
Altered Adipokine Expression in Tumor Microenvironment Promotes Development of Triple Negative Breast Cancer





Abstract
:Simple Summary
Abstract

1. Introduction
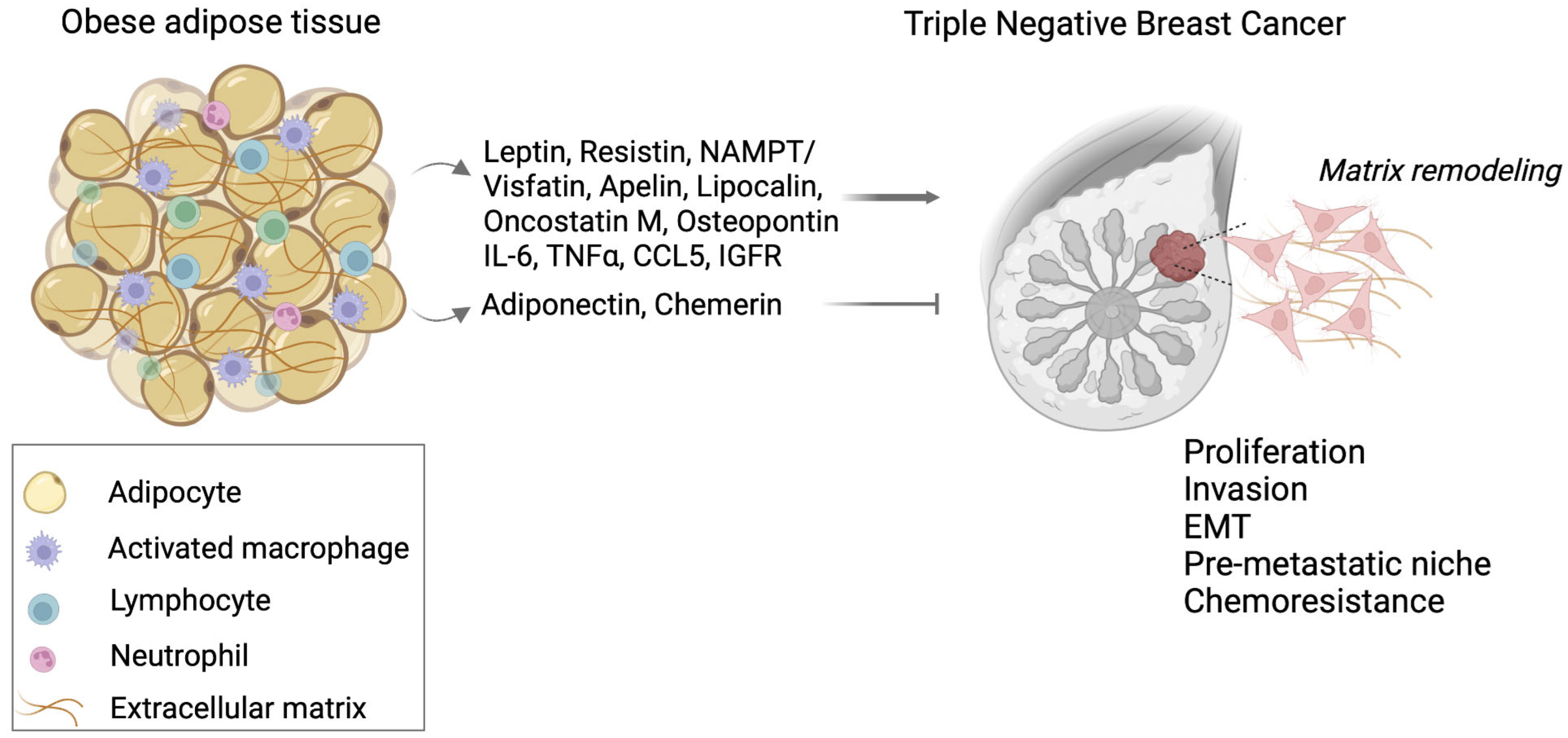
2. Obesity Fat Tissue and Tumor Microenvironment
3. TNBC and Adipose Tissue
4. Adipokines and TNBC
4.1. Leptin
4.2. Adiponectin
4.3. Resistin
4.4. NAMPT/Visfatin
4.5. Lipocalin-2
4.6. Apelin
4.7. Chemerin
4.8. Oncostatin M
4.9. Osteopontin
4.10. Other Adipokines
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pérez-Hernández, A.I.; Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Frühbeck, G. Mechanisms Linking Excess Adiposity and Carcinogenesis Promotion. Front. Endocrinol. 2014, 5, 65. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Nattenmüller, C.J.; Kriegsmann, M.; Sookthai, D.; Fortner, R.T.; Steffen, A.; Walter, B.; Johnson, T.; Kneisel, J.; Katzke, V.; Bergmann, M.; et al. Obesity as risk factor for subtypes of breast cancer: Results from a prospective cohort study. BMC Cancer 2018, 18, 616. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.M.; Press, M.F.; Haile, R.W.; Lynch, C.F.; Glaser, S.L.; Schildkraut, J.; Gammon, M.D.; Douglas Thompson, W.; Bernstein, J.L. Risk factors by molecular subtypes of breast cancer across a population-based study of women 56 years or younger. Breast Cancer Res. Treat. 2011, 130, 587–597. [Google Scholar] [CrossRef]
- Sun, H.; Zou, J.; Chen, L.; Zu, X.; Wen, G.; Zhong, J. Triple-negative breast cancer and its?association with obesity (Review). Mol. Clin. Oncol. 2017, 7, 935–942. [Google Scholar] [CrossRef]
- Kolb, R.; Zhang, W. Obesity and Breast Cancer: A Case of Inflamed Adipose Tissue. Cancers 2020, 12, 1686. [Google Scholar] [CrossRef]
- Ayoub, N.M.; Yaghan, R.J.; Abdo, N.M.; Matalka, I.I.; Akhu-Zaheya, L.M.; Al-Mohtaseb, A.H. Impact of Obesity on Clinicopathologic Characteristics and Disease Prognosis in Pre- and Postmenopausal Breast Cancer Patients: A Retrospective Institutional Study. J. Obes. 2019, 2019, 3820759. [Google Scholar] [CrossRef]
- Calle, E.E.; Thun, M.J. Obesity and cancer. Oncogene 2004, 23, 6365–6378. [Google Scholar] [CrossRef]
- Hao, S.; Liu, Y.; Yu, K.-D.; Chen, S.; Yang, W.-T.; Shao, Z.-M. Overweight as a Prognostic Factor for Triple-Negative Breast Cancers in Chinese Women. PLoS ONE 2015, 10, e0129741. [Google Scholar] [CrossRef] [PubMed]
- Playdon, M.C.; Bracken, M.B.; Sanft, T.B.; Ligibel, J.A.; Harrigan, M.; Irwin, M.L. Weight Gain After Breast Cancer Diagnosis and All-Cause Mortality: Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2015, 107, djv275. [Google Scholar] [CrossRef] [PubMed]
- Allott, E.H.; Hursting, S.D. Obesity and cancer: Mechanistic insights from transdisciplinary studies. Endocr. Relat. Cancer 2015, 22, R365–R386. [Google Scholar] [CrossRef]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer—mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef]
- D’Esposito, V.; Passaretti, F.; Hammarstedt, A.; Liguoro, D.; Terracciano, D.; Molea, G.; Canta, L.; Miele, C.; Smith, U.; Beguinot, F.; et al. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia 2012, 55, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.L.; Shaw, J.E.; Peeters, A.; Cartensen, B.; Magliano, D.J. Cancer Risk Among People With Type 1 and Type 2 Diabetes: Disentangling True Associations, Detection Bias, and Reverse Causation. Diabetes Care 2015, 38, 264–270. [Google Scholar] [CrossRef]
- Rajala, M.W.; Scherer, P.E. Minireview: The Adipocyte—At the Crossroads of Energy Homeostasis, Inflammation, and Atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J.; Muruzábal, F.J.; Burrell, M.A. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Metab. 2001, 280, E827–E847. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, B.S.; Ryu, H.S.; Lee, H.-B.; Lee, M.; Park, I.A.; Kim, J.; Han, W.; Noh, D.-Y.; Moon, H.-G. Transition into inflammatory cancer-associated adipocytes in breast cancer microenvironment requires microRNA regulatory mechanism. PLoS ONE 2017, 12, e0174126. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Piperigkou, Z.; Passi, A.; Götte, M.; Rousselle, P.; Vlodavsky, I. Extracellular matrix-based cancer targeting. Trends Mol. Med. 2021, 27, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Andresen Eguiluz, R.C.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015, 7, 301ra130. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef]
- Bifulco, M.; Pisanti, S. “Adiponcosis”: A New Term to Name the Obesity and Cancer Link. J. Clin. Endocrinol. Metab. 2013, 98, 4664–4665. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef] [PubMed]
- Tworoger, S.S.; Huang, T. Obesity and Ovarian Cancer; Springer: Berlin/Heidelberg, Germany, 2016; pp. 155–176. [Google Scholar]
- Tarasiuk, A.; Mosińska, P.; Fichna, J. The mechanisms linking obesity to colon cancer: An overview. Obes. Res. Clin. Pract. 2018, 12, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Rio, M.-C.; Dali-Youcef, N.; Tomasetto, C. Local adipocyte cancer cell paracrine loop: Can “sick fat” be more detrimental? Horm. Mol. Biol. Clin. Investig. 2015, 21, 43–56. [Google Scholar] [CrossRef]
