
Melanoma Tumour Vascularization and Tissue-Resident Endothelial Progenitor Cells


Abstract
Simple Summary
Abstract
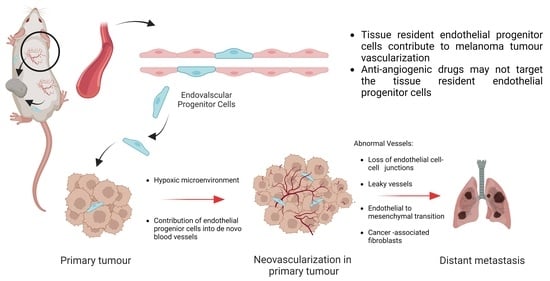
1. Introduction
2. How Do Tumour Blood Vessels Form in Melanoma?
2.1. Sprouting Angiogenesis
2.2. Intussusceptive Angiogenesis
2.3. Vessel Co-Option
2.4. Vasculogenic Mimicry
2.5. Vasculogenesis
3. Role of Endothelial Progenitor Cells in Tumour Vascularization
4. How Do Endothelial Cells Reshape the Tumour Microenvironment?
5. Role of Vascularization in Delivery of Immune Cells and Drugs
6. Targeting the Tumour Vascularization with Antiangiogenic Drugs
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANG2 | Angiopoietin 2 |
| BMP | Bone morphogenic protein |
| CAFs | Cancer-associated Fibroblast |
| CCN2 | Cellular communication network factor 2 |
| CTLA-4 | Cytotoxic T-lymphocyte-associated 4 |
| DLL4 | Delta-like ligand 4 |
| ECFCs | Endothelial colony-forming cells |
| ECM | Extra cellular matrix |
| EndMT | Endothelial to mesenchymal transition |
| EPCs | Endothelial progenitor cells |
| eNOS | Endothelial nitric oxide synthesis |
| EVMM | Extravascular migratory metastasis |
| EVP | Endovascular progenitor cells |
| FGFs | Fibroblast growth factors |
| FSP1 | Fibroblast specific protein 1 |
| HEVs | High endothelial venules |
| HMVECs | Human microvascular endothelial cells |
| ICAM1 | Intercellular adhesion molecule 1 |
| ICIs | Immune checkpoint inhibitors |
| IFI | Interstitial fluid pressure |
| IFN | Interferon |
| MMPs | Matrix metalloproteinases |
| MMPs | Matrix metalloproteases |
| NK cells | Natural killer cells |
| NICD | Notch intracellular domain |
| PD-1 | Programmed cell death protein 1 |
| RBPJ | Recombination signal-binding protein for immunoglobulin kappa J |
| TSP1 | Thrombospondin 1 |
| uPAR | Urokinase-type plasminogen activator receptor |
| VCAM1 | Vascular cell adhesion molecule 1 |
| VE-cadherin | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| vWF | von Willebrand factor |
References
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P. Cutaneous Malignant Melanoma: A Review of Early Diagnosis and Management. World J. Oncol. 2021, 12, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Garrisi, V.M.; Strippoli, S.; De Summa, S.; Pinto, R.; Perrone, A.; Guida, G.; Azzariti, A.; Guida, M.; Tommasi, S. Proteomic profile and in silico analysis in metastatic melanoma with and without BRAF mutation. PLoS ONE 2014, 9, e112025. [Google Scholar] [CrossRef]
- Manganelli, M.; Guida, S.; Ferretta, A.; Pellacani, G.; Porcelli, L.; Azzariti, A.; Guida, G. Behind the Scene: Exploiting MC1R in Skin Cancer Risk and Prevention. Genes 2021, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.I.; Gershenwald, J.E. Evidence-based treatment of early-stage melanoma. J. Surg. Oncol. 2011, 104, 341–353. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Byzova, T.V. Angiogenesis in melanoma. Semin. Oncol. 2007, 34, 555–565. [Google Scholar] [CrossRef]
- Srivastava, A.; Laidler, P.; Hughes, L.E.; Woodcock, J.; Shedden, E.J. Neovascularization in human cutaneous melanoma: A quantitative morphological and Doppler ultrasound study. Eur. J. Cancer Clin. Oncol. 1986, 22, 1205–1209. [Google Scholar] [CrossRef]
- Srivastava, A.; Laidler, P.; Davies, R.P.; Horgan, K.; Hughes, L.E. The prognostic significance of tumor vascularity in intermediate-thickness (0.76–4.0 mm thick) skin melanoma. A quantitative histologic study. Am. J. Pathol. 1988, 133, 419–423. [Google Scholar]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Reynolds, A.R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020, 23, 55–74. [Google Scholar] [CrossRef]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Lenzi, P.; Bocci, G.; Natale, G. John Hunter and the origin of the term “angiogenesis”. Angiogenesis 2016, 19, 255–256. [Google Scholar] [CrossRef]
- Hall, A.P. Review of the pericyte during angiogenesis and its role in cancer and diabetic retinopathy. Toxicol. Pathol. 2006, 34, 763–775. [Google Scholar] [CrossRef]
