
Enhanced Cytotoxicity on Cancer Cells by Combinational Treatment of PARP Inhibitor and 5-Azadeoxycytidine Accompanying Distinct Transcriptional Profiles

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals
2.3. Flow Cytometry
2.4. Cytotoxicity Assay
2.5. Pulsed-Field Gel Electrophoresis
2.6. DNA Ladder Formation Induced by Apoptosis
2.7. Genomic DNA Extraction and Restriction Enzyme Treatment
2.8. Microarray Analysis
2.9. Statistical Analysis
3. Results
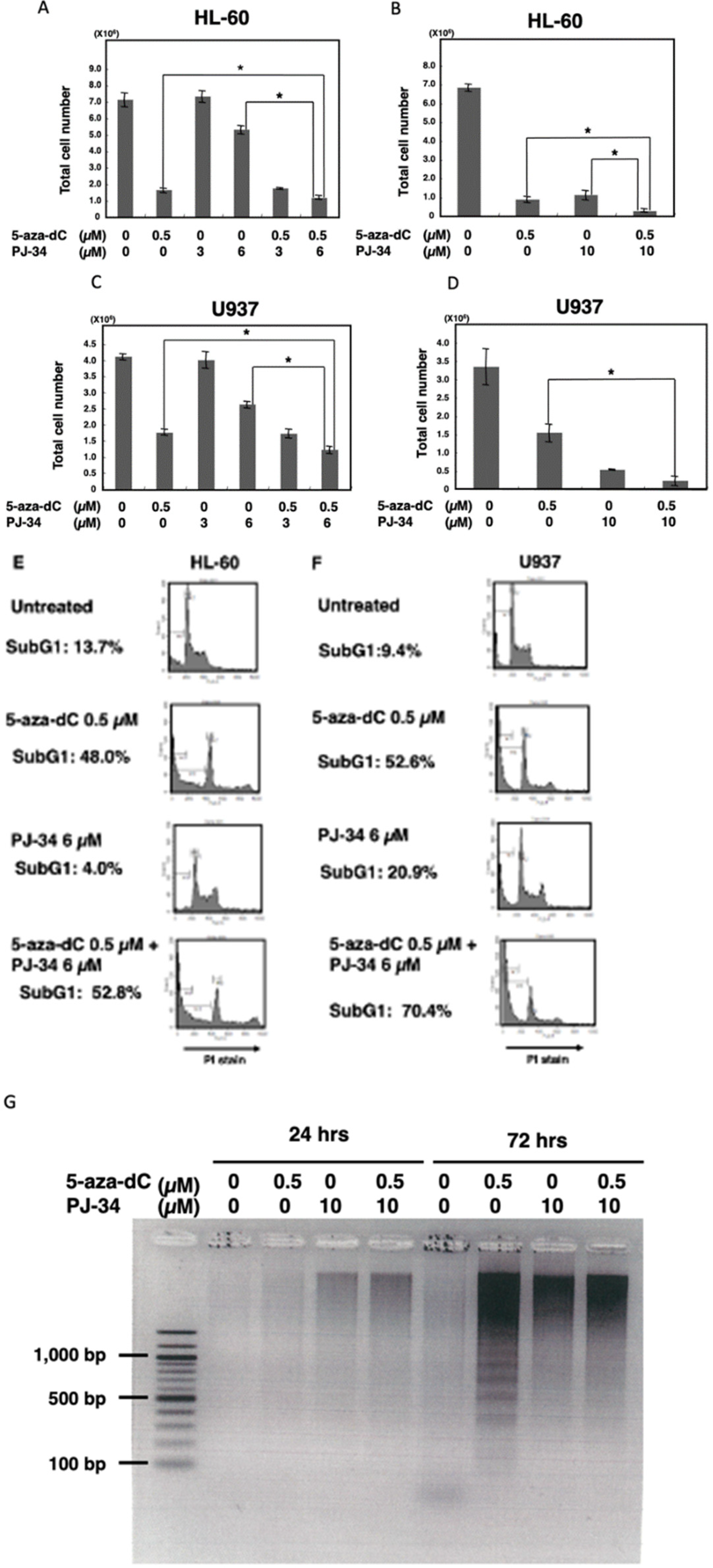
3.1. PARP Inhibitor Enhanced Cytotoxicity of 5-aza-dC in HL-60 and U937 Cells
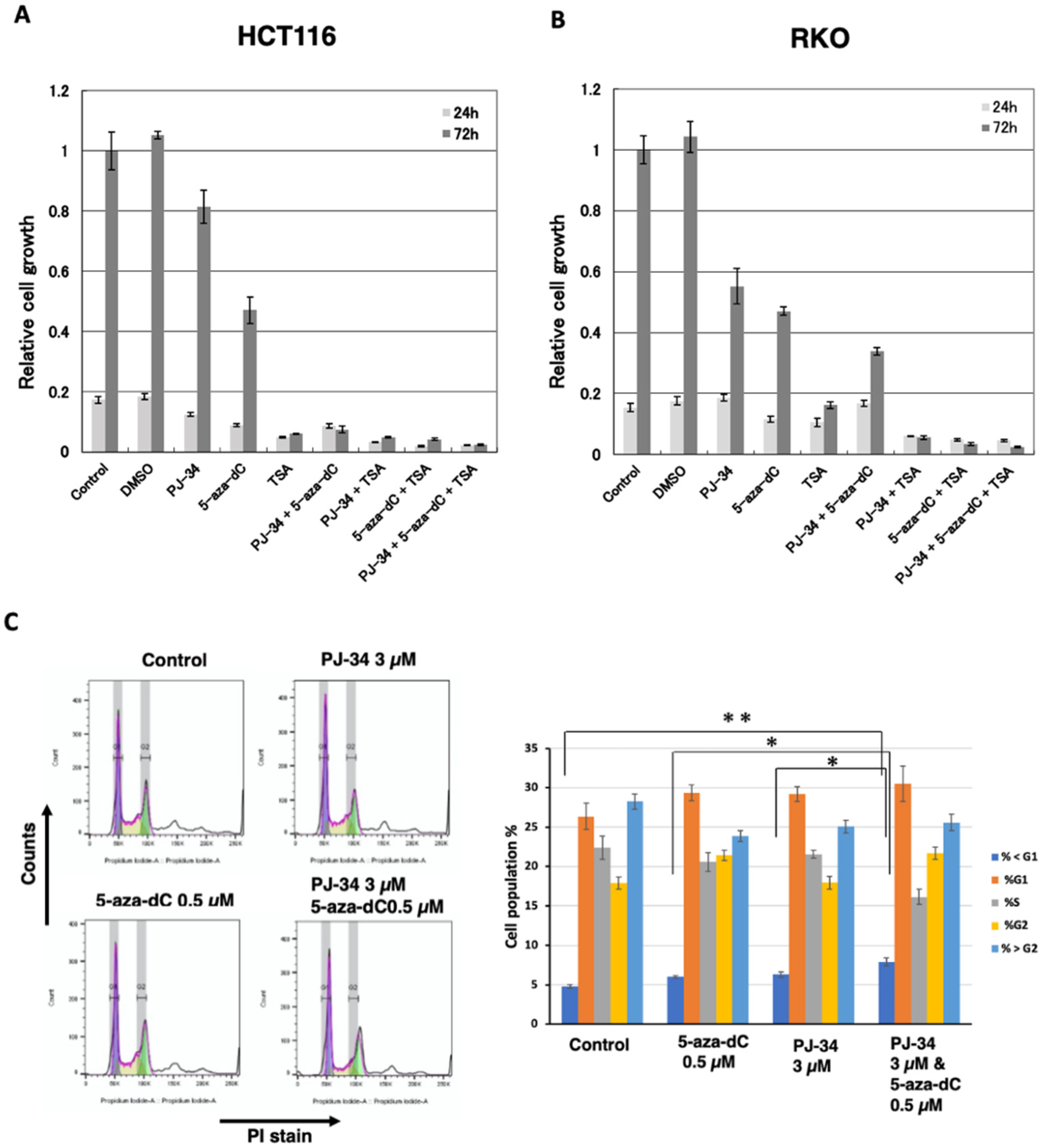
3.2. PARP Inhibitor Enhanced Cytotoxicity of 5-aza-dC in Colon Cancer HCT116 and RKO Cell Lines
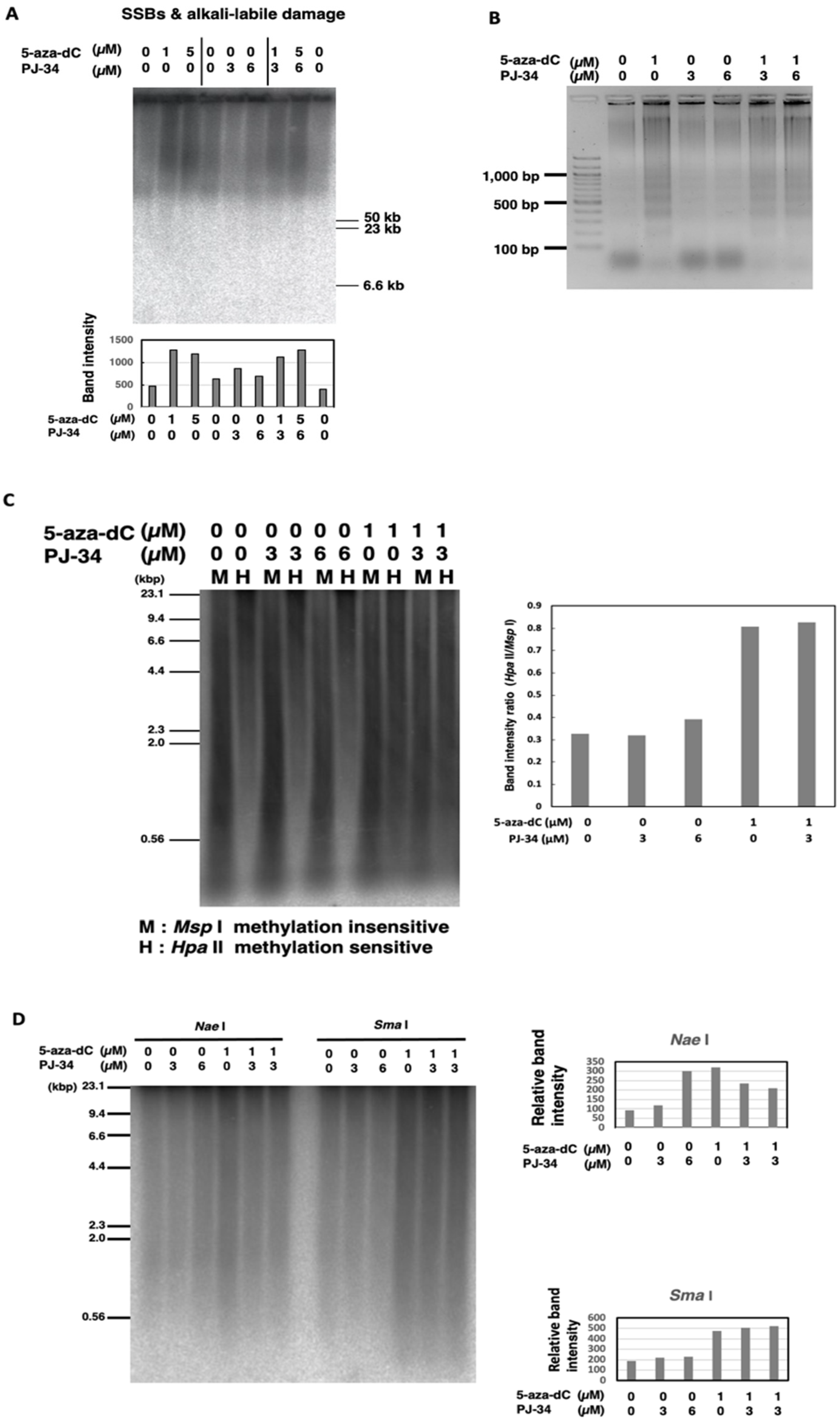
3.3. Treatment with 5-aza-dC but Not PJ-34 Caused SSBs and Alkali-Labile DNA Damage and Apoptotic DNA Ladder Formation in HCT116 Cells
3.4. Global Genome DNA Demethylation was Induced by 5-aza-dC but Not Affected by PJ-34 in HCT116
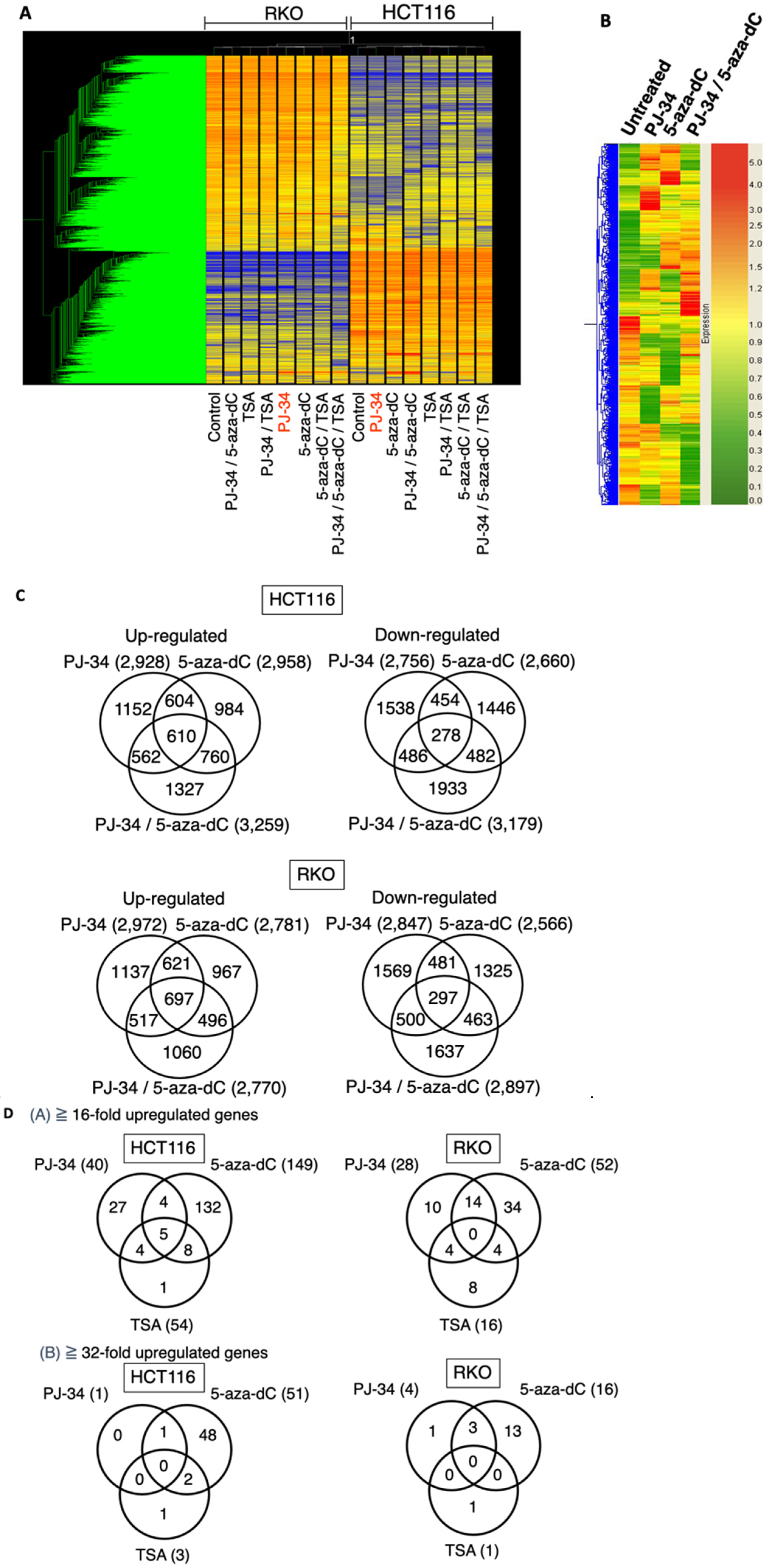
3.5. Induction of Distinct Changes in Transcriptional Profiles by 5-aza-dC, PARP Inhibitor, and Their Combination in HCT116 and RKO Cells
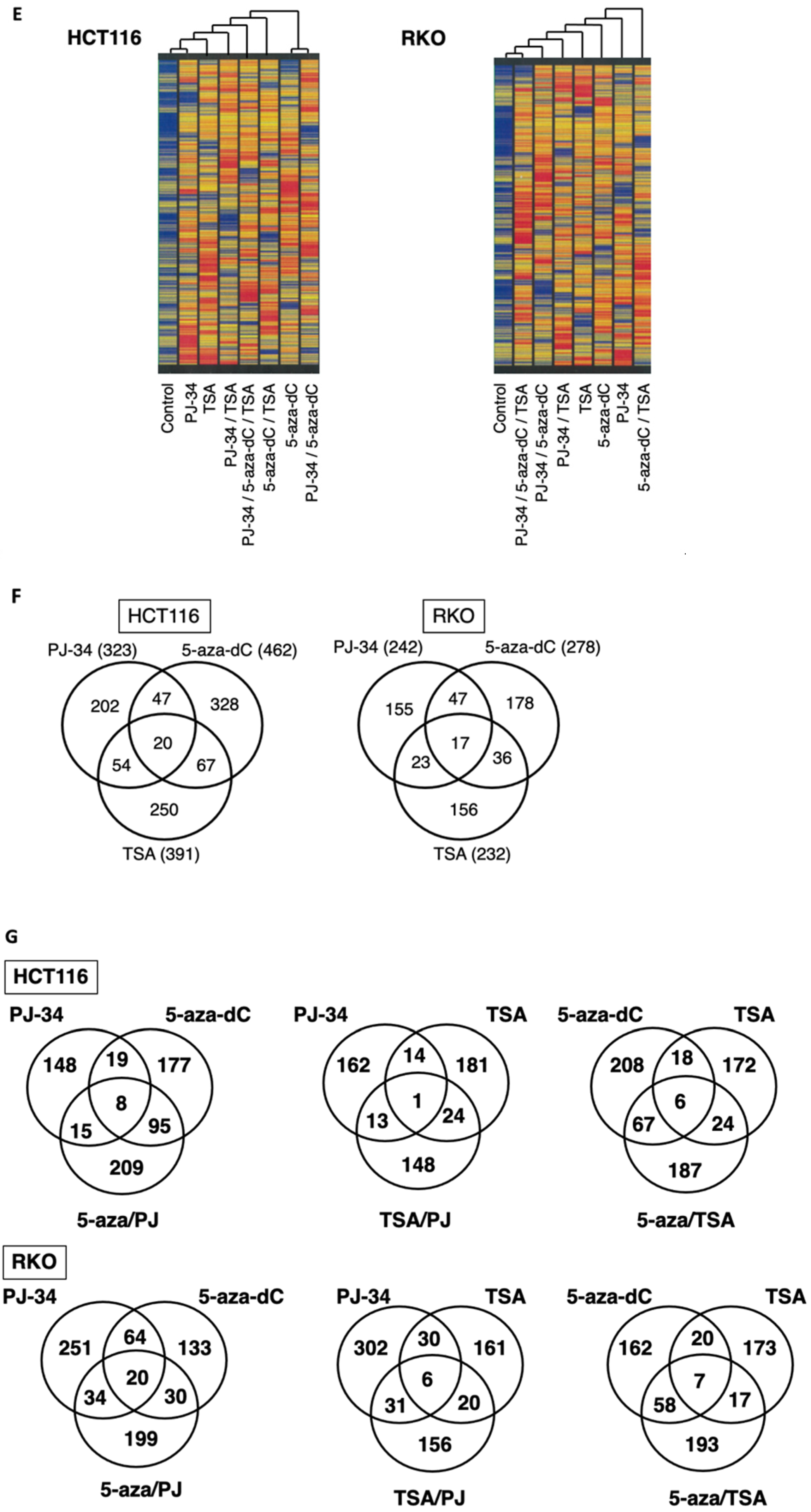
3.6. Distinct Profiles of Reactivation of Silenced Genes by 5-aza-dC, PARP Inhibitor, and Their Combination in HCT116 and RKO Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar]
- Ushijima, T. Epigenetic field for cancerization. J. Biochem. Mol. Biol. 2007, 40, 142–150. [Google Scholar]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for mds and cmml: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar]
- Kim, N.; Norsworthy, K.J.; Subramaniam, S.; Chen, H.; Manning, M.L.; Kitabi, E.; Earp, J.; Ehrlich, L.A.; Okusanya, O.O.; Vallejo, J.; et al. Fda approval summary: Decitabine and cedazuridine tablets for myelodysplastic syndromes. Clin. Cancer Res. 2022, 28, 3411–3416. [Google Scholar]