- Attané, C.; Muller, C. Drilling for Oil: Tumor-Surrounding Adipocytes Fueling Cancer. Trends Cancer 2020, 6, 593–604. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Peng, W.-B.; Zhang, P.; Yang, X.-P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Andarawewa, K.L.; Motrescu, E.R.; Chenard, M.-P.; Gansmuller, A.; Stoll, I.; Tomasetto, C.; Rio, M.-C. Stromelysin-3 Is a Potent Negative Regulator of Adipogenesis Participating to Cancer Cell-Adipocyte Interaction/Crosstalk at the Tumor Invasive Front. Cancer Res. 2005, 65, 10862–10871. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, P. The role of adipokines in chronic inflammation. ImmunoTargets Ther. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Hefetz-Sela, S.; Scherer, P.E. Adipocytes: Impact on tumor growth and potential sites for therapeutic intervention. Pharmacol. Ther. 2013, 138, 197–210. [Google Scholar] [CrossRef]
- Ortiz-Huidobro, R.I.; Velasco, M.; Larqué, C.; Escalona, R.; Hiriart, M. Molecular Insulin Actions Are Sexually Dimorphic in Lipid Metabolism. Front. Endocrinol. 2021, 12, 690484. [Google Scholar] [CrossRef]
- Mair, K.M.; Gaw, R.; MacLean, M.R. Obesity, estrogens and adipose tissue dysfunction—implications for pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 1–21. [Google Scholar] [CrossRef]
- Kousidou, O.C.; Berdiaki, A.; Kletsas, D.; Zafiropoulos, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Estradiol-estrogen receptor: A key interplay of the expression of syndecan-2 and metalloproteinase-9 in breast cancer cells. Mol. Oncol. 2008, 2, 223–232. [Google Scholar] [CrossRef]
- Umar, M.I.; Hassan, W.; Murtaza, G.; Buabeid, M.; Arafa, E.; Irfan, H.M.; Asmawi, M.Z.; Huang, X. The Adipokine Component in the Molecular Regulation of Cancer Cell Survival, Proliferation and Metastasis. Pathol. Oncol. Res. 2021, 27, 1609828. [Google Scholar] [CrossRef]
- Christodoulatos, G.S.; Spyrou, N.; Kadillari, J.; Psallida, S.; Dalamaga, M. The Role of Adipokines in Breast Cancer: Current Evidence and Perspectives. Curr. Obes. Rep. 2019, 8, 413–433. [Google Scholar] [CrossRef]
- Muehlberg, F.L.; Song, Y.-H.; Krohn, A.; Pinilla, S.P.; Droll, L.H.; Leng, X.; Seidensticker, M.; Ricke, J.; Altman, A.M.; Devarajan, E.; et al. Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis 2009, 30, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Welte, G.; Alt, E.; Devarajan, E.; Krishnappa, S.; Jotzu, C.; Song, Y.-H. Interleukin-8 derived from local tissue-resident stromal cells promotes tumor cell invasion. Mol. Carcinog. 2012, 51, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Song, Y.-H.; Krishnappa, S.; Alt, E. Epithelial-mesenchymal transition in breast cancer lines is mediated through PDGF-D released by tissue-resident stem cells. Int. J. Cancer 2012, 131, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, L.; Li, H.; Han, Q.; Li, J.; Qu, X.; Huang, S.; Zhao, R.C. Mesenchymal stem cells play a potential role in regulating the establishment and maintenance of epithelial-mesenchymal transition in MCF7 human breast cancer cells by paracrine and induced autocrine TGF-β. Int. J. Oncol. 2012, 41, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Muir, S.E.; Elliott, S.; Guillot, L.M.; Antoon, J.W.; Penfornis, P.; Tilghman, S.L.; Salvo, V.A.; Fonseca, J.P.; Lacey, M.R.; et al. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res. Treat. 2010, 121, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Chandler, E.M.; Seo, B.R.; Califano, J.P.; Andresen Eguiluz, R.C.; Lee, J.S.; Yoon, C.J.; Tims, D.T.; Wang, J.X.; Cheng, L.; Mohanan, S.; et al. Implanted adipose progenitor cells as physicochemical regulators of breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9786–9791. [Google Scholar] [CrossRef]
- Chandler, E.M.; Saunders, M.P.; Yoon, C.J.; Gourdon, D.; Fischbach, C. Adipose progenitor cells increase fibronectin matrix strain and unfolding in breast tumors. Phys. Biol. 2011, 8, 15008. [Google Scholar] [CrossRef]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef]
- Pallegar, N.K.; Garland, C.J.; Mahendralingam, M.; Viloria-Petit, A.M.; Christian, S.L. A Novel 3-Dimensional Co-culture Method Reveals a Partial Mesenchymal to Epithelial Transition in Breast Cancer Cells Induced by Adipocytes. J. Mammary Gland Biol. Neoplasia 2019, 24, 85–97. [Google Scholar] [CrossRef]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of Leptin Action and Leptin Resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.; Gonzalez-Perez, R. Leptin-Induced JAK/STAT Signaling and Cancer Growth. Vaccines 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Losso, J.N.; Bawadi, H.A. Hypoxia Inducible Factor Pathways as Targets for Functional Foods. J. Agric. Food Chem. 2005, 53, 3751–3768. [Google Scholar] [CrossRef] [PubMed]
- Atoum, M.F.; Alzoughool, F.; Al-Hourani, H. Linkage Between Obesity Leptin and Breast Cancer. Breast Cancer Basic Clin. Res. 2020, 14, 117822341989845. [Google Scholar] [CrossRef]