- Goldmann, E. The Growth of Malignant Disease in Man and the Lower Animals, with special reference to the Vascular System. Proc. R. Soc. Med. 1908, 1, 1–13. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Xu, J.Y.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Bba-Mol. Basis Dis. 2014, 1842, 2106–2119. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix—Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Heissig, B.; Hattori, K.; Friedrich, M.; Rafii, S.; Werb, Z. Angiogenesis: Vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 2003, 10, 136–141. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragon, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Tetzlaff, F.; Adam, M.G.; Feldner, A.; Moll, I.; Menuchin, A.; Rodriguez-Vita, J.; Sprinzak, D.; Fischer, A. MPDZ promotes DLL4-induced Notch signaling during angiogenesis. Elife 2018, 7, e32860. [Google Scholar] [CrossRef]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A Team Effort Coordinated by Notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef]
- Mouillesseaux, K.P.; Wiley, D.S.; Saunders, L.M.; Wylie, L.A.; Kushner, E.J.; Chong, D.C.; Citrin, K.M.; Barber, A.T.; Park, Y.; Kim, J.D.; et al. Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat. Commun. 2016, 7, 13247. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Lu, M.Y.; Onuchic, J.N.; Clementi, C.; Ben-Jacob, E. Jagged-Delta asymmetry in Notch signaling can give rise to a Sender/Receiver hybrid phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E402–E409. [Google Scholar] [CrossRef]
- Koon, Y.L.; Zhang, S.J.; Rahmat, M.B.; Koh, C.G.; Chiam, K.H. Enhanced Delta-Notch Lateral Inhibition Model Incorporating Intracellular Notch Heterogeneity and Tension-Dependent Rate of Delta-Notch Binding that Reproduces Sprouting Angiogenesis Patterns. Sci. Rep. 2018, 8, 9519. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor Endothelial Cells. Csh. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Warren, B.A.; Shubik, P. The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Lab. Investig. 1966, 15, 464–478. [Google Scholar] [CrossRef]
- Montoyo-Pujol, Y.G.; Wang, X.; Bermudez-Sanchez, S.; Martin, A.; Almazan, F.; Lopez-Nevot, M.A. Measurement of 45 cytokine, chemokine and growth factors in established cell culture supernatants and autologous serum from advanced melanoma patients. Carcinogenesis 2021, 42, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, P.; Neshat, A.; Mokhtari, M.; Rajabi, M.A.; Eftekhari, M.; Tavakoli, P. The role of VEGF in melanoma progression. J. Res. Med. Sci. 2012, 17, 534–539. [Google Scholar]
- Gowda, R.; Robertson, B.M.; Iyer, S.; Barry, J.; Dinavahi, S.S.; Robertson, G.P. The role of exosomes in metastasis and progression of melanoma. Cancer Treat. Rev. 2020, 85, 101975. [Google Scholar] [CrossRef]
- Hood, J.L.; Pan, H.; Lanza, G.M.; Wickline, S.A.; Consortium for Translational Research in Advanced Imaging and Nanomedicine. Paracrine induction of endothelium by tumor exosomes. Lab. Investig. 2009, 89, 1317–1328. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serrati, S.; Mocali, A.; et al. uPAR-expressing melanoma exosomes promote angiogenesis by VE-Cadherin, EGFR and uPAR overexpression and rise of ERK1,2 signaling in endothelial cells. Cell. Mol. Life Sci. 2021, 78, 3057–3072. [Google Scholar] [CrossRef]
- Laurenzana, A.; Biagioni, A.; Bianchini, F.; Peppicelli, S.; Chilla, A.; Margheri, F.; Luciani, C.; Pimpinelli, N.; Del Rosso, M.; Calorini, L.; et al. Inhibition of uPAR-TGFbeta crosstalk blocks MSC-dependent EMT in melanoma cells. J. Mol. Med. 2015, 93, 783–794. [Google Scholar] [CrossRef]
- Hugdahl, E.; Bachmann, I.M.; Schuster, C.; Ladstein, R.G.; Akslen, L.A. Prognostic value of uPAR expression and angiogenesis in primary and metastatic melanoma. PLoS ONE 2019, 14, e0210399. [Google Scholar] [CrossRef]
- Cho, W.C.; Jour, G.; Aung, P.P. Role of angiogenesis in melanoma progression: Update on key angiogenic mechanisms and other associated components. Semin. Cancer Biol. 2019, 59, 175–186. [Google Scholar] [CrossRef]
- De Spiegelaere, W.; Casteleyn, C.; Van den Broeck, W.; Plendl, J.; Bahramsoltani, M.; Simoens, P.; Djonov, V.; Cornillie, P. Intussusceptive angiogenesis: A biologically relevant form of angiogenesis. J. Vasc. Res. 2012, 49, 390–404. [Google Scholar] [CrossRef]
- Caduff, J.H.; Fischer, L.C.; Burri, P.H. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat. Rec. 1986, 216, 154–164. [Google Scholar] [CrossRef]