- Xu, K.; Hansen, E. Novel agents for myelodysplastic syndromes. J. Oncol. Pharm. Pract. 2021, 27, 1982–1992. [Google Scholar]
- Shi, H.; Wei, S.H.; Leu, Y.W.; Rahmatpanah, F.; Liu, J.C.; Yan, P.S.; Nephew, K.P.; Huang, T.H. Triple analysis of the cancer epigenome: An integrated microarray system for assessing gene expression, DNA methylation, and histone acetylation. Cancer Res. 2003, 63, 2164–2171. [Google Scholar]
- Yamashita, K.; Upadhyay, S.; Osada, M.; Hoque, M.O.; Xiao, Y.; Mori, M.; Sato, F.; Meltzer, S.J.; Sidransky, D. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell 2002, 2, 485–495. [Google Scholar]
- Luszczek, W.; Cheriyath, V.; Mekhail, T.M.; Borden, E.C. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: Correlation of resistance with ifn-stimulated gene expression. Mol. Cancer Ther. 2010, 9, 2309–2321. [Google Scholar]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, U.T.; Werner, M.; Busch, H.; Boerries, M.; et al. Selective inhibition of esophageal cancer cells by combination of hdac inhibitors and azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(adp-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar]
- Kanai, M.; Tong, W.M.; Sugihara, E.; Wang, Z.Q.; Fukasawa, K.; Miwa, M. Involvement of poly(adp-ribose) polymerase 1 and poly(adp-ribosyl)ation in regulation of centrosome function. Mol. Cell. Biol. 2003, 23, 2451–2462. [Google Scholar]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(adp-ribose) polymerase in tumors from brca mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar]
- Yasukawa, M.; Fujihara, H.; Fujimori, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yamamoto, N.; Kishi, Y.; Hamada, Y.; Masutani, M. Synergetic effects of parp inhibitor azd2281 and cisplatin in oral squamous cell carcinoma in vitro and in vivo. Int. J. Mol. Sci. 2016, 17, 272. [Google Scholar]
