
Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. RNA Sequencing and Data Acquisition
2.3. Gene Set Enrichment Analysis
2.4. RNA-Seq Data Analysis
2.5. Conditioned Media Growth Rate Analysis
2.6. Native ECM Growth Rate Analysis
2.7. Target Gene Selection
2.8. Compound Toxicity Screening
2.9. Synchronized Cell Cycle Analysis
2.10. 3D Cultures
2.11. Live/Dead Analysis
2.12. 3D Culture Structure Analysis
2.13. Nuclear Size Analysis
2.14. Tissue Processing
2.15. Primary Cell Culture
2.16. Western Blot Analysis
3. Results
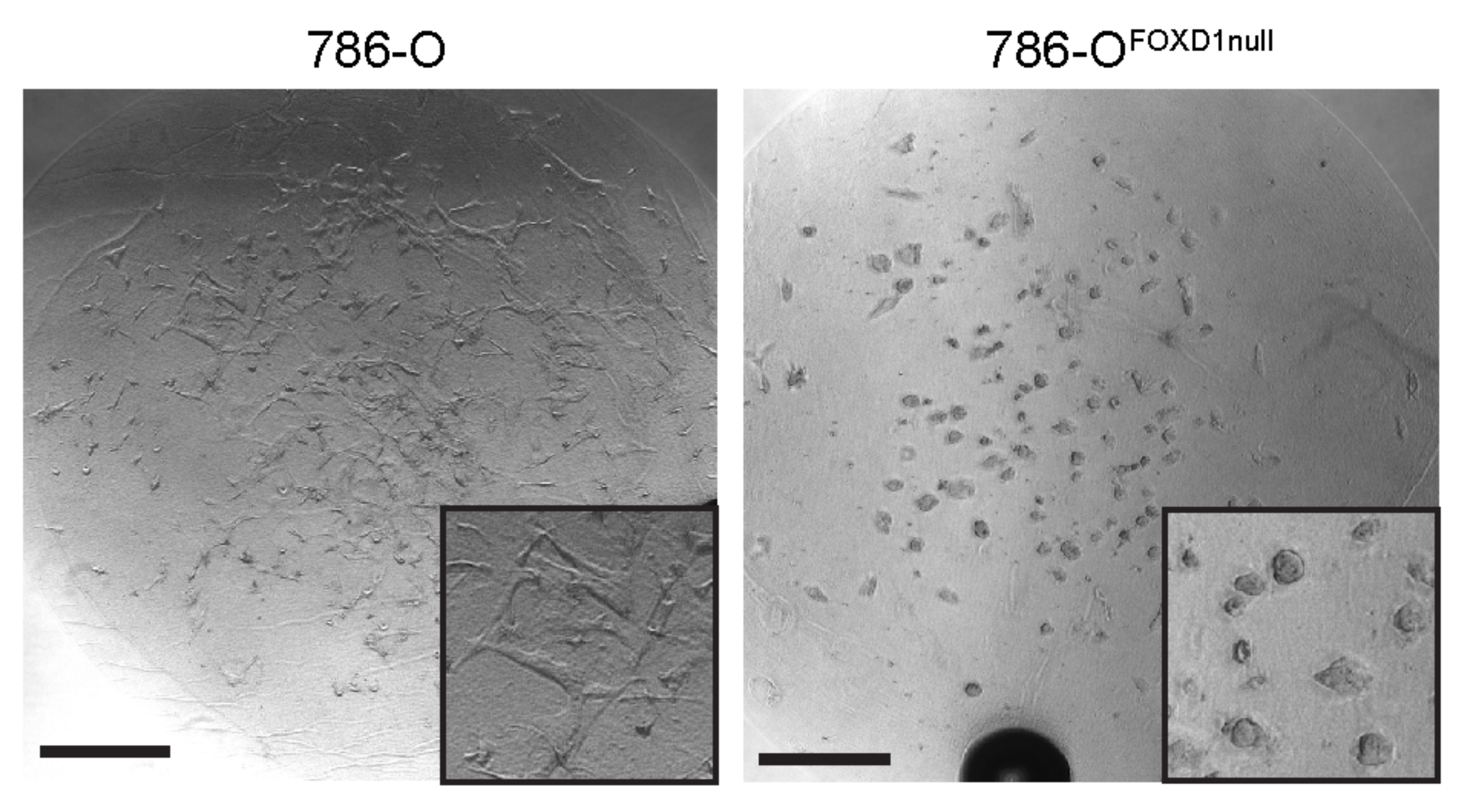
3.1. RNA-Seq Analysis Identifies Novel Targets Downstream of FOXD1
3.2. Exclusion of Secreted Factors
3.3. Exclusion of ECM
3.4. Selection of Activators and Inhibitors
3.5. Inhibition of Cell Growth with Minimal Toxicity by Compounds Targeting Candidate Genes
3.6. Selection of Candidate Compounds That Induce Cell Cycle Arrest Resembling the Effect of FOXD1 Inactivation
3.7. FOXD1 Inactivation Affects Three-Dimensional Growth
3.8. Treatment-Induced Growth Delay Affects 3D Growth and Nuclear Structure of 786-O Cells
3.9. Effects of Blocking FOXD1 G2/M Targets on 3D Patient Tumor Replica Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, L.; Hu, Y.; Song, M.; Liu, Z.; Zhang, W.; Yu, F.X.; Wu, J.; Wang, S.; Izpisua Belmonte, J.C.; Chan, P.; et al. Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol. 2019, 17, e3000201. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larribère, L.; Sun, Q.; Novak, D.; Sachindra, S.; Granados, K.; Umansky, V.; Utikal, J. Loss of neural crest-associated gene FOXD1 impairs melanoma invasion and migration via RAC1B downregulation. Int. J. Cancer 2018, 143, 2962–2972. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F.; Zhao, J.Y.; Yue, H.; Hu, K.S.; Shen, H.; Guo, Z.G.; Su, X.J. FOXD1 promotes breast cancer proliferation and chemotherapeutic drug resistance by targeting p27. Biochem. Biophys. Res. Commun. 2015, 456, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Zhu, T.; Mao, X.Y.; Mao, C.X.; Li, L.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. Silencing of Forkhead box D1 inhibits proliferation and migration in glioma cells. Oncol. Rep. 2017, 37, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Hatini, V.; Huh, S.O.; Herzlinger, D.; Soares, V.C.; Lai, E. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 1996, 10, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Fetting, J.L.; Guay, J.A.; Karolak, M.J.; Iozzo, R.V.; Adams, D.C.; Maridas, D.E.; Brown, A.C.; Oxburgh, L. FOXD1 promotes nephron progenitor differentiation by repressing decorin in the embryonic kidney. Development 2014, 141, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gkolfinopoulos, S.; Psyrri, A.; Bamias, A. Clear-cell renal cell carcinoma—A comprehensive review of agents used in the contemporary management of advanced/metastatic disease. Oncol. Rev. 2021, 15, 530. [Google Scholar] [CrossRef]
- Bond, K.H.; Fetting, J.L.; Lary, C.W.; Emery, I.F.; Oxburgh, L. FOXD1 regulates cell division in clear cell renal cell carcinoma. BMC Cancer 2021, 21, 312. [Google Scholar] [CrossRef]