- Burri, P.H.; Tarek, M.R. A novel mechanism of capillary growth in the rat pulmonary microcirculation. Anat. Rec. 1990, 228, 35–45. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Floris, C.; Mangieri, D.; Piras, F.; Ennas, M.G.; Vacca, A.; Sirigu, P. Microvascular density, vascular endothelial growth factor immunoreactivity in tumor cells, vessel diameter and intussusceptive microvascular growth in primary melanoma. Oncol. Rep. 2005, 14, 81–84. [Google Scholar]
- Esteban, S.; Clemente, C.; Koziol, A.; Gonzalo, P.; Rius, C.; Martinez, F.; Linares, P.M.; Chaparro, M.; Urzainqui, A.; Andres, V.; et al. Endothelial MT1-MMP targeting limits intussusceptive angiogenesis and colitis via TSP1/nitric oxide axis. EMBO Mol. Med. 2020, 12, e10862. [Google Scholar] [CrossRef]
- Pandita, A.; Ekstrand, M.; Bjursten, S.; Zhao, Z.; Fogelstrand, P.; Le Gal, K.; Ny, L.; Bergo, M.O.; Karlsson, J.; Nilsson, J.A.; et al. Intussusceptive Angiogenesis in Human Metastatic Malignant Melanoma. Am. J. Pathol. 2021, 191, 2023–2038. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Pezzella, F.; Di Bacco, A.; Andreola, S.; Nicholson, A.G.; Pastorino, U.; Harris, A.L. Angiogenesis in primary lung cancer and lung secondaries. Eur. J. Cancer 1996, 32A, 2494–2500. [Google Scholar] [CrossRef]
- .Pezzella, F.; Pastorino, U.; Tagliabue, E.; Andreola, S.; Sozzi, G.; Gasparini, G.; Menard, S.; Gatter, K.C.; Harris, A.L.; Fox, S.; et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 1997, 151, 1417–1423. [Google Scholar]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; De Rooij, L.; Cuypers, A.; Rohlenova, K.; Dumas, S.J.; Garcia-Caballero, M.; Meta, E.; Amersfoort, J.; Taverna, F.; Becker, L.M.; et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. 2021, 35, 109253. [Google Scholar] [CrossRef] [PubMed]
- Dome, B.; Paku, S.; Somlai, B.; Timar, J. Vascularization of cutaneous melanoma involves vessel co-option and has clinical significance. J. Pathol. 2002, 197, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Bentolila, L.A.; Prakash, R.; Mihic-Probst, D.; Wadehra, M.; Kleinman, H.K.; Carmichael, T.S.; Peault, B.; Barnhill, R.L.; Lugassy, C. Imaging of Angiotropism/Vascular Co-Option in a Murine Model of Brain Melanoma: Implications for Melanoma Progression along Extravascular Pathways. Sci. Rep. 2016, 6, 23834. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.J.; Erickson, C.A.; Takada, S.; Burrus, L.W. Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev. Biol. 2001, 233, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, C.; Eyden, B.P.; Christensen, L.; Escande, J.P. Angio-tumoral complex in human malignant melanoma characterised by free laminin: Ultrastructural and immunohistochemical observations. J. Submicrosc. Cytol. Pathol. 1997, 29, 19–28. [Google Scholar] [PubMed]
- Lugassy, C.; Barnhill, R.L. Angiotropic melanoma and extravascular migratory metastasis: A review. Adv. Anat. Pathol. 2007, 14, 195–201. [Google Scholar] [CrossRef]
- Barnhill, R.L.; Lemaitre, S.; Levy-Gabrielle, C.; Rodrigues, M.; Desjardins, L.; Dendale, R.; Vincent-Salomon, A.; Roman-Roman, S.; Lugassy, C.; Cassoux, N. Satellite in transit metastase in rapidly fatal conjunctival melanoma: Implications for angiotropism and extravascular migratory metastasis (description of a murine model for conjuctival melanoma). Pathology 2016, 48, 166–176. [Google Scholar] [CrossRef]
- Crowson, A.N.; Magro, C.M.; Mihm, M.C. Prognosticators of melanoma, the melanoma report, and the sentinel lymph node. Mod. Pathol. 2006, 19 (Suppl. 2), S71–S87. [Google Scholar] [CrossRef]
- Hung, T.; Morin, J.; Munday, W.R.; Mackenzie, I.R.; Lugassy, C.; Barnhill, R.L. Angiotropism in primary cutaneous melanoma with brain metastasis: A study of 20 cases. Am. J. Dermatopathol. 2013, 35, 650–654. [Google Scholar] [CrossRef]
- Barnhill, R.; van Dam, P.J.; Vermeulen, P.; Champenois, G.; Nicolas, A.; Rawson, R.V.; Wilmott, J.S.; Thompson, J.F.; Long, G.V.; Cassoux, N.; et al. Replacement and desmoplastic histopathological growth patterns in cutaneous melanoma liver metastases: Frequency, characteristics, and robust prognostic value. J. Pathol. Clin. Res. 2020, 6, 195–206. [Google Scholar] [CrossRef]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Homig-Holzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- Leenders, W.P.; Kusters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef]