- Garofalo, C.; Surmacz, E. Leptin and cancer. J. Cell. Physiol. 2006, 207, 12–22. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Pan, H.; Deng, L.-L.; Cui, J.-Q.; Shi, L.; Yang, Y.-C.; Luo, J.-H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk. Medicine 2018, 97, e11345. [Google Scholar] [CrossRef]
- Frankenberry, K.A.; Skinner, H.; Somasundar, P.; McFadden, D.W.; Vona-Davis, L.C. Leptin receptor expression and cell signaling in breast cancer. Int. J. Oncol. 2006, 28, 985–993. [Google Scholar] [CrossRef]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A.; et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr. Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef]
- Hosney, M.; Sabet, S.; El-Shinawi, M.; Gaafar, K.M.; Mohamed, M.M. Leptin is overexpressed in the tumor microenvironment of obese patients with estrogen receptor positive breast cancer. Exp. Ther. Med. 2017, 13, 2235–2246. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased Expression of Leptin and the Leptin Receptor as a Marker of Breast Cancer Progression: Possible Role of Obesity-Related Stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Taliaferro-Smith, L.; Knight, B.B.; Merlin, D.; Anania, F.A.; O’Regan, R.M.; Sharma, D. Bidirectional Crosstalk between Leptin and Insulin-like Growth Factor-I Signaling Promotes Invasion and Migration of Breast Cancer Cells via Transactivation of Epidermal Growth Factor Receptor. Cancer Res. 2008, 68, 9712–9722. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Dunlap, S.M.; Zhu, J.; Downs-Kelly, E.; Rich, J.; Hursting, S.D.; Berger, N.A.; Reizes, O. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr. Relat. Cancer 2011, 18, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Lipsey, C.C.; Harbuzariu, A.; Robey, R.W.; Huff, L.M.; Gottesman, M.M.; Gonzalez-Perez, R.R. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3794. [Google Scholar] [CrossRef]
- Daley-Brown, D.; Harbuzariu, A.; Kurian, A.A.; Oprea-Ilies, G.; Gonzalez-Perez, R.R. Leptin-induced Notch and IL-1 signaling crosstalk in endometrial adenocarcinoma is associated with invasiveness and chemoresistance. World J. Clin. Oncol. 2019, 10, 222–233. [Google Scholar] [CrossRef]
- Wang, L.; Harlow, B.; Bowers, L.; Hursting, S.; DeGraffenried, L.A.; Brenner, A.J. The role of miR200c in leptin-mediated triple-negative breast cancer progression to an epithelial-to-mesenchymal transition. J. Clin. Oncol. 2019, 37, e12548. [Google Scholar] [CrossRef]
- Otvos, L.; Kovalszky, I.; Riolfi, M.; Ferla, R.; Olah, J.; Sztodola, A.; Nama, K.; Molino, A.; Piubello, Q.; Wade, J.D.; et al. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 2011, 47, 1578–1584. [Google Scholar] [CrossRef]
- Sabol, R.A.; Bowles, A.C.; Côté, A.; Wise, R.; O’Donnell, B.; Matossian, M.D.; Hossain, F.M.; Burks, H.E.; Del Valle, L.; Miele, L.; et al. Leptin produced by obesity-altered adipose stem cells promotes metastasis but not tumorigenesis of triple-negative breast cancer in orthotopic xenograft and patient-derived xenograft models. Breast Cancer Res. 2019, 21, 67. [Google Scholar] [CrossRef]
- Llanos, A.A.M.; Lin, Y.; Chen, W.; Yao, S.; Norin, J.; Chekmareva, M.A.; Omene, C.; Cong, L.; Omilian, A.R.; Khoury, T.; et al. Immunohistochemical analysis of adipokine and adipokine receptor expression in the breast tumor microenvironment: Associations of lower leptin receptor expression with estrogen receptor-negative status and triple-negative subtype. Breast Cancer Res. 2020, 22, 18. [Google Scholar] [CrossRef]
- Naviglio, S. Integrating leptin and cAMP signalling pathways in triple-negative breast cancer cells. Front. Biosci. 2013, 18, 133. [Google Scholar] [CrossRef]
- Avtanski, D.; Garcia, A.; Caraballo, B.; Thangeswaran, P.; Marin, S.; Bianco, J.; Lavi, A.; Poretsky, L. Resistin induces breast cancer cells epithelial to mesenchymal transition (EMT) and stemness through both adenylyl cyclase-associated protein 1 (CAP1)-dependent and CAP1-independent mechanisms. Cytokine 2019, 120, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Kim, N.; Lee, H.J.; Lee, Y.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Resistin, a fat-derived secretory factor, promotes metastasis of MDA-MB-231 human breast cancer cells through ERM activation. Sci. Rep. 2016, 6, 18923. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, X.; He, Q.; Gimple, R.C.; Liao, Y.; Wang, L.; Wu, R.; Xie, Q.; Rich, J.N.; Shen, K.; et al. Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. USA 2020, 117, 33295–33304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, N.; Liu, Y.; Su, P.; Liang, Y.; Li, Y.; Wang, X.; Chen, T.; Song, X.; Sang, Y.; et al. Epigenetic Regulation of NAMPT by NAMPT-AS Drives Metastatic Progression in Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3347–3359. [Google Scholar] [CrossRef]
- Bajrami, I.; Kigozi, A.; Van Weverwijk, A.; Brough, R.; Frankum, J.; Lord, C.J.; Ashworth, A. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 2012, 4, 1087–1096. [Google Scholar] [CrossRef]