- Ikejima, M.; Noguchi, S.; Yamashita, R.; Ogura, T.; Sugimura, T.; Gill, D.M.; Miwa, M. The zinc fingers of human poly(adp-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. Other structures recognize intact DNA. J. Biol. Chem. 1990, 265, 21907–21913. [Google Scholar]
- Thomas, C.; Ji, Y.; Wu, C.; Datz, H.; Boyle, C.; MacLeod, B.; Patel, S.; Ampofo, M.; Currie, M.; Harbin, J.; et al. Hit and run versus long-term activation of parp-1 by its different domains fine-tunes nuclear processes. Proc. Natl. Acad. Sci. USA 2019, 116, 9941–9946. [Google Scholar]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent parp-1 activation by phosphorylated erk2 increases elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar]
- Idogawa, M.; Yamada, T.; Honda, K.; Sato, S.; Imai, K.; Hirohashi, S. Poly(adp-ribose) polymerase-1 is a component of the oncogenic t-cell factor-4/beta-catenin complex. Gastroenterology 2005, 128, 1919–1936. [Google Scholar]
- Gadad, S.S.; Camacho, C.V.; Malladi, V.; Hutti, C.R.; Nagari, A.; Kraus, W.L. Parp-1 regulates estrogen-dependent gene expression in estrogen receptor alpha-positive breast cancer cells. Mol. Cancer Res. 2021, 19, 1688–1698. [Google Scholar]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. Adp-ribosyltransferases parp1 and parp7 safeguard pluripotency of es cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(adp-ribosyl)ation regulates ctcf-dependent chromatin insulation. Nat. Genet. 2004, 36, 1105–1110. [Google Scholar]