- Velez, D.O.; Tsui, B.; Goshia, T.; Chute, C.L.; Han, A.; Carter, H.; Fraley, S.I. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 2017, 8, 1651. [Google Scholar] [CrossRef]
- Clarijs, R.; Otte-Holler, I.; Ruiter, D.J.; de Waal, R.M. Presence of a fluid-conducting meshwork in xenografted cutaneous and primary human uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 912–918. [Google Scholar]
- Smetsers, T.F.; van de Westerlo, E.M.; ten Dam, G.B.; Clarijs, R.; Versteeg, E.M.; van Geloof, W.L.; Veerkamp, J.H.; van Muijen, G.N.; van Kuppevelt, T.H. Localization and characterization of melanoma-associated glycosaminoglycans: Differential expression of chondroitin and heparan sulfate epitopes in melanoma. Cancer Res. 2003, 63, 2965–2970. [Google Scholar]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef]
- Radnot, M.; Antal, M. Vessels of intraocular malignant melanomas. Am. J. Ophthalmol. 1979, 88, 472–478. [Google Scholar] [CrossRef]
- Bissell, M.J. Tumor plasticity allows vasculogenic mimicry, a novel form of angiogenic switch. A rose by any other name? Am. J. Pathol. 1999, 155, 675–679. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef]
- Bartolome, R.A.; Torres, S.; Isern de Val, S.; Escudero-Paniagua, B.; Calvino, E.; Teixido, J.; Casal, J.I. VE-cadherin RGD motifs promote metastasis and constitute a potential therapeutic target in melanoma and breast cancers. Oncotarget 2017, 8, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Mihic-Probst, D.; Ikenberg, K.; Tinguely, M.; Schraml, P.; Behnke, S.; Seifert, B.; Civenni, G.; Sommer, L.; Moch, H.; Dummer, R. Tumor cell plasticity and angiogenesis in human melanomas. PLoS ONE 2012, 7, e33571. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, P.; Meng, A.; Zhang, R.; Zhou, Y. Down-regulating Myoferlin inhibits the vasculogenic mimicry of melanoma via decreasing MMP-2 and inducing mesenchymal-to-epithelial transition. J. Cell. Mol. Med. 2018, 22, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Martini, C.; DeNichilo, M.; King, D.P.; Cockshell, M.P.; Ebert, B.; Dale, B.; Ebert, L.M.; Woods, A.; Bonder, C.S. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer 2021, 21, 765. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zong, Y.; Gao, Y.; Sun, X.; Zhao, H.; Luo, W.; Jia, S. VEGF Induce Vasculogenic Mimicry of Choroidal Melanoma through the PI3k Signal Pathway. Biomed. Res. Int. 2019, 2019, 3909102. [Google Scholar] [CrossRef]
- Yuan, Y.; Geng, B.; Xu, X.; Zhao, H.; Bai, J.; Dou, Z.; Jia, S.; Yu, X.; Luo, W. Dual VEGF/PDGF knockdown suppresses vasculogenic mimicry formation in choroidal melanoma cells via the Wnt5a/beta-catenin/AKT signaling pathway. Acta Histochem. 2022, 124, 151842. [Google Scholar] [CrossRef]
- Flamme, I.; Risau, W. Induction of vasculogenesis and hematopoiesis in vitro. Development 1992, 116, 435–439. [Google Scholar] [CrossRef]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Asahara, T.; Takahashi, T.; Masuda, H.; Kalka, C.; Chen, D.; Iwaguro, H.; Inai, Y.; Silver, M.; Isner, J.M. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999, 18, 3964–3972. [Google Scholar] [CrossRef]
- Kaushal, S.; Amiel, G.E.; Guleserian, K.J.; Shapira, O.M.; Perry, T.; Sutherland, F.W.; Rabkin, E.; Moran, A.M.; Schoen, F.J.; Atala, A.; et al. Functional small-diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat. Med. 2001, 7, 1035–1040. [Google Scholar] [CrossRef]
- Quirici, N.; Soligo, D.; Caneva, L.; Servida, F.; Bossolasco, P.; Deliliers, G.L. Differentiation and expansion of endothelial cells from human bone marrow CD133(+) cells. Br. J. Haematol. 2001, 115, 186–194. [Google Scholar] [CrossRef]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef]
- Purhonen, S.; Palm, J.; Rossi, D.; Kaskenpaa, N.; Rajantie, I.; Yla-Herttuala, S.; Alitalo, K.; Weissman, I.L.; Salven, P. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 6620–6625. [Google Scholar] [CrossRef]
- Bailey, A.S.; Willenbring, H.; Jiang, S.; Anderson, D.A.; Schroeder, D.A.; Wong, M.H.; Grompe, M.; Fleming, W.H. Myeloid lineage progenitors give rise to vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 13156–13161. [Google Scholar] [CrossRef]
