
5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action



 ,
, 
Abstract
:Simple Summary
Abstract
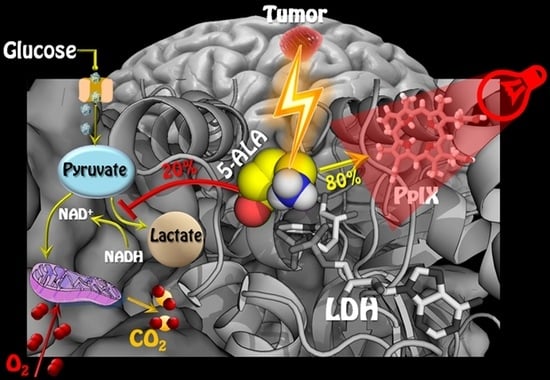
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Metabolic Assays
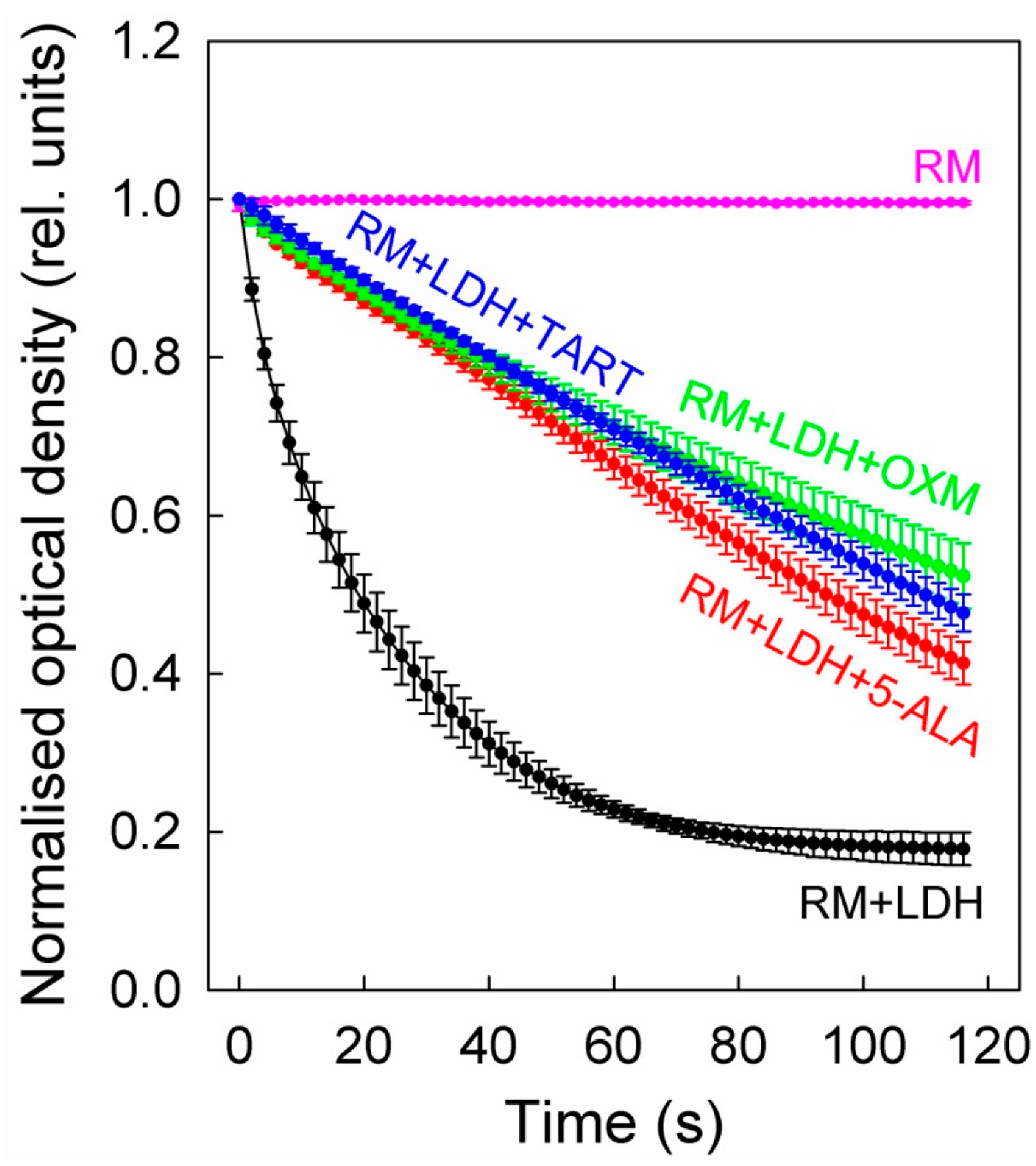
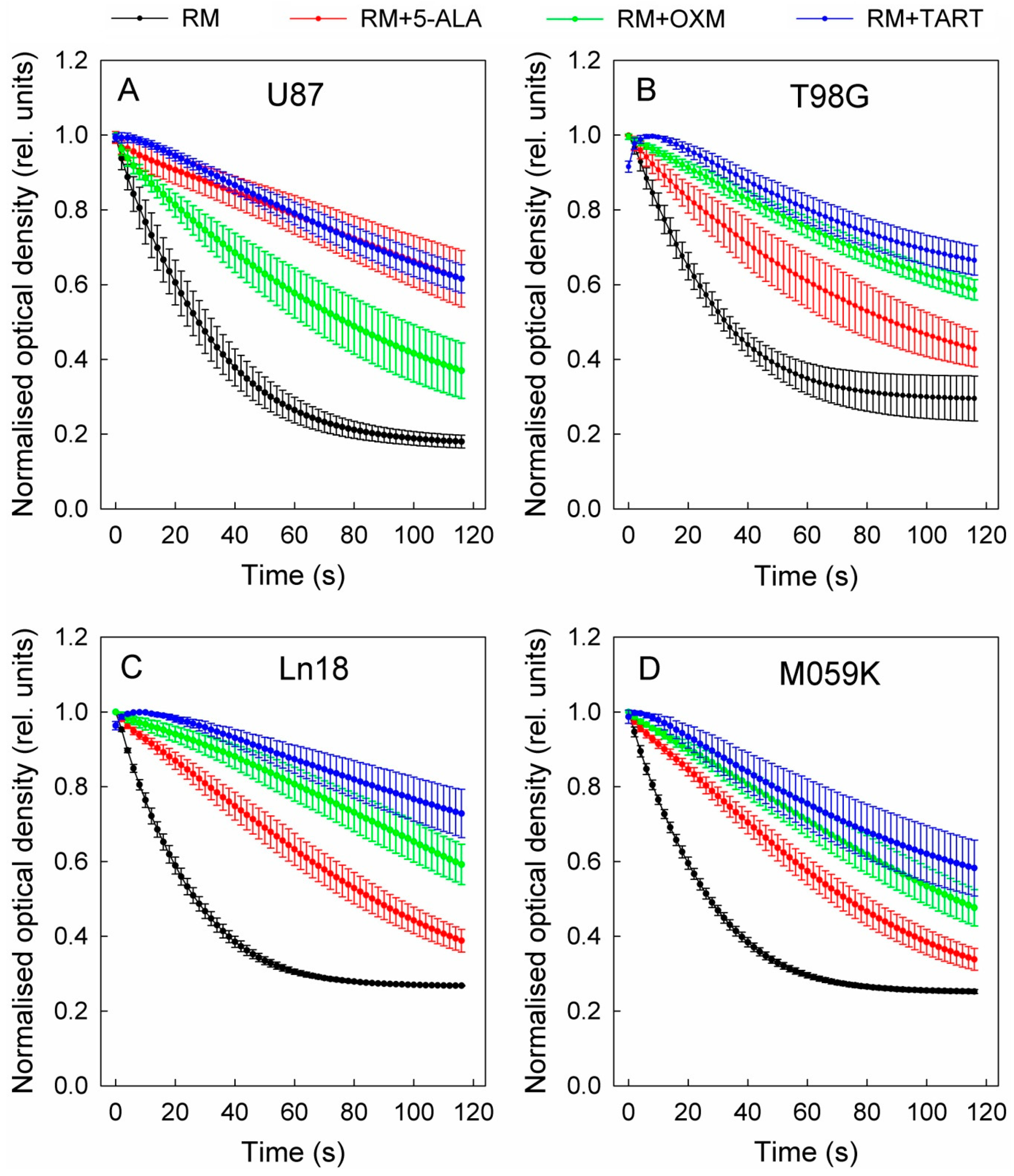
2.4. Spectrophotometric Assesment of LDH Kinetics with or without Inhibitors, in Purified Enzyme Assays or Cell Lysates
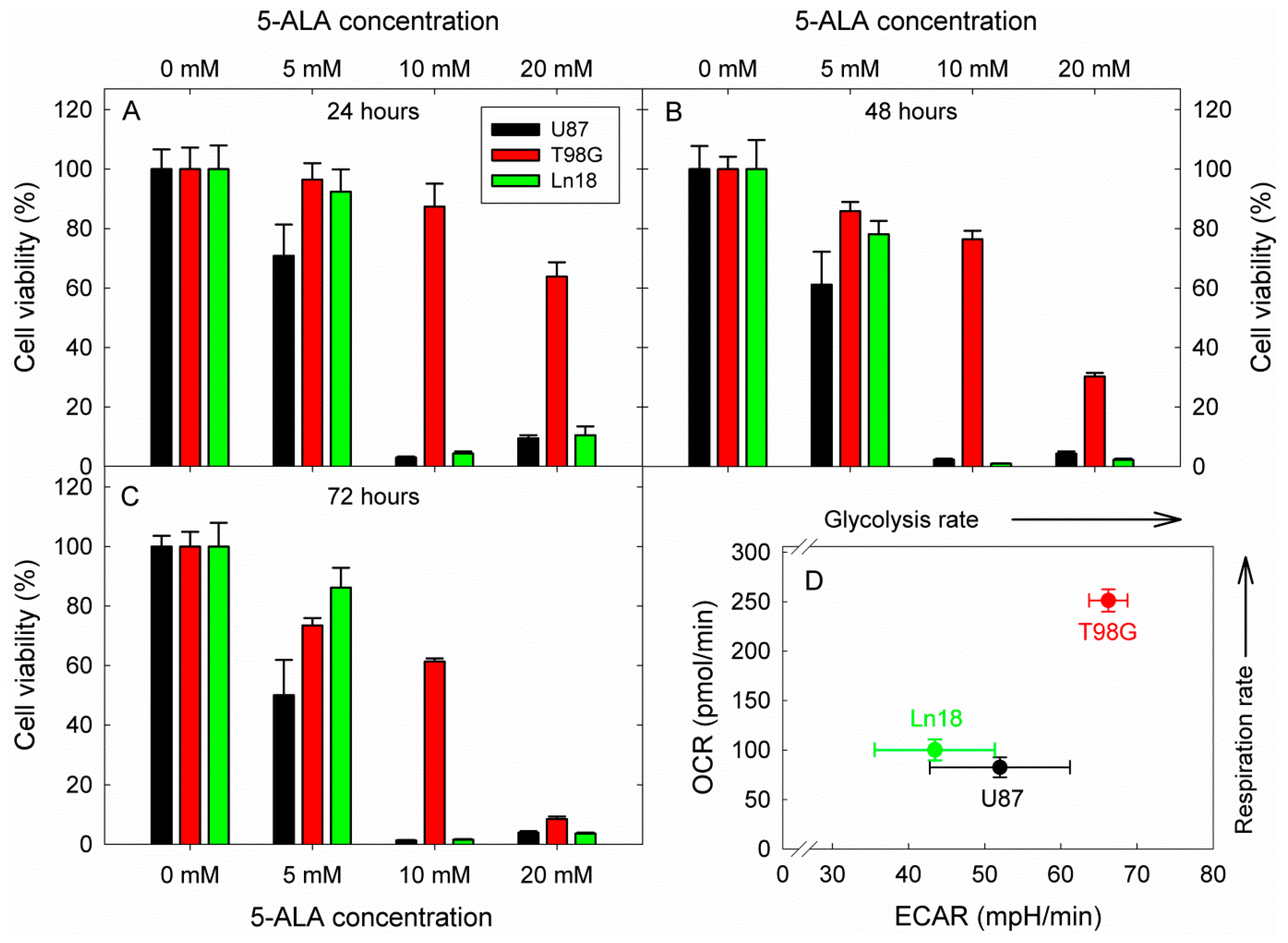
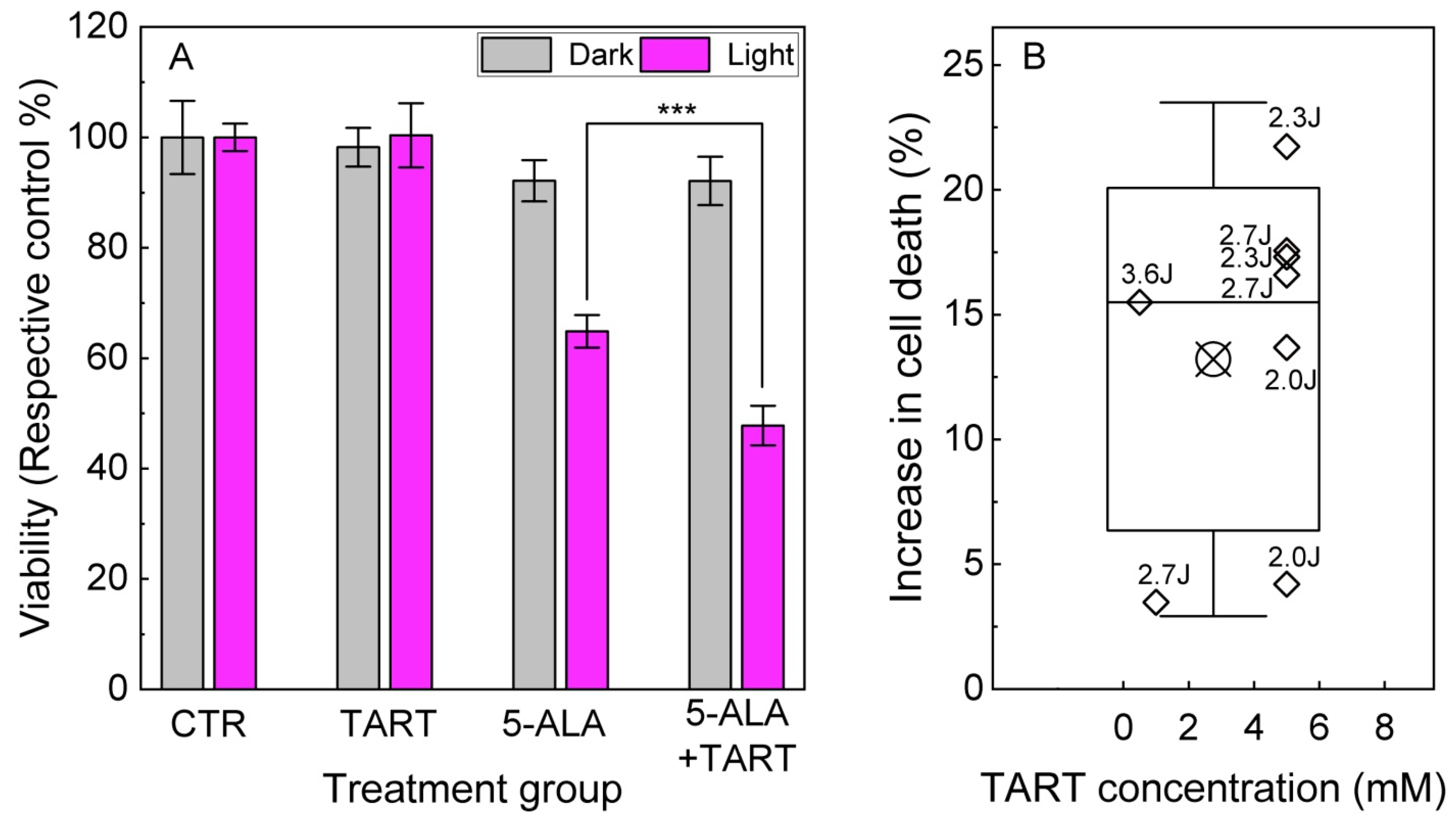
2.5. Cytotoxicity by 5-ALA Induced Glycolysis Inhibition
2.6. PDT Experiments
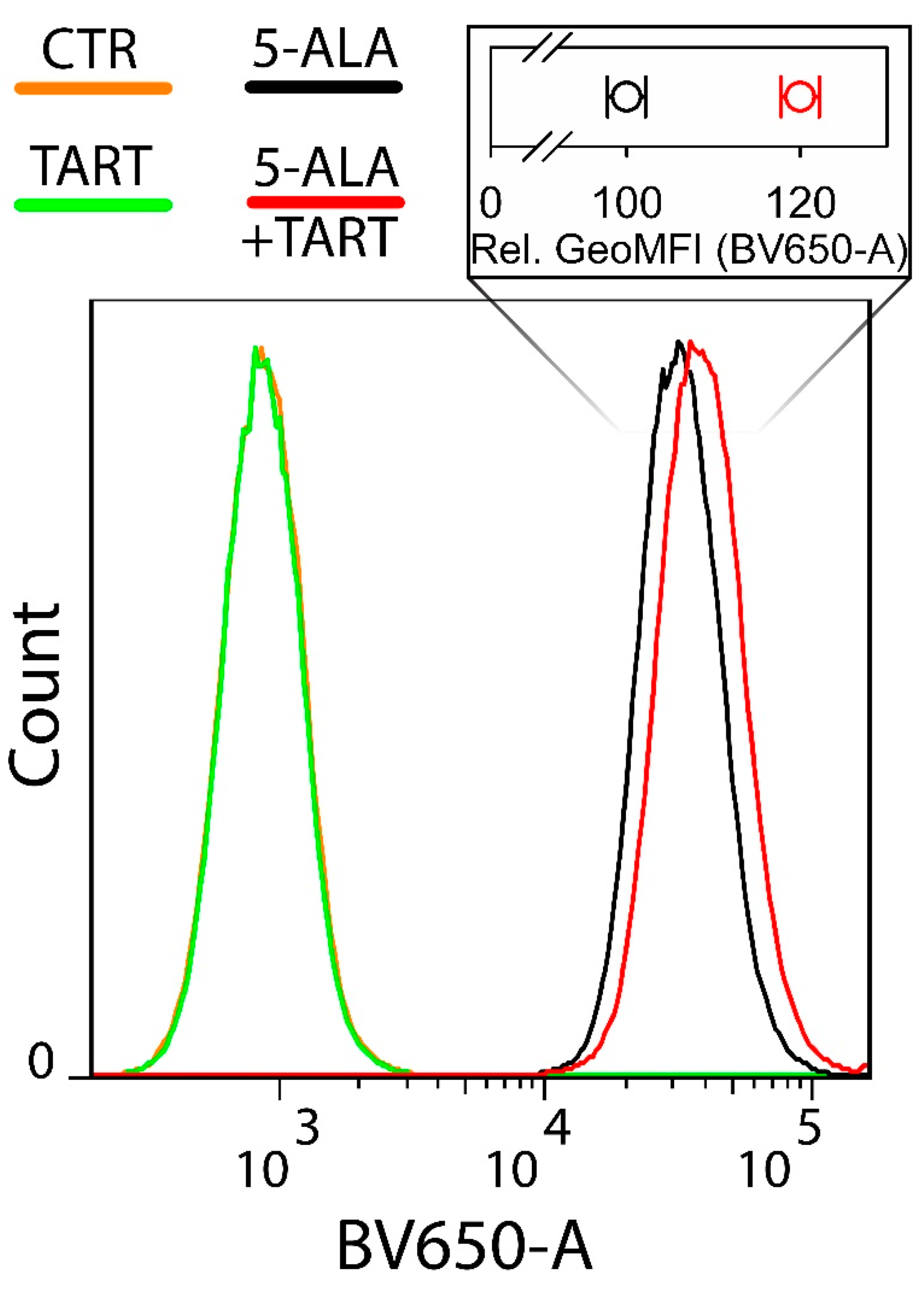
2.7. Flow Cytometry
2.8. Computational Methods
2.9. NMR Experiments
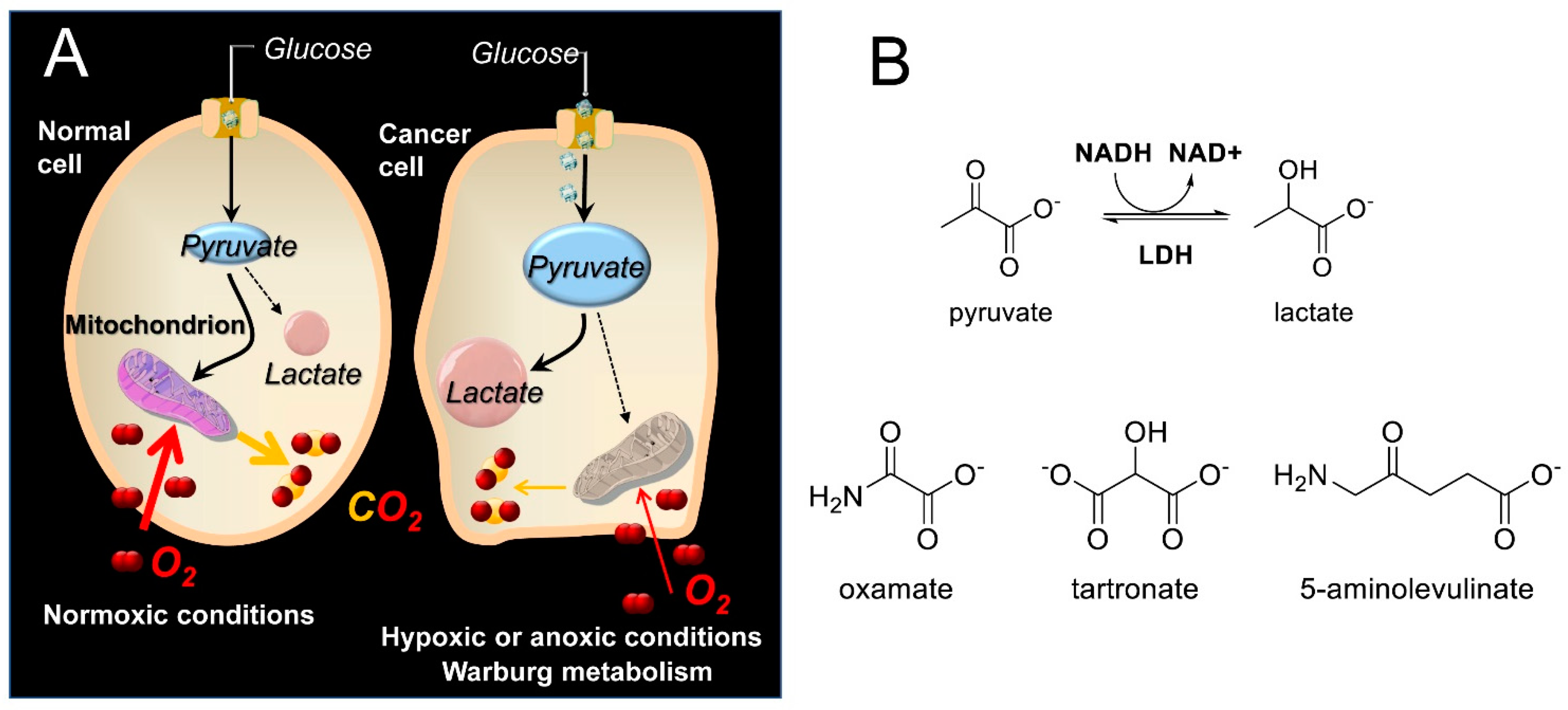
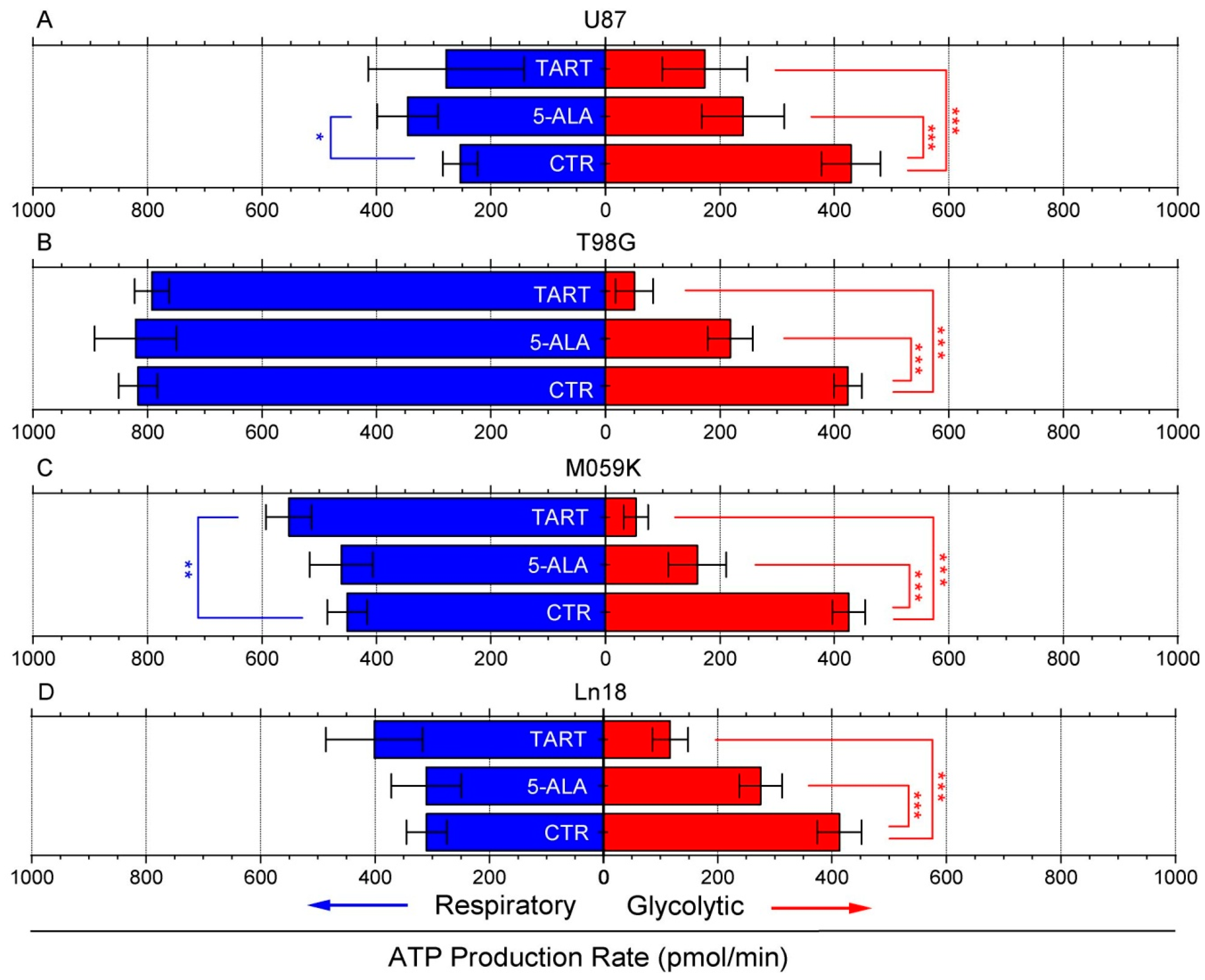
3. Results
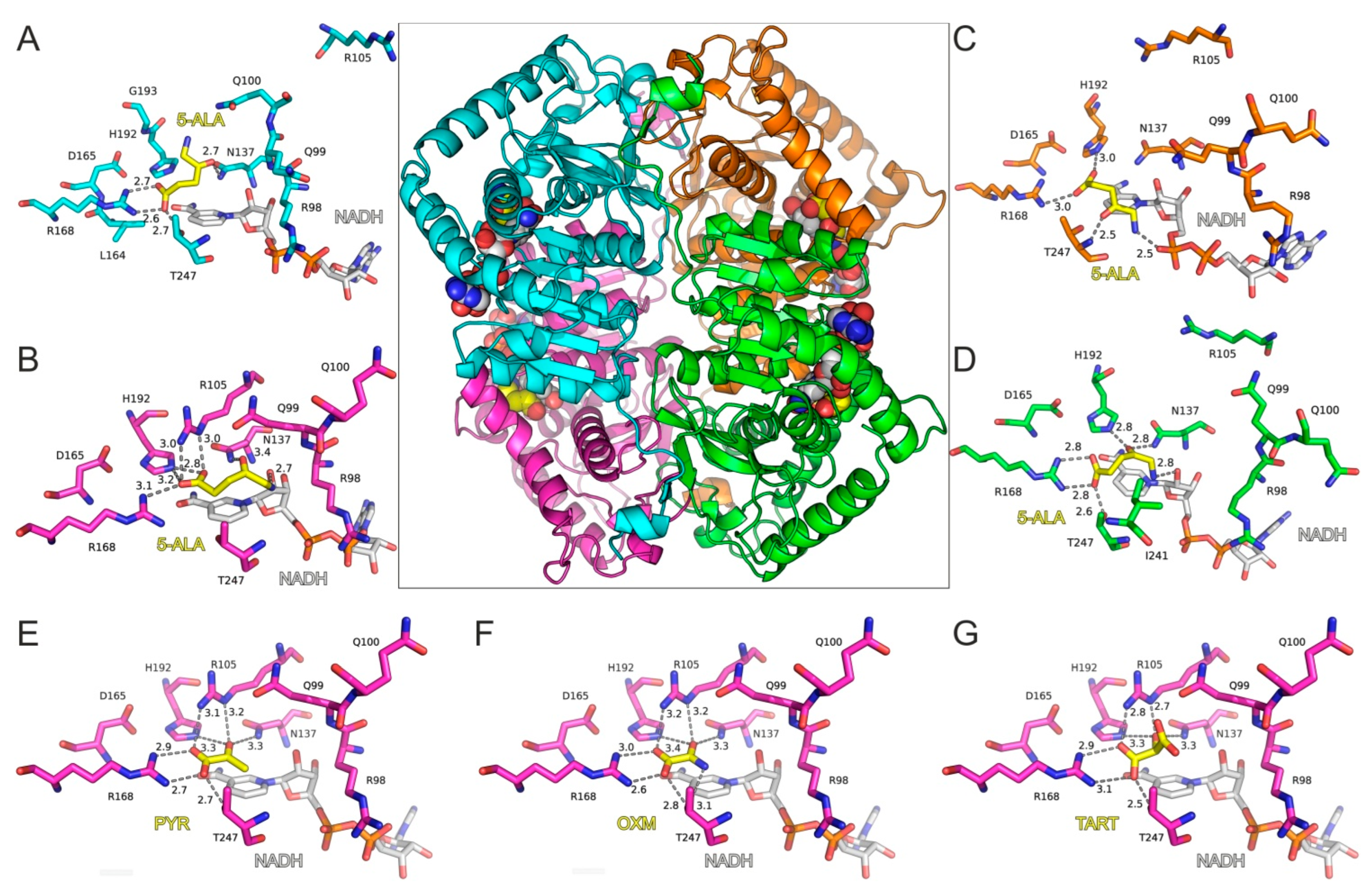
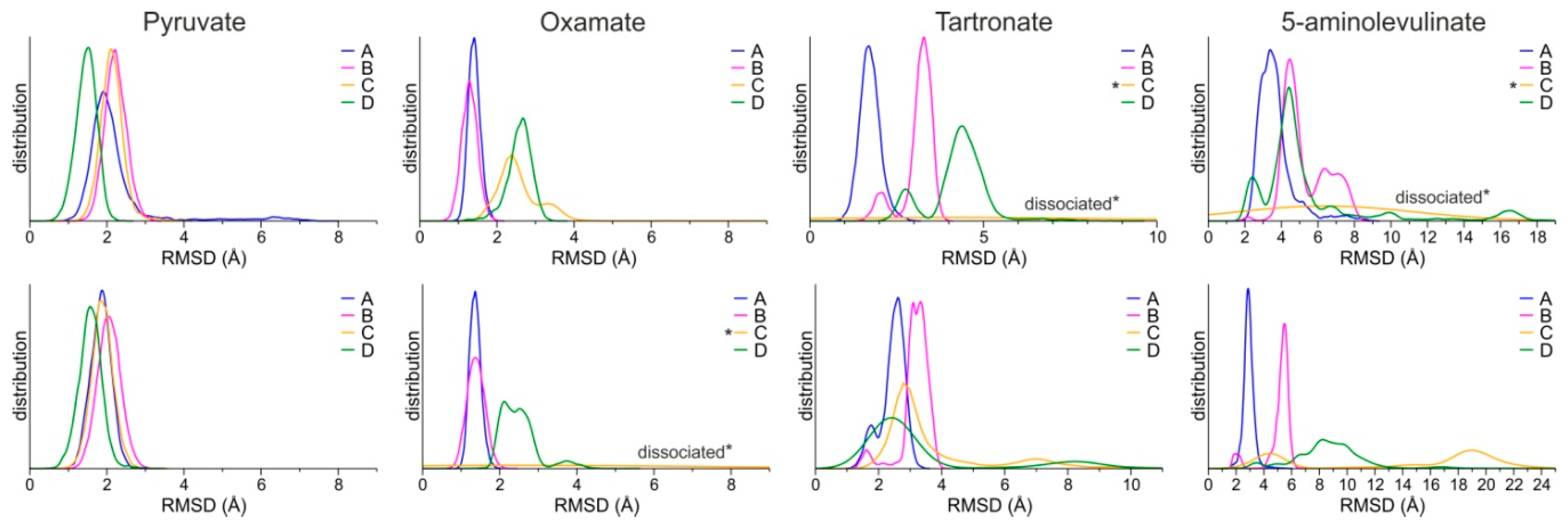
Molecular Modeling of 5-ALA in Complex with LDH-A
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melkonian, E.A.; Schury, M.P. Biochemistry, Anaerobic Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase A in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Ward, R.A.; Brassington, C.; Breeze, A.L.; Caputo, A.; Critchlow, S.; Davies, G.; Goodwin, L.; Hassall, G.; Greenwood, R.; Holdgate, G.A.; et al. Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J. Med. Chem. 2012, 55, 3285–3306. [Google Scholar] [CrossRef]
- Kohlmann, A.; Zech, S.G.; Li, F.; Zhou, T.; Squillace, R.M.; Commodore, L.; Greenfield, M.T.; Lu, X.; Miller, D.P.; Huang, W.S.; et al. Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J. Med. Chem. 2013, 56, 1023–1040. [Google Scholar] [CrossRef]