- Cordover, E.; Wei, J.; Patel, C.; Shan, N.L.; Gionco, J.; Sargsyan, D.; Wu, R.; Cai, L.; Kong, A.-N.; Jacinto, E.; et al. KPT-9274, an Inhibitor of PAK4 and NAMPT, Leads to Downregulation of mTORC2 in Triple Negative Breast Cancer Cells. Chem. Res. Toxicol. 2020, 33, 482–491. [Google Scholar] [CrossRef]
- Podsednik, A.; Jiang, J.; Jacob, A.; Li, L.Z.; Xu, H.N. Optical Redox Imaging of Treatment Responses to Nampt Inhibition and Combination Therapy in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5563. [Google Scholar] [CrossRef]
- Cheng, G.; Sun, X.; Wang, J.; Xiao, G.; Wang, X.; Fan, X.; Zu, L.; Hao, M.; Qu, Q.; Mao, Y.; et al. HIC1 Silencing in Triple-Negative Breast Cancer Drives Progression through Misregulation of LCN2. Cancer Res. 2014, 74, 862–872. [Google Scholar] [CrossRef]
- Guo, P.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Gote, V.; Pal, D. Octreotide-Targeted Lcn2 siRNA PEGylated Liposomes as a Treatment for Metastatic Breast Cancer. Bioengineering 2021, 8, 44. [Google Scholar] [CrossRef]
- Valashedi, M.R.; Roushandeh, A.M.; Tomita, K.; Kuwahara, Y.; Pourmohammadi-Bejarpasi, Z.; Kozani, P.S.; Sato, T.; Roudkenar, M.H. CRISPR/Cas9-mediated knockout of Lcn2 in human breast cancer cell line MDA-MB-231 ameliorates erastin-mediated ferroptosis and increases cisplatin vulnerability. Life Sci. 2022, 304, 120704. [Google Scholar] [CrossRef] [PubMed]
- Gourgue, F.; Mignion, L.; Van Hul, M.; Dehaen, N.; Bastien, E.; Payen, V.; Leroy, B.; Joudiou, N.; Vertommen, D.; Bouzin, C.; et al. Obesity and triple-negative-breast-cancer: Is apelin a new key target? J. Cell. Mol. Med. 2020, 24, 10233–10244. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Parashar, D.; Geethadevi, A.; Aure, M.R.; Mishra, J.; George, J.; Chen, C.; Mishra, M.K.; Tahiri, A.; Zhao, W.; Nair, B.; et al. miRNA551b-3p Activates an Oncostatin Signaling Module for the Progression of Triple-Negative Breast Cancer. Cell Rep. 2019, 29, 4389–4406.e10. [Google Scholar] [CrossRef]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Martínez, F.; Perez-Balaguer, A.; Ciprián, D.; Andrés, L.; Ponce, J.; Adrover, E.; Sánchez-Payá, J.; Aranda, F.I.; Lerma, E.; Peiró, G. Association of increased osteopontin and splice variant-c mRNA expression with HER2 and triple-negative/basal-like breast carcinomas subtypes and recurrence. Hum. Pathol. 2014, 45, 504–512. [Google Scholar] [CrossRef]
- Suarez-Cuervo, C.; Harris, K.W.; Kallman, L.; Kalervo Väänänen, H.; Selander, K.S. Tumor Necrosis Factor-α Induces Interleukin-6 Production via Extracellular-Regulated Kinase 1 Activation in Breast Cancer Cells. Breast Cancer Res. Treat. 2003, 80, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. [Google Scholar] [CrossRef]
- Rahimi, N.; Saulnier, R.; Nakamura, T.; Park, M.; Elliott, B. Role of Hepatocyte Growth Factor in Breast Cancer: A Novel Mitogenic Factor Secreted by Adipocytes. DNA Cell Biol. 1994, 13, 1189–1197. [Google Scholar] [CrossRef]
- Sung, V.Y.C.; Knight, J.F.; Johnson, R.M.; Stern, Y.E.; Saleh, S.M.; Savage, P.; Monast, A.; Zuo, D.; Duhamel, S.; Park, M. Co-dependency for MET and FGFR1 in basal triple-negative breast cancers. Npj Breast Cancer 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.-S.; Lodish, H.F.; Fruebis, J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur. J. Pharmacol. 2002, 440, 213–221. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T.; Kubota, N. The physiological and pathophysiological role of adiponectin and adiponectin receptors in the peripheral tissues and CNS. FEBS Lett. 2008, 582, 74–80. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The Role of Adiponectin in Cancer: A Review of Current Evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Woo, Y.C.; Wang, Y.; Yeung, C.Y.; Xu, A.; Lam, K.S.L. Obesity, adipokines and cancer: An update. Clin. Endocrinol. 2015, 83, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical Decrease of an Adipose-Specific Protein, Adiponectin, in Obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in Obesity and Type 2 Diabetes: Close Association with Insulin Resistance and Hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma Concentrations of a Novel, Adipose-Specific Protein, Adiponectin, in Type 2 Diabetic Patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-Dependent Signaling Pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Gu, L.; Cao, C.; Fu, J.; Li, Q.; Li, D.-H.; Chen, M.-Y. Serum adiponectin in breast cancer. Medicine 2018, 97, e11433. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-Y.; Wang, M.; Ma, Z.-B.; Yu, L.-X.; Zhang, Q.; Gao, D.-Z.; Wang, F.; Yu, Z.-G. The Role of Adiponectin in Breast Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e73183. [Google Scholar] [CrossRef]
- Orozco-Arguelles, L.; De los Santos, S.; Tenorio-Torres, A.; Méndez, J.P.; Leal-García, M.; Coral-Vázquez, R.; Vega-García, C.; Bautista-Piña, V.; Tejeda, M.E.; Cárdenas-Cárdenas, E.; et al. Adiponectin and adiponectin receptor 1 expression proteins levels are modified in breast cancer in postmenopausal women with obesity. J. Clin. Pathol. 2021, 74, 571–576. [Google Scholar] [CrossRef]