- Romagnani, P.; Annunziato, F.; Liotta, F.; Lazzeri, E.; Mazzinghi, B.; Frosali, F.; Cosmi, L.; Maggi, L.; Lasagni, L.; Scheffold, A.; et al. CD14+CD34low cells with stem cell phenotypic and functional features are the major source of circulating endothelial progenitors. Circ. Res. 2005, 97, 314–322. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Tasev, D.; Koolwijk, P.; van Hinsbergh, V.W. Therapeutic Potential of Human-Derived Endothelial Colony-Forming Cells in Animal Models. Tissue Eng. Part B Rev. 2016, 22, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Tura-Ceide, O.; Hunter, A.; Mitchell, A.; Vesey, A.; Medine, C.; Gallogly, S.; Hadoke, P.W.F.; Keith, C.; Sproul, A.; et al. Endothelial Progenitor Cells Do Not Originate From the Bone Marrow. Circulation 2019, 140, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef]
- Patel, J.; Seppanen, E.; Chong, M.S.; Yeo, J.S.; Teo, E.Y.; Chan, J.K.; Fisk, N.M.; Khosrotehrani, K. Prospective surface marker-based isolation and expansion of fetal endothelial colony-forming cells from human term placenta. Stem Cells Transl. Med. 2013, 2, 839–847. [Google Scholar] [CrossRef]
- Dight, J.; Zhao, J.; Styke, C.; Khosrotehrani, K.; Patel, J. Resident vascular endothelial progenitor definition and function: The age of reckoning. Angiogenesis 2022, 25, 15–33. [Google Scholar] [CrossRef]
- Fang, S.; Wei, J.; Pentinmikko, N.; Leinonen, H.; Salven, P. Generation of functional blood vessels from a single c-kit+ adult vascular endothelial stem cell. PLoS Biol. 2012, 10, e1001407. [Google Scholar] [CrossRef]
- Patel, J.; Seppanen, E.J.; Rodero, M.P.; Wong, H.Y.; Donovan, P.; Neufeld, Z.; Fisk, N.M.; Francois, M.; Khosrotehrani, K. Functional Definition of Progenitors Versus Mature Endothelial Cells Reveals Key SoxF-Dependent Differentiation Process. Circulation 2017, 135, 786–805. [Google Scholar] [CrossRef]
- Lukowski, S.W.; Patel, J.; Andersen, S.B.; Sim, S.L.; Wong, H.Y.; Tay, J.; Winkler, I.; Powell, J.E.; Khosrotehrani, K. Single-Cell Transcriptional Profiling of Aortic Endothelium Identifies a Hierarchy from Endovascular Progenitors to Differentiated Cells. Cell Rep. 2019, 27, 2748–2758.e3. [Google Scholar] [CrossRef]
- Donovan, P.; Patel, J.; Dight, J.; Wong, H.Y.; Sim, S.L.; Murigneux, V.; Francois, M.; Khosrotehrani, K. Endovascular progenitors infiltrate melanomas and differentiate towards a variety of vascular beds promoting tumor metastasis. Nat. Commun. 2019, 10, 18. [Google Scholar] [CrossRef]
- Francois, M.; Caprini, A.; Hosking, B.; Orsenigo, F.; Wilhelm, D.; Browne, C.; Paavonen, K.; Karnezis, T.; Shayan, R.; Downes, M.; et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008, 456, 643–647. [Google Scholar] [CrossRef]
- Duong, T.; Proulx, S.T.; Luciani, P.; Leroux, J.C.; Detmar, M.; Koopman, P.; Francois, M. Genetic ablation of SOX18 function suppresses tumor lymphangiogenesis and metastasis of melanoma in mice. Cancer Res. 2012, 72, 3105–3114. [Google Scholar] [CrossRef]
- Overman, J.; Fontaine, F.; Moustaqil, M.; Mittal, D.; Sierecki, E.; Sacilotto, N.; Zuegg, J.; Robertson, A.A.; Holmes, K.; Salim, A.A.; et al. Pharmacological targeting of the transcription factor SOX18 delays breast cancer in mice. Elife 2017, 6, e21221. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dominiak, A.; Chelstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Laurenzana, A.; Biagioni, A.; D’Alessio, S.; Bianchini, F.; Chilla, A.; Margheri, F.; Luciani, C.; Mazzanti, B.; Pimpinelli, N.; Torre, E.; et al. Melanoma cell therapy: Endothelial progenitor cells as shuttle of the MMP12 uPAR-degrading enzyme. Oncotarget 2014, 5, 3711–3727. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Liu, Q.; Yang, M.; Zhou, Z.; Ye, Y.; Zhou, Z.; He, X.; Wang, L. Myeloid-derived suppressor cells accumulate among myeloid cells contributing to tumor growth in matrix metalloproteinase 12 knockout mice. Cell. Immunol. 2018, 327, 1–12. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, W.; Torphy, R.J.; Yao, S.; Zhu, G.; Lin, R.; Lugano, R.; Miller, E.N.; Fujiwara, Y.; Bian, L.; et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci. Transl. Med. 2021, 13, eabc8922. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Q.; Hong, Y. Tumor Vessel Normalization: A Window to Enhancing Cancer Immunotherapy. Technol. Cancer Res. Treat. 2020, 19, 1533033820980116. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Krizbai, I.A.; Gasparics, A.; Nagyoszi, P.; Fazakas, C.; Molnar, J.; Wilhelm, I.; Bencs, R.; Rosivall, L.; Sebe, A. Endothelial-mesenchymal transition of brain endothelial cells: Possible role during metastatic extravasation. PLoS ONE 2015, 10, e0123845. [Google Scholar] [CrossRef]