- Pavri, R.; Lewis, B.; Kim, T.K.; Dilworth, F.J.; Erdjument-Bromage, H.; Tempst, P.; de Murcia, G.; Evans, R.; Chambon, P.; Reinberg, D. Parp-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol. Cell 2005, 18, 83–96. [Google Scholar]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly(adp-ribose) polymerase-1 by p300/creb-binding protein regulates coactivation of nf-kappab-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar]
- Tulin, A.; Spradling, A. Chromatin loosening by poly(adp)-ribose polymerase (parp) at drosophila puff loci. Science 2003, 299, 560–562. [Google Scholar]
- Osada, T.; Ryden, A.M.; Masutani, M. Poly(adp-ribosylation) regulates chromatin organization through histone h3 modification and DNA methylation of the first cell cycle of mouse embryos. Biochem. Biophys. Res. Commun. 2013, 434, 15–21. [Google Scholar]
- Osada, T.; Nozaki, T.; Masutani, M. Parp1 deficiency confers defects in chromatin surveillance and remodeling during reprogramming by nuclear transfer. Curr. Protein Pept. Sci. 2016, 17, 693–704. [Google Scholar]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.; et al. Rational combination therapy with parp and mek inhibitors capitalizes on therapeutic liabilities in ras mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-cpg-binding protein mecp2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar]
- Zhang, Y.; Fatima, N.; Dufau, M.L. Coordinated changes in DNA methylation and histone modifications regulate silencing/derepression of luteinizing hormone receptor gene transcription. Mol. Cell. Biol. 2005, 25, 7929–7939. [Google Scholar]
- Zampieri, M.; Guastafierro, T.; Calabrese, R.; Ciccarone, F.; Bacalini, M.G.; Reale, A.; Perilli, M.; Passananti, C.; Caiafa, P. Adp-ribose polymers localized on ctcf-parp1-dnmt1 complex prevent methylation of ctcf target sites. Biochem. J. 2012, 441, 645–652. [Google Scholar]