- Mociño-Rodríguez, M.D.; Santillán-Benítez, J.G.; Dozal-Domínguez, D.S.; Hernández-Navarro, M.D.; Flores-Merino, M.V.; Sandoval-Cabrera, A.; García Vázquez, F.J. Expression of AdipoR1 and AdipoR2 Receptors as Leptin-Breast Cancer Regulation Mechanisms. Dis. Markers 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Kataki, A.C.; Borthakur, B.B.; Basumatary, T.K.; Bose, S. Imbalance in leptin-adiponectin levels and leptin receptor expression as chief contributors to triple negative breast cancer progression in Northeast India. Gene 2017, 621, 51–58. [Google Scholar] [CrossRef]
- Oh, S.W.; Park, C.-Y.; Lee, E.S.; Yoon, Y.S.; Lee, E.S.; Park, S.S.; Kim, Y.; Sung, N.J.; Yun, Y.H.; Lee, K.S.; et al. Adipokines, insulin resistance, metabolic syndrome, and breast cancer recurrence: A cohort study. Breast Cancer Res. 2011, 13, R34. [Google Scholar] [CrossRef]
- Chung, S.J.; Purnachandra, G.; Nagalingam, A.; Muniraj, N. BASIC RESEARCH PAPER ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11 / LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, Y.Y.; Yu, B.Y.; Yang, B.-S.; Cho, K.-H.; Yoon, D.K.; Roh, Y.K. Adiponectin induces growth arrest and apoptosis of MDA-MB-231 breast cancer cell. Arch. Pharm. Res. 2005, 28, 1263–1269. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Naimo, G.D.; Gelsomino, L.; Catalano, S.; Mauro, L.; Andò, S. Interfering Role of ERα on Adiponectin Action in Breast Cancer. Front. Endocrinol. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, J.-H.; Lee, S.K.; Song, N.-Y.; Son, S.H.; Kim, K.R.; Chung, W.-Y. Chemerin Treatment Inhibits the Growth and Bone Invasion of Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2871. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, S.; Zhang, H.; Nice, E.C.; Huang, C. Nicotinamide phosphoribosyltransferase (Nampt) in carcinogenesis: New clinical opportunities. Expert Rev. Anticancer Ther. 2016, 16, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Deshmukh, B.; Ramteke, P.; Bhati, F.K.; Bhat, M.K. Resistin: A journey from metabolism to cancer. Transl. Oncol. 2021, 14, 101178. [Google Scholar] [CrossRef] [PubMed]
- Calabro, P.; Samudio, I.; Willerson, J.T.; Yeh, E.T. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. 2004, 110, 3335–3340. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, Y.S.; Won, E.H.; Chang, I.H.; Kim, T.H.; Park, E.S.; Kim, M.K.; Kim, W.; Myung, S.C. Expression of resistin in the prostate and its stimulatory effect on prostate cancer cell proliferation. BJU Int. 2011, 108, E77–E83. [Google Scholar] [CrossRef]
- Wang, C.-H.; Wang, P.-J.; Hsieh, Y.-C.; Lo, S.; Lee, Y.-C.; Chen, Y.-C.; Tsai, C.-H.; Chiu, W.-C.; Chu-Sung Hu, S.; Lu, C.-W.; et al. Resistin facilitates breast cancer progression via TLR4-mediated induction of mesenchymal phenotypes and stemness properties. Oncogene 2018, 37, 589–600. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, A.; Song, D.; Han, B.; Zhang, Z.; Ma, L.; Liu, D.; Fan, Z. Resistin confers resistance to doxorubicin-induced apoptosis in human breast cancer cells through autophagy induction. Am. J. Cancer Res. 2017, 7, 574–583. [Google Scholar]
- Mentoor, I.; Nell, T.; Emjedi, Z.; van Jaarsveld, P.J.; de Jager, L.; Engelbrecht, A.-M. Decreased Efficacy of Doxorubicin Corresponds With Modifications in Lipid Metabolism Markers and Fatty Acid Profiles in Breast Tumors From Obese vs. Lean Mice. Front. Oncol. 2020, 10, 306. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Chen, Y.-J.; Wu, C.-C.; Lo, S.; Hou, M.-F.; Yuan, S.-S.F. Resistin expression in breast cancer tissue as a marker of prognosis and hormone therapy stratification. Gynecol. Oncol. 2012, 125, 742–750. [Google Scholar] [CrossRef]
- Gong, W.-J.; Zheng, W.; Xiao, L.; Tan, L.-M.; Song, J.; Li, X.-P.; Xiao, D.; Cui, J.-J.; Li, X.; Zhou, H.-H.; et al. Circulating resistin levels and obesity-related cancer risk: A meta-analysis. Oncotarget 2016, 7, 57694–57704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, P.A.; Luks, J.; Roycik, M.D.; Sang, Q.-X.A.; Zhang, J. Differentially Expressed Transcripts and Dysregulated Signaling Pathways and Networks in African American Breast Cancer. PLoS ONE 2013, 8, e82460. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Christodoulatos, G.S.; Mantzoros, C.S. The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: Diagnostic and therapeutic perspectives and challenges. Metabolism 2018, 82, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A Protein Secreted by Visceral Fat That Mimics the Effects of Insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- Park, H.-J.; Kim, S.-R.; Kim, S.S.; Wee, H.-J.; Bae, M.-K.; Ryu, M.H.; Bae, S.-K. Visfatin promotes cell and tumor growth by upregulating Notch1 in breast cancer. Oncotarget 2014, 5, 5087–5099. [Google Scholar] [CrossRef]