- Platel, V.; Lechevalier, D.; Bourreau, C.; Renault, S.; Soborova, I.; Jeanniere, C.; Martin, L.; Herault, O.; Corre, I.; Clere, N. NOX1 and NOX2: Two enzymes that promote endothelial-to-mesenchymal transition induced by melanoma conditioned media. Pharmacol. Res. 2022, 177, 106097. [Google Scholar] [CrossRef]
- Nagai, N.; Ohguchi, H.; Nakaki, R.; Matsumura, Y.; Kanki, Y.; Sakai, J.; Aburatani, H.; Minami, T. Downregulation of ERG and FLI1 expression in endothelial cells triggers endothelial-to-mesenchymal transition. PLoS Genet. 2018, 14, e1007826. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61. [Google Scholar] [CrossRef]
- Cai, G.; Yu, X.; Youn, C.; Zhou, J.; Xiao, F. SCANNER: A web platform for annotation, visualization and sharing of single cell RNA-seq data. Database 2022, 2022, baab086. [Google Scholar] [CrossRef]
- Garcia-Mulero, S.; Alonso, M.H.; Del Carpio, L.P.; Sanz-Pamplona, R.; Piulats, J.M. Additive Role of Immune System Infiltration and Angiogenesis in Uveal Melanoma Progression. Int. J. Mol. Sci. 2021, 22, 2669. [Google Scholar] [CrossRef]
- Szalai, E.; Wells, J.R.; Ward, L.; Grossniklaus, H.E. Uveal Melanoma Nuclear BRCA1-Associated Protein-1 Immunoreactivity Is an Indicator of Metastasis. Ophthalmology 2018, 125, 203–209. [Google Scholar] [CrossRef]
- Bronkhorst, I.H.; Vu, T.H.; Jordanova, E.S.; Luyten, G.P.; Burg, S.H.; Jager, M.J. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5370–5378. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment—New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8, 766. [Google Scholar] [CrossRef]
- Griffioen, A.W. Anti-angiogenesis: Making the tumor vulnerable to the immune system. Cancer Immunol. Immunother. 2008, 57, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Weston, C.J.; Oo, Y.H.; Westerlund, N.; Stamataki, Z.; Youster, J.; Hubscher, S.G.; Salmi, M.; Jalkanen, S.; Lalor, P.F.; et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal. endothelium. J. Immunol. 2011, 186, 4147–4155. [Google Scholar] [CrossRef] [PubMed]
- Melder, R.J.; Koenig, G.C.; Witwer, B.P.; Safabakhsh, N.; Munn, L.L.; Jain, R.K. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat. Med. 1996, 2, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Ager, A.; May, M.J. Understanding high endothelial venules: Lessons for cancer immunology. Oncoimmunology 2015, 4, e1008791. [Google Scholar] [CrossRef]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.P. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology 2012, 1, 829–839. [Google Scholar] [CrossRef]
- Asrir, A.; Tardiveau, C.; Coudert, J.; Laffont, R.; Blanchard, L.; Bellard, E.; Veerman, K.; Bettini, S.; Lafouresse, F.; Vina, E.; et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell 2022, 40, 318–334.e9. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Simonsen, T.G.; Gaustad, J.V.; Leinaas, M.N.; Rofstad, E.K. High interstitial fluid pressure is associated with tumor-line specific vascular abnormalities in human melanoma xenografts. PLoS ONE 2012, 7, e40006. [Google Scholar] [CrossRef]
- Navalitloha, Y.; Schwartz, E.S.; Groothuis, E.N.; Allen, C.V.; Levy, R.M.; Groothuis, D.R. Therapeutic implications of tumor interstitial fluid pressure in subcutaneous RG-2 tumors. Neuro Oncol. 2006, 8, 227–233. [Google Scholar] [CrossRef]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popovic, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.C.; Jin, P.R.; Chu, L.A.; Hsu, F.F.; Wang, M.R.; Chang, C.C.; Chiou, S.J.; Qiu, J.T.; Gao, D.Y.; Lin, C.C.; et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat. Nanotechnol. 2019, 14, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Kumper, M.; Hessenthaler, S.; Zamek, J.; Niland, S.; Pach, E.; Mauch, C.; Zigrino, P. Loss of Endothelial Cell Matrix Metalloproteinase 14 Reduces Melanoma Growth and Metastasis by Increasing Tumor Vessel Stability. J. Investig. Dermatol. 2021, 142, 1923–1933.e5. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Ting, K.K.; Chen, J.; Coleman, P.; Liu, K.; Wan, L.; Moller, T.; Vadas, M.A.; Gamble, J.R. The VE-Cadherin/beta-catenin signalling axis regulates immune cell infiltration into tumours. Cancer Lett. 2021, 496, 1–15. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Mills, I.G. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef]