- Bond, K.H.; Chiba, T.; Wynne, K.P.H.; Vary, C.P.H.; Sims-Lucas, S.; Coburn, J.M.; Oxburgh, L. The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth. Cancers 2021, 13, 5873. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Gearing, L.J.; Cumming, H.E.; Chapman, R.; Finkel, A.M.; Woodhouse, I.B.; Luu, K.; Gould, J.A.; Forster, S.C.; Hertzog, P.J. CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS ONE 2019, 14, e0215495. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.M.; Raitman, I.; Schwarzbauer, J.E. Cell-derived decellularized extracellular matrices. Methods Cell Biol. 2018, 143, 97–114. [Google Scholar]
- Lorca, T.; Manfredi, J.J.; Orlic-Milacic, M. Mitotic G2-G2/M phases. Reactome Reactome, 2018. [Google Scholar]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.T.; Naba, A. MatrisomeDB: The ECM-protein knowledge database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kel, A.E.; Goößling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCHTM: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef]
- Arnone, M.I.; Davidson, E.H. The hardwiring of development: Organization and function of genomic regulatory systems. Development 1997, 124, 1851–1864. [Google Scholar] [CrossRef]
- Taneja, N.; Fenix, A.M.; Rathbun, L.; Millis, B.A.; Tyska, M.J.; Hehnly, H.; Burnette, D.T. Focal adhesions control cleavage furrow shape and spindle tilt during mitosis. Sci. Rep. 2016, 6, 29846. [Google Scholar] [CrossRef]
- Meierjohann, S.; Hufnagel, A.; Wende, E.; Kleinschmidt, M.A.; Wolf, K.; Friedl, P.; Gaubatz, S.; Schartl, M. MMP13 mediates cell cycle progression in melanocytes and melanoma cells: In vitro studies of migration and proliferation. Mol. Cancer 2010, 9, 201. [Google Scholar] [CrossRef]
- Kominsky, S.L.; Doucet, M.; Thorpe, M.; Weber, K.L. MMP-13 is over-expressed in renal cell carcinoma bone metastasis and is induced by TGF-beta1. Clin. Exp. Metastasis 2008, 25, 865–870. [Google Scholar] [CrossRef]
- Jaffré, F.; Friedman, A.E.; Hu, Z.; Mackman, N.; Blaxall, B.C. β-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation 2012, 125, 2993–3003. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, J.; He, Z.; Xiao, Y. SUZ12 depletion suppresses the proliferation of gastric cancer cells. Cell Physiol. Biochem. 2013, 31, 778–784. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef]
- Ströbel, T.; Madlener, S.; Tuna, S.; Vose, S.; Lagerweij, T.; Wurdinger, T.; Vierlinger, K.; Wöhrer, A.; Price, B.D.; Demple, B.; et al. Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma. Sci. Rep. 2017, 7, 9674. [Google Scholar] [CrossRef]
- Segura-Collar, B.; Gargini, R.; Tovar-Ambel, E.; Hernández-SanMiguel, E.; Epifano, C.; Pérez de Castro, I.; Hernández-Laín, A.; Casas-Tintó, S.; Sánchez-Gómez, P. The EGFR-TMEM167A-p53 Axis Defines the Aggressiveness of Gliomas. Cancers 2020, 12, 208. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, Y.; Zhang, Z.; Li, Z.; Kong, C. UNC5B mediates G2/M phase arrest of bladder cancer cells by binding to CDC14A and P53. Cancer Gene Ther. 2020, 27, 934–947. [Google Scholar] [CrossRef]