- Gholinejad, Z.; Kheiripour, N.; Nourbakhsh, M.; Ilbeigi, D.; Behroozfar, K.; Hesari, Z.; Golestani, A.; Shabani, M.; Einollahi, N. Extracellular NAMPT/Visfatin induces proliferation through ERK1/2 and AKT and inhibits apoptosis in breast cancer cells. Peptides 2017, 92, 9–15. [Google Scholar] [CrossRef]
- Dalamaga, M.; Archondakis, S.; Sotiropoulos, G.; Karmaniolas, K.; Pelekanos, N.; Papadavid, E.; Lekka, A. Could serum visfatin be a potential biomarker for postmenopausal breast cancer? Maturitas 2012, 71, 301–308. [Google Scholar] [CrossRef]
- Hung, A.C.; Lo, S.; Hou, M.-F.; Lee, Y.-C.; Tsai, C.-H.; Chen, Y.-Y.; Liu, W.; Su, Y.-H.; Lo, Y.-H.; Wang, C.-H.; et al. Extracellular Visfatin-Promoted Malignant Behavior in Breast Cancer Is Mediated Through c-Abl and STAT3 Activation. Clin. Cancer Res. 2016, 22, 4478–4490. [Google Scholar] [CrossRef]
- Moi, S.-H.; Lee, Y.-C.; Chuang, L.-Y.; Yuan, S.-S.F.; Ou-Yang, F.; Hou, M.-F.; Yang, C.-H.; Chang, H.-W. Cumulative receiver operating characteristics for analyzing interaction between tissue visfatin and clinicopathologic factors in breast cancer progression. Cancer Cell Int. 2018, 18, 19. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Yang, Y.-H.; Su, J.-H.; Chang, H.-L.; Hou, M.-F.; Yuan, S.-S.F. High Visfatin Expression in Breast Cancer Tissue Is Associated with Poor Survival. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1892–1901. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Weiskirchen, S.; Weiskirchen, R. Lipocalin 2 (LCN2) Expression in Hepatic Malfunction and Therapy. Front. Physiol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.-T.; Tsai, I.-T.; Wu, C.-C.; Hung, W.-C.; Hsuan, C.-F.; Yu, T.-H.; Hsu, C.-C.; Houng, J.-Y.; Chung, F.-M.; Lee, Y.-J.; et al. Elevated plasma level of neutrophil gelatinase-associated lipocalin (NGAL) in patients with breast cancer. Int. J. Med. Sci. 2021, 18, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Moses, M.A. Lipocalin 2: A multifaceted modulator of human cancer. Cell Cycle 2009, 8, 2347–2352. [Google Scholar] [CrossRef]
- Fernández, C.A.; Yan, L.; Louis, G.; Yang, J.; Kutok, J.L.; Moses, M.A. The Matrix Metalloproteinase-9/Neutrophil Gelatinase-Associated Lipocalin Complex Plays a Role in Breast Tumor Growth and Is Present in the Urine of Breast Cancer Patients. Clin. Cancer Res. 2005, 11, 5390–5395. [Google Scholar] [CrossRef]
- Bauer, M.; Eickhoff, J.C.; Gould, M.N.; Mundhenke, C.; Maass, N.; Friedl, A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008, 108, 389–397. [Google Scholar] [CrossRef]
- Villodre, E.S.; Hu, X.; Larson, R.; Finetti, P.; Gomez, K.; Balema, W.; Stecklein, S.R.; Santiago-Sanchez, G.; Krishnamurthy, S.; Song, J.; et al. Lipocalin 2 promotes inflammatory breast cancer tumorigenesis and skin invasion. Mol. Oncol. 2021, 15, 2752–2765. [Google Scholar] [CrossRef] [PubMed]
- Meurer, S.K.; Tezcan, O.; Lammers, T.; Weiskirchen, R. Differential regulation of Lipocalin 2 (LCN2) in doxorubicin-resistant 4T1 triple negative breast cancer cells. Cell. Signal. 2020, 74, 109731. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, S. Apelin. In Handbook of Hormones; Elsevier: Amsterdam, The Netherlands, 2021; pp. 525–527. [Google Scholar]
- Peng, X.; Li, F.; Wang, P.; Jia, S.; Sun, L.; Huo, H. Apelin-13 induces MCF-7 cell proliferation and invasion via phosphorylation of ERK1/2. Int. J. Mol. Med. 2015, 36, 733–738. [Google Scholar] [CrossRef]
- Sorli, S.C.; Le Gonidec, S.; Knibiehler, B.; Audigier, Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene 2007, 26, 7692–7699. [Google Scholar] [CrossRef]
- Hu, D.; Zhu, W.F.; Shen, W.C.; Xia, Y.; Wu, X.F.; Zhang, H.J.; Liu, W.; Cui, Z.L.; Zheng, X.W.; Chen, G. Expression of Apelin and Snail protein in breast cancer and their prognostic significance. Zhonghua Bing Li Xue Za Zhi = Chin. J. Pathol. 2018, 47, 743–746. [Google Scholar] [CrossRef]
- Hu, D.; Cui, Z.; Peng, W.; Wang, X.; Chen, Y.; Wu, X. Apelin is associated with clinicopathological parameters and prognosis in breast cancer patients. Arch. Gynecol. Obstet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gourgue, F.; Derouane, F.; van Marcke, C.; Villar, E.; Dano, H.; Desmet, L.; Bouzin, C.; Duhoux, F.P.; Cani, P.D.; Jordan, B.F. Tumor apelin and obesity are associated with reduced neoadjuvant chemotherapy response in a cohort of breast cancer patients. Sci. Rep. 2021, 11, 9922. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, J.; Jafarzadeh, A.; Tavakoli, T.; Basirjafar, P.; Zandvakili, R.; Javan, M.R.; Taghipour, Z.; Moazzeni, S.M. Inhibition of apelin/APJ axis enhances the potential of dendritic cell-based vaccination to modulate TH1 and TH2 cell-related immune responses in an animal model of metastatic breast cancer. Adv. Med. Sci. 2022, 67, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Everaert, N.; Decuypere, E.; Buyse, J. Adipose tissue and lipid metabolism. In Sturkie’s Avian Physiology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 647–660. [Google Scholar]