- Rai, G.; Brimacombe, K.R.; Mott, B.T.; Urban, D.J.; Hu, X.; Yang, S.M.; Lee, T.D.; Cheff, D.M.; Kouznetsova, J.; Benavides, G.A.; et al. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Impairment of aerobic glycolysis by inhibitors of lactic dehydrogenase hinders the growth of human hepatocellular carcinoma cell lines. Pharmacology 2010, 86, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget 2014, 5, 11886–11896. [Google Scholar] [CrossRef]
- Rodriguez-Paez, L.; Chena-Taboada, M.A.; Cabrera-Hernandez, A.; Cordero-Martinez, J.; Wong, C. Oxamic acid analogues as LDH-C4-specific competitive inhibitors. J. Enzym. Inhib. Med. Chem. 2011, 26, 579–586. [Google Scholar] [CrossRef]
- Granchi, C.; Bertini, S.; Macchia, M.; Minutolo, F. Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials. Curr. Med. Chem. 2010, 17, 672–697. [Google Scholar] [CrossRef]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Shi, Y.; Pinto, B.M. Human lactate dehydrogenase a inhibitors: A molecular dynamics investigation. PLoS ONE 2014, 9, e86365. [Google Scholar] [CrossRef]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the first highly selective inhibitor of human lactate dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Allemailem, K.S.; Alhumaydhi, F.A.; Gowder, S.J.T.; Rahmani, A.H. The Biochemical and Clinical Perspectives of Lactate Dehydrogenase: An Enzyme of Active Metabolism. Endocr. Metab. Immune Disord. Drug Targets. 2020, 20, 855–868. [Google Scholar] [CrossRef]
- Forkasiewicz, A.; Dorociak, M.; Stach, K.; Szelachowski, P.; Tabola, R.; Augoff, K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell Mol. Biol. Lett. 2020, 25, 35. [Google Scholar] [CrossRef]
- Jacobs, J.W.; Bastarache, L.; Thompson, M.A. Laboratory Predictors of Hemolytic Anemia in Patients With Systemic Loxoscelism. Am. J. Clin. Pathol. 2021, 157, 566–572. [Google Scholar] [CrossRef]