- Sutherland, J.C.; Monteleone, D.C.; Mugavero, J.H.; Trunk, J. Unidirectional pulsed-field electrophoresis of single- and double-stranded DNA in agarose gels: Analytical expressions relating mobility and molecular length and their application in the measurement of strand breaks. Anal. Biochem. 1987, 162, 511–520. [Google Scholar]
- Freeman, S.E.; Blackett, A.D.; Monteleone, D.C.; Setlow, R.B.; Sutherland, B.M.; Sutherland, J.C. Quantitation of radiation-, chemical-, or enzyme-induced single strand breaks in nonradioactive DNA by alkaline gel electrophoresis: Application to pyrimidine dimers. Anal. Biochem. 1986, 158, 119–129. [Google Scholar]
- Palve, V.; Knezevic, C.E.; Bejan, D.S.; Luo, Y.; Li, X.; Novakova, S.; Welsh, E.A.; Fang, B.; Kinose, F.; Haura, E.B.; et al. The non-canonical target parp16 contributes to polypharmacology of the parp inhibitor talazoparib and its synergy with wee1 inhibitors. Cell Chem. Biol. 2022, 29, 202–214.e207. [Google Scholar]
- Orta, M.L.; Höglund, A.; Calderón-Montaño, J.M.; Domínguez, I.; Burgos-Morón, E.; Visnes, T.; Pastor, N.; Ström, C.; López-lázaro, M.; Helleday, T. The parp inhibitor olaparib disrupts base excision repair of 5-aza-2’-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar]
- Chen, J.; Yuan, W.; Wu, L.; Tang, Q.; Xia, Q.; Ji, J.; Liu, Z.; Ma, Z.; Zhou, Z.; Cheng, Y.; et al. Pdgf-d promotes cell growth, aggressiveness, angiogenesis and emt transformation of colorectal cancer by activation of notch1/twist1 pathway. Oncotarget 2017, 8, 9961–9973. [Google Scholar]
- Qin, C.; Zhang, Q.; Wu, G. Ranbp9 suppresses tumor proliferation in colorectal cancer. Oncol. Lett. 2019, 17, 4409–4416. [Google Scholar]
- Duan, Z.; Duan, Y.; Lamendola, D.E.; Yusuf, R.Z.; Naeem, R.; Penson, R.T.; Seiden, M.V. Overexpression of mage/gage genes in paclitaxel/doxorubicin-resistant human cancer cell lines. Clin. Cancer Res. 2003, 9, 2778–2785. [Google Scholar]