- Helfer, G.; Wu, Q.-F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef]
- El-Sagheer, G.; Gayyed, M.; Ahmad, A.; Abd El-Fattah, A.; Mohamed, M. Expression of chemerin correlates with a poor prognosis in female breast cancer patients. Breast Cancer Targets Ther. 2018, 10, 169–176. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Wang, P.; Salazar, N.; Zheng, Y.; Nease, L.; Rosalez, J.; Leong, W.-I.; Virdi, G.; Rennier, K.; Shin, W.J.; et al. Chemerin Suppresses Breast Cancer Growth by Recruiting Immune Effector Cells Into the Tumor Microenvironment. Front. Immunol. 2019, 10, 983. [Google Scholar] [CrossRef]
- Stephens, J.M.; Elks, C.M. Oncostatin M: Potential Implications for Malignancy and Metabolism. Curr. Pharm. Des. 2017, 23, 3645–3657. [Google Scholar] [CrossRef]
- Sanchez-Infantes, D.; Stephens, J.M. Adipocyte Oncostatin Receptor Regulates Adipose Tissue Homeostasis and Inflammation. Front. Immunol. 2021, 11, 612013. [Google Scholar] [CrossRef]
- Masjedi, A.; Hajizadeh, F.; Beigi Dargani, F.; Beyzai, B.; Aksoun, M.; Hojjat-Farsangi, M.; Zekiy, A.; Jadidi-Niaragh, F. Oncostatin M: A mysterious cytokine in cancers. Int. Immunopharmacol. 2021, 90, 107158. [Google Scholar] [CrossRef]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef]
- Kazanecki, C.C.; Uzwiak, D.J.; Denhardt, D.T. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J. Cell. Biochem. 2007, 102, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Kodama, K.; Wei, K.; Tolentino, L.L.; Choi, O.; Engleman, E.G.; Butte, A.J.; McLaughlin, T. The receptor CD44 is associated with systemic insulin resistance and proinflammatory macrophages in human adipose tissue. Diabetologia 2015, 58, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Sodek, J.; Ganss, B.; McKee, M.D. Osteopontin. Crit. Rev. Oral Biol. Med. 2000, 11, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Tardelli, M.; Zeyda, K.; Moreno-Viedma, V.; Wanko, B.; Grün, N.G.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol. Metab. 2016, 5, 1131–1137. [Google Scholar] [CrossRef]
- Bandopadhyay, M.; Bulbule, A.; Butti, R.; Chakraborty, G.; Ghorpade, P.; Ghosh, P.; Gorain, M.; Kale, S.; Kumar, D.; Kumar, S.; et al. Osteopontin as a therapeutic target for cancer. Expert Opin. Ther. Targets 2014, 18, 883–895. [Google Scholar] [CrossRef]
- Rudland, P.S.; Platt-Higgins, A.; El-Tanani, M.; De Silva Rudland, S.; Barraclough, R.; Winstanley, J.H.R.; Howitt, R.; West, C.R. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 2002, 62, 3417–3427. [Google Scholar]
- Wang, X.; Chao, L.; Ma, G.; Chen, L.; Tian, B.; Zang, Y.; Sun, J. Increased expression of osteopontin in patients with triple-negative breast cancer. Eur. J. Clin. Investig. 2008, 38, 438–446. [Google Scholar] [CrossRef]
- Anborgh, P.H.; Lee, D.J.; Stam, P.F.; Tuck, A.B.; Chambers, A.F. Role of osteopontin as a predictive biomarker for anti-EGFR therapy in triple-negative breast cancer. Expert Opin. Ther. Targets 2018, 22, 727–734. [Google Scholar] [CrossRef]
- Cha, Y.J.; Koo, J.S. Adipokines as therapeutic targets in breast cancer treatment. Expert Opin. Ther. Targets 2018, 22, 941–953. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Razmkhah, M.; Jaberipour, M.; Hosseini, A.; Safaei, A.; Khalatbari, B.; Ghaderi, A. Expression profile of IL-8 and growth factors in breast cancer cells and adipose-derived stem cells (ASCs) isolated from breast carcinoma. Cell. Immunol. 2010, 265, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-Y.; Qu, R.-M.; Lin, X.-S.; Liu, T.-X.; Sun, Q.-Q.; Yang, C.; Li, X.-H.; Lu, W.; Hu, X.-F.; Dai, J.-X.; et al. IGF-1 from Adipose-Derived Mesenchymal Stem Cells Promotes Radioresistance of Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 15, 10115–10119. [Google Scholar] [CrossRef] [PubMed]
- Eterno, V.; Zambelli, A.; Pavesi, L.; Villani, L.; Zanini, V.; Petrolo, G.; Manera, S.; Tuscano, A.; Amato, A. Adipose-derived mesenchymal stem cells (ASCs) may favour breast cancer recurrence via HGF/c-Met signaling. Oncotarget 2014, 5, 613–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Adipokines/Receptors | Expression/Actions in TNBC | References |
|---|---|---|
| Leptin/LEPR | Leptin and LEPR are essentially overexpressed in TNBC | [72] |
| A significant crosstalk between leptin and IGF-I increase the activation of EGFR and LEPR and drive TNBC progression | [73] | |
| Leptin induces BC stemness and resistance to chemotherapy | [75] | |
| Elevated leptin levels develop EMT | [77] | |
| Leptin receptor antagonist peptide Allo-aca inhibited leptin-induced proliferation of MDA-MB-231 cells | [78] | |
| Leptin secreted by obesity altered adipose stem cells induced the EMT and CSC reprogramming | [79] | |
| Reduced IHC expression of LEPR was correlated with ER-status and TN subtype | [80] | |
| Leptin induces apoptosis in TNBC cells when used in combination with cAMP elevating agents | [81] | |
| Resistin | Resistin increases the malignant potential of MCF-7 and MB-231 cells through EMT initiation and stemness | [82,83] |