- Vasudevan, G.; Mercer, D.W.; Varat, M.A. Lactic dehydrogenase isoenzyme determination in the diagnosis of acute myocardial infarction. Circulation 1978, 57, 1055–1057. [Google Scholar] [CrossRef]
- Cui, J.; Xiong, J.; Zhang, Y.; Peng, T.; Huang, M.; Lin, Y.; Guo, Y.; Wu, H.; Wang, C. Serum lactate dehydrogenase is predictive of persistent organ failure in acute pancreatitis. J. Crit. Care 2017, 41, 161–165. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Becker, K. Renal Cortical Lactate Dehydrogenase: A Useful, Accurate, Quantitative Marker of In Vivo Tubular Injury and Acute Renal Failure. PLoS ONE 2013, 8, e66776. [Google Scholar] [CrossRef]
- Kotoh, K.; Kato, M.; Kohjima, M.; Tanaka, M.; Miyazaki, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Lactate dehydrogenase production in hepatocytes is increased at an early stage of acute liver failure. Exp. Ther. Med. 2011, 2, 195–199. [Google Scholar] [CrossRef]
- Kikuchi, G.; Kumar, A.; Talmage, P.; Shemin, D. The enzymatic synthesis of delta-aminolevulinic acid. J. Biol. Chem. 1958, 233, 1214–1219. [Google Scholar] [CrossRef]
- Macrobert, A.J.; Theodossiou, T. Photodynamic Therapy of Cancer. Encycl. Mod. Opt. 2005, 1, 53–62. [Google Scholar]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Kennedy, J.C.; Pottier, R.H. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J. Photochem. Photobiol. B 1992, 14, 275–292. [Google Scholar] [CrossRef]
- Batlle, A.M. Porphyrins, porphyrias, cancer and photodynamic therapy—A model for carcinogenesis. J. Photochem. Photobiol. B 1993, 20, 5–22. [Google Scholar] [CrossRef]