- Dong, X.; Wang, J.; Tang, B.; Hao, Y.X.; Li, P.Y.; Li, S.Y.; Yu, P.W. The role and gene expression profile of socs3 in colorectal carcinoma. Oncotarget 2018, 9, 15984–15996. [Google Scholar]
- Suzuki, H.; Watkins, D.N.; Jair, K.W.; Schuebel, K.E.; Markowitz, S.D.; Chen, W.D.; Pretlow, T.P.; Yang, B.; Akiyama, Y.; Van Engeland, M.; et al. Epigenetic inactivation of sfrp genes allows constitutive wnt signaling in colorectal cancer. Nat. Genet. 2004, 36, 417–422. [Google Scholar]
- Christou, N.; Perraud, A.; Blondy, S.; Jauberteau, M.O.; Battu, S.; Mathonnet, M. E-cadherin: A potential biomarker of colorectal cancer prognosis. Oncol. Lett. 2017, 13, 4571–4576. [Google Scholar]
- Fang, J.Y.; Yang, L.; Zhu, H.Y.; Chen, Y.X.; Lu, J.; Lu, R.; Cheng, Z.H.; Xiao, S.D. 5-aza-2′-deoxycitydine induces demethylation and up-regulates transcription of p16ink4a gene in human gastric cancer cell lines. Chin. Med. J. (Engl.) 2004, 117, 99–103. [Google Scholar]
- Stepnik, M.; Spryszynska, S.; Gorzkiewicz, A.; Ferlinska, M. Cytotoxicity of anticancer drugs and pj-34 (poly(adp-ribose)polymerase-1 (parp-1) inhibitor) on hl-60 and jurkat cells. Adv. Clin. Exp. Med. 2017, 26, 379–385. [Google Scholar]
- Ogino, H.; Nozaki, T.; Gunji, A.; Maeda, M.; Suzuki, H.; Ohta, T.; Murakami, Y.; Nakagama, H.; Sugimura, T.; Masutani, M. Loss of parp-1 affects gene expression profile in a genome-wide manner in es cells and liver cells. BMC Genomics 2007, 8, 41. [Google Scholar]
- Liang, Y.C.; Hsu, C.Y.; Yao, Y.L.; Yang, W.M. Parp-2 regulates cell cycle-related genes through histone deacetylation and methylation independently of poly(adp-ribosyl)ation. Biochem. Biophys. Res. Commun. 2013, 431, 58–64. [Google Scholar]
- Tang, B.; Luo, H.; Xie, S.; Pan, C.; Fu, J. Deubiquitination of tnks1 regulates wnt/beta-catenin to affect the expression of usp25 to promote the progression of glioma. Dis. Markers 2022, 2022, 9087190. [Google Scholar]