| Higher expression of both TAZ and resistin in adipose tissue of TNBC tumors correlated with higher stage and poor prognosis, giving the idea of targeted therapy | [84] | |
| NAMPT/visfatin | NAMPT inhibits autophagy and apoptosis and induces cell survival and invasiveness through mTOR activation in TNBC cells | [85] |
| The combined use of NAMPT inhibitor FK866 with olaparib inhibited TN breast tumor growth in vivo | [86] | |
| Treatment with the small molecule KPT-9274, inhibitor of PAK4 and NAMPT, abrogates TNBC cell proliferation and eventually leads to cell death | [87] | |
| NAMPT inhibitor FK866 combined with FX11 (lactate dehydrogenase A inhibitor) and paclitaxel caused significant growth restriction of MDA-MB-231 cells | [88] | |
| Lipocalin2 (NGAL) | In TNBC cells, secretion of LCN2, induced by HIC1 loss, activated the AKT pathway and caused tumor progression | [89] |
| Lcn2 knockdown by ICAM-LCN2-LP led to a significant reduction of VEGF in MDA-MB-231 cells, which led to reduced angiogenesis both in vitro and in vivo | [90] | |
| Silencing of Lcn2 mRNA by OCT-Lcn2-Lipo display anti-angiogenic results in MCF-7 and MDA-MB-231 cells by diminishing VEGF-A and endothelial migration | [91] | |
| Targeting Lcn2 by CRISPR/Cas9 reduced cancer cell malignant potential and increased cell susceptibility of MDA-MB-231 cells to cisplatin | [92] | |
| Apelin | The administration of apelin to lean mice elevated TNBC growth and brain metastases. The apelinergic antagonist F13A could reduce TNBC growth and be a novel therapeutic strategy for TNBC in obese conditions | [93] |
| Oncostatin M | OSM is thoroughly expressed in the TNBC subtype in comparison with other molecular breast cancer subtypes | [94] |
| The KM plotter survival analysis portal demonstrated that higher expression of OSM ER-negative patients was associated with poor outcomes | [95] | |
| TME cytokines OSM and IFN-b express antagonistic roles in CSC plasticity coordination in TNBC | [96] | |
| miR551b-3p named as “Oncostatin signaling module” translocates from the cytoplasm to nucleus and upregulates the expression of OSM receptor IL31 receptor as well as their ligands OSM and IL31 | [95] | |
| Osteopontin | OPN mRNA is upregulated in triple negative/basal like tumors | [97] |
| Other | TNF-a induces IL-6 in MDA-MB-231 cells via ERK1 activation | [98] |
| IL-6 and CCL5 promote TNBC tumor growth via cancer cell-lymphatic vessels cross talk | [99] | |
| Adipocytes enhanced MDA-MB-231 cancer cell invasiveness, through CCL5 signaling, which negatively correlated with OS | [60] | |
| The IGF-I/IGF-IR signaling promotes the FAK-YAP cascade activation and triggers TNBC growth | [100] | |
| Hepatocyte growth factor (HGF) is a mitogenic factor released by adipocytes and its receptor c-met is expressed at high levels on breast cancer cells, at the adipose-cancer interface, highlighting the importance of stromal–tumor cell interactions in breast cancer growth. In TNBCs, elevated levels of the MET receptor predict poor clinical outcome | [101,102] |
| Adipokines/Receptors | Expression/Actions in TNBC | References |
|---|---|---|
| Adiponectin/ AdipoR1/AdipoR2 | Reduced adiponectin: leptin is correlated with the diagnosis of TNBC | [116] |
| In ER/PR-negative BC cells, it inhibits cell growth, invasion, migration, and vascular proliferation and induces apoptosis and autophagic cell death | [117,118] | |
| Normal adiponectin amounts significantly suppress the proliferation of MDA-MB-231 cancer cells | [119] | |
| Diminished adiponectin is thoroughly correlated with TNBC development and progression, regardless of obesity and insulin resistance | [120] | |
| Chemerin | Chemerin restricts the growth and invasion of breast cancer cells and prevents bone loss resulting from MDA-MB-231 cell growth | [122] |
| Treatment with peptide LRH7-G5 significantly decreased TNBC cell growth demonstrating chemerin/GPR1 as a novel therapeutic target for TNBC | [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papakonstantinou, E.; Piperigkou, Z.; Karamanos, N.K.; Zolota, V. Altered Adipokine Expression in Tumor Microenvironment Promotes Development of Triple Negative Breast Cancer. Cancers 2022, 14, 4139. https://doi.org/10.3390/cancers14174139
Papakonstantinou E, Piperigkou Z, Karamanos NK, Zolota V. Altered Adipokine Expression in Tumor Microenvironment Promotes Development of Triple Negative Breast Cancer. Cancers. 2022; 14(17):4139. https://doi.org/10.3390/cancers14174139
Chicago/Turabian StylePapakonstantinou, Efthymia, Zoi Piperigkou, Nikos K. Karamanos, and Vasiliki Zolota. 2022. "Altered Adipokine Expression in Tumor Microenvironment Promotes Development of Triple Negative Breast Cancer" Cancers 14, no. 17: 4139. https://doi.org/10.3390/cancers14174139
APA StylePapakonstantinou, E., Piperigkou, Z., Karamanos, N. K., & Zolota, V. (2022). Altered Adipokine Expression in Tumor Microenvironment Promotes Development of Triple Negative Breast Cancer. Cancers, 14(17), 4139. https://doi.org/10.3390/cancers14174139