- Wu, G.; Cheng Zhang, C. Membrane protein CAR promotes hematopoietic regeneration upon stress. Haematologica 2021, 106, 2180–2190. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, L.; Lu, L.; Zhang, L.; Zhang, G.; Wang, Q.; Yang, P. Role and mechanism of the alkylglycerone phosphate synthase in suppressing the invasion potential of human glioma and hepatic carcinoma cells in vitro. Oncol. Rep. 2014, 32, 431–436. [Google Scholar] [CrossRef]
- Stazi, G.; Battistelli, C.; Piano, V.; Mazzone, R.; Marrocco, B.; Marchese, S.; Louie, S.M.; Zwergel, C.; Antonini, L.; Patsilinakos, A.; et al. Development of alkyl glycerone phosphate synthase inhibitors: Structure-activity relationship and effects on ether lipids and epithelial-mesenchymal transition in cancer cells. Eur. J. Med. Chem. 2019, 163, 722–735. [Google Scholar] [CrossRef]
- Morita, K.; He, S.; Nowak, R.P.; Wang, J.; Zimmerman, M.W.; Fu, C.; Durbin, A.D.; Martel, M.W.; Prutsch, N.; Gray, N.S.; et al. Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 2020, 181, 702–715. [Google Scholar] [CrossRef]
- Chen, X.; Müller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef]
- Xue, Y.J.; Xiao, R.H.; Long, D.Z.; Zou, X.F.; Wang, X.N.; Zhang, G.X.; Yuan, Y.H.; Wu, G.Q.; Yang, J.; Wu, Y.T.; et al. Overexpression of FoxM1 is associated with tumor progression in patients with clear cell renal cell carcinoma. J. Transl. Med. 2012, 10, 200. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef]
- Akashi, E.; Fujihara, S.; Morishita, A.; Tadokoro, T.; Chiyo, T.; Fujikawa, K.; Kobara, H.; Mori, H.; Iwama, H.; Okano, K.; et al. Effects of galectin-9 on apoptosis, cell cycle and autophagy in human esophageal adenocarcinoma cells. Oncol. Rep. 2017, 38, 506–514. [Google Scholar] [CrossRef]
- Jikuya, R.; Kishida, T.; Sakaguchi, M.; Yokose, T.; Yasui, M.; Hashizume, A.; Tatenuma, T.; Mizuno, N.; Muraoka, K.; Umemoto, S.; et al. Galectin-9 expression as a poor prognostic factor in patients with renal cell carcinoma. Cancer Immunol. Immunother. CII 2020, 69, 2041–2051. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamamoto, H.; Akutsu, N.; Nosho, K.; Adachi, Y.; Imai, K.; Shinomura, Y. Transcriptional silencing of hedgehog-interacting protein by CpG hypermethylation and chromatic structure in human gastrointestinal cancer. J. Pathol. 2007, 213, 131–139. [Google Scholar] [CrossRef]
- Kotulak-Chrzaszcz, A.; Klacz, J.; Matuszewski, M.; Kmiec, Z.; Wierzbicki, P.M. Expression of the Sonic Hedgehog pathway components in clear cell renal cell carcinoma. Oncol. Lett. 2019, 18, 5801–5810. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Wang, G.; Barsukov, I.L.; Gross, S.R.; Smith, R.; Rudland, P.S. Direct interaction of metastasis-inducing S100P protein with tubulin causes enhanced cell migration without changes in cell adhesion. Biochem. J. 2020, 477, 1159–1178. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Sun, D.; Peng, Z.; Pal, A.; Maxwell, D.S.; Bornmann, W.G.; Logsdon, C.D. Designing and Developing S100P Inhibitor 5-Methyl Cromolyn for Pancreatic Cancer Therapy. Mol. Cancer Ther. 2013, 12, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Stojic, L.; Lun, A.T.L.; Mascalchi, P.; Ernst, C.; Redmond, A.M.; Mangei, J.; Barr, A.R.; Bousgouni, V.; Bakal, C.; Marioni, J.C.; et al. A high-content RNAi screen reveals multiple roles for long noncoding RNAs in cell division. Nat. Commun. 2020, 11, 1851. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Sun, R.; Zhang, Y.; Fu, Y.; Zheng, Z.; Ji, Z.; Zhao, D. ANGPTL4 Promotes the Proliferation of Papillary Thyroid Cancer via AKT Pathway. OncoTargets Ther. 2020, 13, 2299–2309. [Google Scholar] [CrossRef]
- Cui, D.; Zhao, Y.; Xu, J. Activation of CXCL5-CXCR2 axis promotes proliferation and accelerates G1 to S phase transition of papillary thyroid carcinoma cells and activates JNK and p38 pathways. Cancer Biol. Ther. 2019, 20, 608–616. [Google Scholar] [CrossRef]
- Tominaga, K.; Minato, H.; Murayama, T.; Sasahara, A.; Nishimura, T.; Kiyokawa, E.; Kanauchi, H.; Shimizu, S.; Sato, A.; Nishioka, K.; et al. Semaphorin signaling via MICAL3 induces symmetric cell division to expand breast cancer stem-like cells. Proc. Natl. Acad. Sci. 2019, 116, 625–630. [Google Scholar] [CrossRef]
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757. [Google Scholar] [CrossRef]
- Sinha, R.; Winer, A.G.; Chevinsky, M.; Jakubowski, C.; Chen, Y.B.; Dong, Y.; Tickoo, S.K.; Reuter, V.E.; Russo, P.; Coleman, J.A.; et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat. Commun. 2017, 8, 15165. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Pal-Ghosh, R.; Xue, D.; Warburton, R.; Hill, N.; Polgar, P.; Wilson, J.L. CDC2 Is an Important Driver of Vascular Smooth Muscle Cell Proliferation via FOXM1 and PLK1 in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 6943. [Google Scholar] [CrossRef]
- Jeong, A.L.; Yang, Y. PP2A function toward mitotic kinases and substrates during the cell cycle. BMB Rep. 2013, 46, 289–294. [Google Scholar] [CrossRef]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. CCS 2018, 16, 57. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834. [Google Scholar] [CrossRef]
- Wang, S.P.; Wu, S.Q.; Huang, S.H.; Tang, Y.X.; Meng, L.Q.; Liu, F.; Zhu, Q.H.; Xu, Y.G. FDI-6 inhibits the expression and function of FOXM1 to sensitize BRCA-proficient triple-negative breast cancer cells to Olaparib by regulating cell cycle progression and DNA damage repair. Cell Death Dis. 2021, 12, 1138. [Google Scholar] [CrossRef]
- Xia, X.; Gholkar, A.; Senese, S.; Torres, J.Z. A LCMT1-PME-1 methylation equilibrium controls mitotic spindle size. Cell Cycle 2015, 14, 1938–1947. [Google Scholar] [CrossRef][Green Version]
- Guo, S.; Bai, X.; Liu, Y.; Shi, S.; Wang, X.; Zhan, Y.; Kang, X.; Chen, Y.; An, H. Inhibition of TMEM16A by Natural Product Silibinin: Potential Lead Compounds for Treatment of Lung Adenocarcinoma. Front. Pharmacol. 2021, 12, 643489. [Google Scholar] [CrossRef]
- Portela, M.; Segura-Collar, B.; Argudo, I.; Sáiz, A.; Gargini, R.; Sánchez-Gómez, P.; Casas-Tintó, S. Oncogenic dependence of glioma cells on kish/TMEM167A regulation of vesicular trafficking. Glia 2019, 67, 404–417. [Google Scholar] [CrossRef]