- Ohgari, Y.; Nakayasu, Y.; Kitajima, S.; Sawamoto, M.; Mori, H.; Shimokawa, O.; Matsui, H.; Taketani, S. Mechanisms involved in delta-aminolevulinic acid (ALA)-induced photosensitivity of tumor cells: Relation of ferrochelatase and uptake of ALA to the accumulation of protoporphyrin. Biochem. Pharmacol. 2005, 71, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Krieg, R.C.; Fickweiler, S.; Wolfbeis, O.S.; Knuechel, R. Cell-type specific protoporphyrin IX metabolism in human bladder cancer in vitro. Photochem. Photobiol. 2000, 72, 226–233. [Google Scholar] [CrossRef]
- Navone, N.M.; Polo, C.F.; Frisardi, A.L.; Andrade, N.E.; Battle, A.M. Heme biosynthesis in human breast cancer--mimetic “in vitro” studies and some heme enzymic activity levels. Int. J. Biochem. 1990, 22, 1407–1411. [Google Scholar] [CrossRef]
- Hinnen, P.; de Rooij, F.W.; van Velthuysen, M.L.; Edixhoven, A.; van Hillegersberg, R.; Tilanus, H.W.; Wilson, J.H.; Siersema, P.D. Biochemical basis of 5-aminolaevulinic acid-induced protoporphyrin IX accumulation: A study in patients with (pre)malignant lesions of the oesophagus. Br. J. Cancer 1998, 78, 679–682. [Google Scholar] [CrossRef]
- Peng, Q.; Warloe, T.; Berg, K.; Moan, J.; Kongshaug, M.; Giercksky, K.E.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy. Clinical research and future challenges. Cancer 1997, 79, 2282–2308. [Google Scholar] [CrossRef]
- Inoue, K.; Fukuhara, H.; Shimamoto, T.; Kamada, M.; Iiyama, T.; Miyamura, M.; Kurabayashi, A.; Furihata, M.; Tanimura, M.; Watanabe, H.; et al. Comparison between intravesical and oral administration of 5-aminolevulinic acid in the clinical benefit of photodynamic diagnosis for nonmuscle invasive bladder cancer. Cancer 2012, 118, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Inoue, K.; Satake, H.; Tamura, K.; Karashima, T.; Yamasaki, I.; Tatsuo, I.; Kurabayashi, A.; Furihata, M.; Shuin, T. Photodynamic diagnosis of positive margin during radical prostatectomy: Preliminary experience with 5-aminolevulinic