Implications for personalized pharmacological treatment. FEBS J. 2020, 288, 2332–2346. [Google Scholar] [CrossRef]
- Salazar, J.; Le, A. The heterogeneity of liver cancer metabolism. Adv. Exp. Med. Biol. 2021, 1311, 127–136. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, W.-N.; Chen, X.; Peng, X.-D.; Jeon, S.-M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, A.; Sun, H. Power of metabolomics in diagnosis and biomarker discovery of hepatocellular carcinoma. Hepatology 2013, 57, 2072–2077. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Age | Sex | Diagnosis | BMI | Steatosis [%] | ASH | NASH | Fibrosis | Cirrhosis |
|---|---|---|---|---|---|---|---|---|---|
| D1 | 74 | male | CRLM * | 23.6 | 10 | no | no | yes | no |
| D2 | 39 | male | CRLM * | 27.8 | 15 | no | no | yes | no |
| D3 | 46 | female | CRLM * | 20.3 | no | no | no | yes | no |
| D4 | 28 | female | Adenoma | 27.5 | 5 | no | no | yes | no |
| DH1 | 66 | male | HCC G1 | 34.5 | 10–60 | no | yes | no | yes |
| DH2 | 63 | male | HCC G1 | 33.6 | 5 | no | no | yes | no |
| DH3 | 79 | female | HCC G1 | 28 | 60 | yes | no | no | yes |
| DH4 | 66 | male | HCC G1 | 28 | 5 | no | no | yes | no |
| DH5 | 63 | male | HCC G1 | 25 | 60 | no | yes | no | yes |
| DH6 | 79 | male | HCC G2 | 27 | 20 | no | yes | yes | no |
| DH7 | 76 | male | HCC G2 | 24.8 | 25 | yes | no | yes | yes |
| DH8 | 71 | male | HCC G1 | 26.1 | 20 | no | yes | yes | yes |
| DH9 | 50 | female | HCC G2 | 62.1 | 35 | no | yes | yes | no |
| DH10 | 67 | female | HCC G1 | 43.9 | 60 | no | yes | no | yes |
| Gene Name | Type | Primer Sequence |
|---|---|---|
| FGF7 | forward reverse | GAAGGAGGGGATATAAGAGTGAG ATTCTTCATCTCTTGGGTCCC |
| GPC3 | forward reverse | TGTGCCCATTCTCAACAACG AGCAAAGGGTGTCGTTTTCC |
| HK2 | forward reverse | TACCTGGGTGAGATTGTCCG CAAGCCCTAAGTGTTGCAGG |
| KPNA2 | forward reverse | AGGAAAACCGCAACAACCAG TTTCGGAATCAAACCAGCCC |
| SPINK1 | forward reverse | AGAGACGTGGTAAGTGCGG ATTTGGCCTCTCTTCCCAGG |
| SPP1 | forward reverse | CACACATGGAAAGCGAGGAG TGGAATTCACGGCTGACTTTG |
| TWIST2 | forward reverse | CTACAGCAAGAAGTCGAGCG CTTGCTCAGCTTGTCAGAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schicht, G.; Seidemann, L.; Haensel, R.; Seehofer, D.; Damm, G. Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma. Cancers 2022, 14, 4227. https://doi.org/10.3390/cancers14174227
Schicht G, Seidemann L, Haensel R, Seehofer D, Damm G. Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma. Cancers. 2022; 14(17):4227. https://doi.org/10.3390/cancers14174227
Chicago/Turabian StyleSchicht, Gerda, Lena Seidemann, Rene Haensel, Daniel Seehofer, and Georg Damm. 2022. "Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma" Cancers 14, no. 17: 4227. https://doi.org/10.3390/cancers14174227
APA StyleSchicht, G., Seidemann, L., Haensel, R., Seehofer, D., & Damm, G. (2022). Critical Investigation of the Usability of Hepatoma Cell Lines HepG2 and Huh7 as Models for the Metabolic Representation of Resectable Hepatocellular Carcinoma. Cancers, 14(17), 4227. https://doi.org/10.3390/cancers14174227