- Comelli, M.C.; Mengs, U.; Schneider, C.; Prosdocimi, M. Toward the definition of the mechanism of action of silymarin: Activities related to cellular protection from toxic damage induced by chemotherapy. Integr. Cancer Ther. 2007, 6, 120–129. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Stage | Grade | Race | Gender/Age | |
|---|---|---|---|---|---|
| Tp17-s322 | Unclassified renal neoplasm | NA | NA | White | F/70–79 |
| Tp17-s438 | Unclassified renal neoplasm | 2 | 3 | White | F/30–39 |
| Tp18-S453 | Clear cell renal cell carcinoma | 1 | 3 | White | M/80–86 |
| Tp18-s109 | Clear cell renal cell carcinoma | 3 | 2 | White | M/60–60 |
| Tp18-s114 | Clear cell renal cell carcinoma | 3 | 3 | White | M/80–89 |
| Tp20-s199 | Clear cell renal cell carcinoma | 3 | 2 | NA | M/74 |
| Tp20-s277 | Leiomyosarcoma | 3 | NA | White | M/69 |
| # | Compound | Target | Action | Reference |
|---|---|---|---|---|
| 1 | WAY 170523 | MMP13 | INHIBIT | [23,24,25] |
| 2 | EED226 | SUZ12 | INHIBIT | [26,27] |
| 3 | APE1 Inhibitor III | APEX1 | INHIBIT | [28] |
| 4 | Silibinin | TMEM167A | INHIBIT | [29] |
| 5 | Recombinant Netrin | UNC5B | ACTIVATE | [30] |
| 6 | CINPA 1 | CXADR | INHIBIT | [31] |
| 7 | BITC | AGPS | INHIBIT | [32,33] |
| 8 | AMZ-30 | PME1 | INHIBIT | [34] |
| 9 | FDI-6 | FOXM1 | INHIBIT | [35,36] |
| 10 | CPI-637 | EP300 | INHIBIT | [37] |
| 11 | Anti-Galectin-9 Antibody | LGALS9 | INHIBIT | [38,39] |
| 12 | Recombinant Sonic Hedgehog/Shh | HHIP | INHIBIT | [40,41] |
| 13 | C5OH | S100P | INHIBIT | [25,42,43] |
| 14 | Recombinant C1qTNF1 | C1QTNF1 | ACTIVATE | [44] |
| 15 | Recombinant Angiopoietin-like 4 | ANGPTL4 | ACTIVATE | [45] |
| 16 | Recombinant CXCL5/ENA-7 | CXCL5 | ACTIVATE | [46] |
| 17 | Recombinant Semaphorin 3C | SEMA3C | ACTIVATE | [47] |
| 18 | Recombinant LTBP1 | LTBP1 | ACTIVATE | [48] |
| 786-O | FOXD1 null | +AMZ30 | +Silibinin | +FDI6 | |
|---|---|---|---|---|---|
| # of Structures | + | ++ | + | +++ | ++ |
| Colony Size | ++ | +++ | +++ | ++ | + |
| Colony Morphology | Invasive | Round | Round | Round and Vacuolated | Irregular and protruding |
| Nuclear Defects | + Normal | ++ Large, mitotic defects prevalent | ++ Small and irregular | ++ Large and swollen | +++ Very small, fragmented |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bond, K.H.; Sims-Lucas, S.; Oxburgh, L. Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways. Cancers 2022, 14, 3958. https://doi.org/10.3390/cancers14163958
Bond KH, Sims-Lucas S, Oxburgh L. Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways. Cancers. 2022; 14(16):3958. https://doi.org/10.3390/cancers14163958
Chicago/Turabian StyleBond, Kyle H., Sunder Sims-Lucas, and Leif Oxburgh. 2022. "Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways" Cancers 14, no. 16: 3958. https://doi.org/10.3390/cancers14163958
APA StyleBond, K. H., Sims-Lucas, S., & Oxburgh, L. (2022). Targets for Renal Carcinoma Growth Control Identified by Screening FOXD1 Cell Proliferation Pathways. Cancers, 14(16), 3958. https://doi.org/10.3390/cancers14163958