acid. Int. J. Urol. 2011, 18, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Stocker, S.; Wagner, S.; Stepp, H.; Fritsch, C.; Goetz, C.; Goetz, A.E.; Kiefmann, R.; Reulen, H.J. Intraoperative detection of malignant gliomas by 5-aminolevulinic acid-induced porphyrin fluorescence. Neurosurgery 1998, 42, 518–525; discussion 525–526. [Google Scholar] [CrossRef]
- Stummer, W.; Novotny, A.; Stepp, H.; Goetz, C.; Bise, K.; Reulen, H.J. Fluorescence-guided resection of glioblastoma multiforme by using 5-aminolevulinic acid-induced porphyrins: A prospective study in 52 consecutive patients. J. Neurosurg. 2000, 93, 1003–1013. [Google Scholar] [CrossRef]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; Group, A.L.-G.S. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Ennis, S.R.; Novotny, A.; Xiang, J.; Shakui, P.; Masada, T.; Stummer, W.; Smith, D.E.; Keep, R.F. Transport of 5-aminolevulinic acid between blood and brain. Brain Res. 2003, 959, 226–234. [Google Scholar] [CrossRef]
- Ogura, S.; Maruyama, K.; Hagiya, Y.; Sugiyama, Y.; Tsuchiya, K.; Takahashi, K.; Abe, F.; Tabata, K.; Okura, I.; Nakajima, M.; et al. The effect of 5-aminolevulinic acid on cytochrome c oxidase activity in mouse liver. BMC Res. Notes 2011, 4, 66. [Google Scholar] [CrossRef]
- Frank, J.; Lornejad-Schafer, M.R.; Schoffl, H.; Flaccus, A.; Lambert, C.; Biesalski, H.K. Inhibition of heme oxygenase-1 increases responsiveness of melanoma cells to ALA-based photodynamic therapy. Int. J. Oncol. 2007, 31, 1539–1545. [Google Scholar] [CrossRef]
- Hara, T.; Koda, A.; Nozawa, N.; Ota, U.; Kondo, H.; Nakagawa, H.; Kamiya, A.; Miyashita, K.; Itoh, H.; Nakajima, M.; et al. Combination of 5-aminolevulinic acid and ferrous ion reduces plasma glucose and hemoglobin A1c levels in Zucker diabetic fatty rats. FEBS Open Bio 2016, 6, 515–528. [Google Scholar] [CrossRef]