- Wu, Q.; Xuan, Y.F.; Su, A.L.; Bao, X.B.; Miao, Z.H.; Wang, Y.Q. Tnks inhibitors potentiate proliferative inhibition of bet inhibitors via reducing beta-catenin in colorectal cancer cells. Am. J. Cancer Res. 2022, 12, 1069–1087. [Google Scholar]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase i clinical trial of DNA methyltransferase inhibitor decitabine and parp inhibitor talazoparib combination therapy in relapsed/refractory acute myeloid leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group b. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase iii study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50 * | IC30 * |
|---|---|---|
| 5-aza-dC | >0.7 μM | 0.47 μM |
| PJ-34 | >3.4 μM | >3.4 μM |
| 5-aza-dC/PJ-34 (7:34) # | 5-aza-dC 0.61 μM | 5-aza-dC 0.44 μM |
| PJ-34 3.0 μM | PJ-34 2.1 μM | |
| 5-aza-dC | >0.7 μM | 0.57 μM |
| Olaparib | >3.8 μM | 3.3 μM |
| 5-aza-dC/Olaparib (7:38) # | 5-aza-dC 0.28 μM | 5-aza-dC 0.13 μM |
| Olaparib 1.5 μM | Olaparib 0.74 μM | |
| 5-aza-dC | >0.7 | 0.61 |
| Talazoparib | 0.095 | 0.032 |
| 5-aza-dC/Talazoparib (7:1) # | 5-aza-dC 0.14 μM | 5-aza-dC 0.64 μM |
| Talazoparib 0.02 μM | Talazoparib 0.0092 μM |
| (i) Both single and combinational treatments show reactivation, and the combinational treatment shows higher levels of reactivation than the reactivation by the single treatment. | ||
| Combination | HCT116 | RKO |
| PJ-34/5-aza-dC | 48 | 51 |
| PJ-34/TSA | 29 | 31 |
| 5-aza-dC/TSA | 51 | 45 |
| (ii) Either of single treatments shows reactivation, and the combinational treatment shows higher levels of reactivation than the reactivation by the single treatment | ||
| Combination (classified in groups with the level of reactivation in single treatment) | HCT116 | RKO |