- Folkman, J.; Ingber, D. Inhibition of angiogenesis. Semin. Cancer Biol. 1992, 3, 89–96. [Google Scholar]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Dirkx, A.E.; oude Egbrink, M.G.; Castermans, K.; van der Schaft, D.W.; Thijssen, V.L.; Dings, R.P.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Bouma-ter Steege, J.C.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. Faseb J. 2006, 20, 621–630. [Google Scholar] [CrossRef]
- Brouty-Boye, D.; Zetter, B.R. Inhibition of cell motility by interferon. Science 1980, 208, 516–518. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Ingber, D.; Fujita, T.; Kishimoto, S.; Sudo, K.; Kanamaru, T.; Brem, H.; Folkman, J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 1990, 348, 555–557. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Muhsin, M.; Graham, J.; Kirkpatrick, P. Bevacizumab. Nat. Rev. Drug Discov. 2004, 3, 995–996. [Google Scholar] [CrossRef]
- Gross-Goupil, M.; Francois, L.; Quivy, A.; Ravaud, A. Axitinib: A review of its safety and efficacy in the treatment of adults with advanced renal cell carcinoma. Clin. Med. Insights Oncol. 2013, 7, 269–277. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef]
- Algazi, A.P.; Cha, E.; Ortiz-Urda, S.M.; McCalmont, T.; Bastian, B.C.; Hwang, J.; Pampaloni, M.H.; Behr, S.; Chong, K.; Cortez, B.; et al. The combination of axitinib followed by paclitaxel/carboplatin yields extended survival in advanced BRAF wild-type melanoma: Results of a clinical/correlative prospective phase II clinical trial. Br. J. Cancer 2015, 112, 1326–1331. [Google Scholar] [CrossRef]
- Yan, X.; Sheng, X.; Chi, Z.; Si, L.; Cui, C.; Kong, Y.; Tang, B.; Mao, L.; Wang, X.; Lian, B.; et al. Randomized Phase II Study of Bevacizumab in Combination With Carboplatin Plus Paclitaxel in Patients With Previously Untreated Advanced Mucosal Melanoma. J. Clin. Oncol. 2021, 39, 881–889. [Google Scholar] [CrossRef]
- Taylor, M.H.; Lee, C.H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 1154–1163. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; El-Masry, M.; Osann, K.; Parmakhtiar, B.; Yamamoto, M.; Jakowatz, J.G. Phase II study of pazopanib in combination with paclitaxel in patients with metastatic melanoma. Cancer Chemother. Pharmacol. 2018, 82, 353–360. [Google Scholar] [CrossRef]
- Hussain, R.N.; Heimann, H.; Damato, B. Neoadjuvant intravitreal ranibizumab treatment in high-risk ocular melanoma patients: A two-stage single-centre phase II single-arm study. Melanoma Res. 2020, 30, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Ibrahim, N.; Lawrence, D.P.; Aldridge, J.; Giobbie-Hurder, A.; Hodi, F.S.; Flaherty, K.T.; Conley, C.; Mier, J.W.; Atkins, M.B.; et al. A Phase I Trial of Bortezomib and Sorafenib in Advanced Malignant Melanoma. Oncologist 2015, 20, 617–618. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flaherty, K.T.; Lee, S.J.; Zhao, F.; Schuchter, L.M.; Flaherty, L.; Kefford, R.; Atkins, M.B.; Leming, P.; Kirkwood, J.M. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J. Clin. Oncol. 2013, 31, 373–379. [Google Scholar] [CrossRef]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.; De Divitiis, C.; Ottaiano, A.; von Arx, C.; Scala, S.; Tatangelo, F.; Delrio, P.; Tafuto, S. Lenvatinib, a molecule with versatile application: From preclinical evidence to future development in anti-cancer treatment. Cancer Manag. Res. 2019, 11, 3847–3860. [Google Scholar] [CrossRef] [PubMed]
- Varker, K.A.; Biber, J.E.; Kefauver, C.; Jensen, R.; Lehman, A.; Young, D.; Wu, H.; Lesinski, G.B.; Kendra, K.; Chen, H.X.; et al. A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Ann. Surg. Oncol. 2007, 14, 2367–2376. [Google Scholar] [CrossRef]