- Rodriguez, B.L.; Curb, J.D.; Davis, J.; Shintani, T.; Perez, M.H.; Apau-Ludlum, N.; Johnson, C.; Harrigan, R.C. Use of the dietary supplement 5-aminiolevulinic acid (5-ALA) and its relationship with glucose levels and hemoglobin A1C among individuals with prediabetes. Clin. Transl. Sci. 2012, 5, 314–320. [Google Scholar] [CrossRef]
- Higashikawa, F.; Noda, M.; Awaya, T.; Tanaka, T.; Sugiyama, M. 5-aminolevulinic acid, a precursor of heme, reduces both fasting and postprandial glucose levels in mildly hyperglycemic subjects. Nutrition 2013, 29, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Al-Saber, F.; Aldosari, W.; Alselaiti, M.; Khalfan, H.; Kaladari, A.; Khan, G.; Harb, G.; Rehani, R.; Kudo, S.; Koda, A.; et al. The Safety and Tolerability of 5-Aminolevulinic Acid Phosphate with Sodium Ferrous Citrate in Patients with Type 2 Diabetes Mellitus in Bahrain. J. Diabetes Res. 2016, 2016, 8294805. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Goodsell, D.; Halliday, R.; Huey, R.; Hart, W.; Belew, R.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: A metabolic survival role for tumor-associated stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Brand, K.A.; Hermfisse, U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. FASEB J. 1997, 11, 388–395. [Google Scholar] [CrossRef]
- Allison, S.J.; Knight, J.R.; Granchi, C.; Rani, R.; Minutolo, F.; Milner, J.; Phillips, R.M. Identification of LDH-A as a therapeutic target for cancer cell killing via (i) p53/NAD(H)-dependent and (ii) p53-independent pathways. Oncogenesis 2014, 3, e102. [Google Scholar] [CrossRef]
- Gadmar, O.B.; Moan, J.; Scheie, E.; Ma, L.W.; Peng, Q. The stability of 5-aminolevulinic acid in solution. J. Photochem. Photobiol. B 2002, 67, 187–193. [Google Scholar] [CrossRef]