| PJ-34 > 5-aza-dC | 35 | 29 |
| 5-aza-dC > PJ-34 | 31 | 89 |
| PJ-34 > TSA | 43 | 23 |
| TSA > PJ-34 | 27 | 33 |
| 5-aza-dC > TSA | 56 | 26 |
| TSA > 5-aza-dC | 33 | 33 |
| (iii) The combinational treatment only shows reactivation | ||
| Combination | HCT116 | RKO |
| PJ-34/5-aza-dC | 94 | 92 |
| PJ-34/TSA | 59 | 47 |
| 5-aza-dC/TSA | 77 | 79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araki, T.; Hamada, K.; Myat, A.B.; Ogino, H.; Hayashi, K.; Maeda, M.; Tong, Y.; Murakami, Y.; Nakao, K.; Masutani, M. Enhanced Cytotoxicity on Cancer Cells by Combinational Treatment of PARP Inhibitor and 5-Azadeoxycytidine Accompanying Distinct Transcriptional Profiles. Cancers 2022, 14, 4171. https://doi.org/10.3390/cancers14174171
Araki T, Hamada K, Myat AB, Ogino H, Hayashi K, Maeda M, Tong Y, Murakami Y, Nakao K, Masutani M. Enhanced Cytotoxicity on Cancer Cells by Combinational Treatment of PARP Inhibitor and 5-Azadeoxycytidine Accompanying Distinct Transcriptional Profiles. Cancers. 2022; 14(17):4171. https://doi.org/10.3390/cancers14174171
Chicago/Turabian StyleAraki, Tomonori, Kensuke Hamada, Aung Bhone Myat, Hideki Ogino, Kohei Hayashi, Miho Maeda, Ying Tong, Yasufumi Murakami, Kazuhiko Nakao, and Mitsuko Masutani. 2022. "Enhanced Cytotoxicity on Cancer Cells by Combinational Treatment of PARP Inhibitor and 5-Azadeoxycytidine Accompanying Distinct Transcriptional Profiles" Cancers 14, no. 17: 4171. https://doi.org/10.3390/cancers14174171
APA StyleAraki, T., Hamada, K., Myat, A. B., Ogino, H., Hayashi, K., Maeda, M., Tong, Y., Murakami, Y., Nakao, K., & Masutani, M. (2022). Enhanced Cytotoxicity on Cancer Cells by Combinational Treatment of PARP Inhibitor and 5-Azadeoxycytidine Accompanying Distinct Transcriptional Profiles. Cancers, 14(17), 4171. https://doi.org/10.3390/cancers14174171