- Corrie, P.G.; Marshall, A.; Nathan, P.D.; Lorigan, P.; Gore, M.; Tahir, S.; Faust, G.; Kelly, C.G.; Marples, M.; Danson, S.J.; et al. Adjuvant bevacizumab for melanoma patients at high risk of recurrence: Survival analysis of the AVAST-M trial. Ann. Oncol. 2018, 29, 1843–1852. [Google Scholar] [CrossRef]
- Helfrich, I.; Scheffrahn, I.; Bartling, S.; Weis, J.; von Felbert, V.; Middleton, M.; Kato, M.; Ergun, S.; Augustin, H.G.; Schadendorf, D. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 2010, 207, 491–503. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef]
- Mouriaux, F.; Servois, V.; Parienti, J.J.; Lesimple, T.; Thyss, A.; Dutriaux, C.; Neidhart-Berard, E.M.; Penel, N.; Delcambre, C.; Peyro Saint Paul, L.; et al. Sorafenib in metastatic uveal melanoma: Efficacy, toxicity and health-related quality of life in a multicentre phase II study. Br. J. Cancer 2016, 115, 20–24. [Google Scholar] [CrossRef]
- Song, Y.; Fu, Y.; Xie, Q.; Zhu, B.; Wang, J.; Zhang, B. Anti-angiogenic Agents in Combination With Immune Checkpoint Inhibitors: A Promising Strategy for Cancer Treatment. Front. Immunol. 2020, 11, 1956. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Ott, P.A.; Nazzaro, M.; Pfaff, K.L.; Gjini, E.; Felt, K.D.; Wolff, J.O.; Buchbinder, E.I.; Haq, R.; Sullivan, R.J.; Lawrence, D.P.; et al. Combining CTLA-4 and angiopoietin-2 blockade in patients with advanced melanoma: A phase I trial. J. Immunother. Cancer 2021, 9, e003318. [Google Scholar] [CrossRef]
- Li, S.; Wu, X.; Yan, X.; Zhou, L.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Mao, L.; Lian, B.; et al. Toripalimab plus axitinib in patients with metastatic mucosal melanoma: 3-year survival update and biomarker analysis. J. Immunother. Cancer 2022, 10, e004036. [Google Scholar] [CrossRef]
- Collet, G.; Szade, K.; Nowak, W.; Klimkiewicz, K.; El Hafny-Rahbi, B.; Szczepanek, K.; Sugiyama, D.; Weglarczyk, K.; Foucault-Collet, A.; Guichard, A.; et al. Endothelial precursor cell-based therapy to target the pathologic angiogenesis and compensate tumor hypoxia. Cancer Lett. 2016, 370, 345–357. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-Angiogenic Agent | Mode of Action/TARGET | Type of Melanoma | Clinical Indications | Reference |
|---|---|---|---|---|
| Axitinib (Inlyta®) | Tyrosine kinase inhibitor of VEGFR1, -2,-3, c-Kit, and PDGFR [155] | Human mucosal melanoma; advanced BRAF wild-type melanoma | Combination with toripalimab; combination with paclitaxel/carboplatin | [156,157] |
| Bevacizumab (Avastin®) | Monoclonal antibody to VEGFA | Human mucosal melanoma, metastatic melanoma | Combination with carboplatin plus paclitaxel | [158] |
| Lenvatinib mesylate (Lenvima®) | Kinase inhibitor against VEGFR1, -2, -3, PDGFR, RET, FGFR, c-Kit | Melanoma | Combination with pembrolizumab | [159] |
| Pazopanib (Votrient®) | Tyrosine kinase inhibitor of VEGFR1, -2, -3, PDGFR, FGFR, and c-Kit | Metastatic melanoma | Combination with paclitaxel | [160] |
| Ranibizumab (Lucentis®) | Monoclonal antibody to VEGFA | Uveal melanoma | As a single agent | [161] |
| Sorafenib (Nexavar®) | Kinase inhibitor against VEGFR, PDGFR, BRAF, RAF-1, c-Kit, and RET | Metastatic melanoma | As a single agent and in combination with bortezomib, carboplatin, and paclitaxel | [162,163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashemi, G.; Dight, J.; Khosrotehrani, K.; Sormani, L. Melanoma Tumour Vascularization and Tissue-Resident Endothelial Progenitor Cells. Cancers 2022, 14, 4216. https://doi.org/10.3390/cancers14174216
Hashemi G, Dight J, Khosrotehrani K, Sormani L. Melanoma Tumour Vascularization and Tissue-Resident Endothelial Progenitor Cells. Cancers. 2022; 14(17):4216. https://doi.org/10.3390/cancers14174216
Chicago/Turabian StyleHashemi, Ghazaleh, James Dight, Kiarash Khosrotehrani, and Laura Sormani. 2022. "Melanoma Tumour Vascularization and Tissue-Resident Endothelial Progenitor Cells" Cancers 14, no. 17: 4216. https://doi.org/10.3390/cancers14174216
APA StyleHashemi, G., Dight, J., Khosrotehrani, K., & Sormani, L. (2022). Melanoma Tumour Vascularization and Tissue-Resident Endothelial Progenitor Cells. Cancers, 14(17), 4216. https://doi.org/10.3390/cancers14174216