- Elfsson, B.; Wallin, I.; Eksborg, S.; Rudaeus, K.; Ros, A.M.; Ehrsson, H. Stability of 5-aminolevulinic acid in aqueous solution. Eur. J. Pharm. Sci. 1999, 7, 87–91. [Google Scholar] [CrossRef]
- Wu, J.T.; Wu, L.H.; Knight, J.A. Stability of NADPH: Effect of various factors on the kinetics of degradation. Clin. Chem. 1986, 32, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelopoulou, M.; Grigalavicius, M.; Raabe, T.H.; Skarpen, E.; Juzenas, P.; Peng, Q.; Berg, K.; Theodossiou, T.A. Predictive biomarkers for 5-ALA-PDT can lead to personalized treatments and overcome tumor-specific resistances. Cancer Rep. 2020, e1278. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.P.; Wardhaugh, T.; Patrick, L.; Turner, M.; Phillips, J.B.; Bruce, J.I.; Kimani, S.G. Targeting tumour energy metabolism potentiates the cytotoxicity of 5-aminolevulinic acid photodynamic therapy. Br. J. Cancer 2013, 109, 976–982. [Google Scholar] [CrossRef]
- Floridi, A.; Paggi, M.G.; D’Atri, S.; De Martino, C.; Marcante, M.L.; Silvestrini, B.; Caputo, A. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res. 1981, 41, 4661–4666. [Google Scholar] [PubMed]
- Sugiyama, Y.; Hagiya, Y.; Nakajima, M.; Ishizuka, M.; Tanaka, T.; Ogura, S. The heme precursor 5-aminolevulinic acid disrupts the Warburg effect in tumor cells and induces caspase-dependent apoptosis. Oncol. Rep. 2014, 31, 1282–1286. [Google Scholar] [CrossRef]
- Daniele, S.; Giacomelli, C.; Zappelli, E.; Granchi, C.; Trincavelli, M.L.; Minutolo, F.; Martini, C. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci. Rep. 2015, 5, 15556. [Google Scholar] [CrossRef]
- Dalton, J.T.; Yates, C.R.; Yin, D.; Straughn, A.; Marcus, S.L.; Golub, A.L.; Meyer, M.C. Clinical pharmacokinetics of 5-aminolevulinic acid in healthy volunteers and patients at high risk for recurrent bladder cancer. J. Pharmacol. Exp. Ther. 2002, 301, 507–512. [Google Scholar] [CrossRef]
- Stepp, H.; Beck, T.; Pongratz, T.; Meinel, T.; Kreth, F.W.; Tonn, J.; Stummer, W. ALA and malignant glioma: Fluorescence-guided resection and photodynamic treatment. J. Environ. Pathol. Toxicol. Oncol. 2007, 26, 157–164. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Cell Lysates | ||||
|---|---|---|---|---|---|
| U87 | T98G | Ln18 | M059K | ||
| No inhib. | −2.28 | −1.43 | −1.20 | −1.48 | −1.47 |
| +5-ALA | −0.51 | −0.32 | −0.57 | −0.47 | −0.53 |
| +OXM | −0.44 | −0.73 | −0.25 | −0.24 | −0.32 |
| +TART | −0.32 | −0.24 | −0.26 | −0.17 | −0.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigalavicius, M.; Ezzatpanah, S.; Papakyriakou, A.; Raabe, T.T.H.; Yannakopoulou, K.; Theodossiou, T.A. 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers 2022, 14, 4003. https://doi.org/10.3390/cancers14164003
Grigalavicius M, Ezzatpanah S, Papakyriakou A, Raabe TTH, Yannakopoulou K, Theodossiou TA. 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers. 2022; 14(16):4003. https://doi.org/10.3390/cancers14164003
Chicago/Turabian StyleGrigalavicius, Mantas, Somayeh Ezzatpanah, Athanasios Papakyriakou, Tine Therese Henriksen Raabe, Konstantina Yannakopoulou, and Theodossis A. Theodossiou. 2022. "5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action" Cancers 14, no. 16: 4003. https://doi.org/10.3390/cancers14164003
APA StyleGrigalavicius, M., Ezzatpanah, S., Papakyriakou, A., Raabe, T. T. H., Yannakopoulou, K., & Theodossiou, T. A. (2022). 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers, 14(16), 4003. https://doi.org/10.3390/cancers14164003