
Targeting MDM4 as a Novel Therapeutic Approach in Prostate Cancer Independent of p53 Status

 , , , , ,
, , , , ,  ,
,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Prostate Cancer Specimens
2.2. Immunohistochemistry
2.3. Cell Culture
2.4. Inducible Lentiviral Short Hairpin RNA (shRNA) Sequences and Viral Production
2.5. Transduction of PC-3 (p53null) with p53-R273H
2.6. p53 Immunofluorescence Assay of PC-3 (p53R273H)
2.7. Incucyte® Cell Count Proliferation and Caspase 3/7 Apoptosis Assays
2.8. Small Molecule Preparation and Treatments
2.9. Colourimetric Assays
2.10. Immunoblotting
2.11. Flow Cytometry
2.12. RNA Extraction and Polymerase Chain Reactions (PCRs)
2.13. In Vivo Mice Experiments
2.14. Statistical Analyses
3. Results
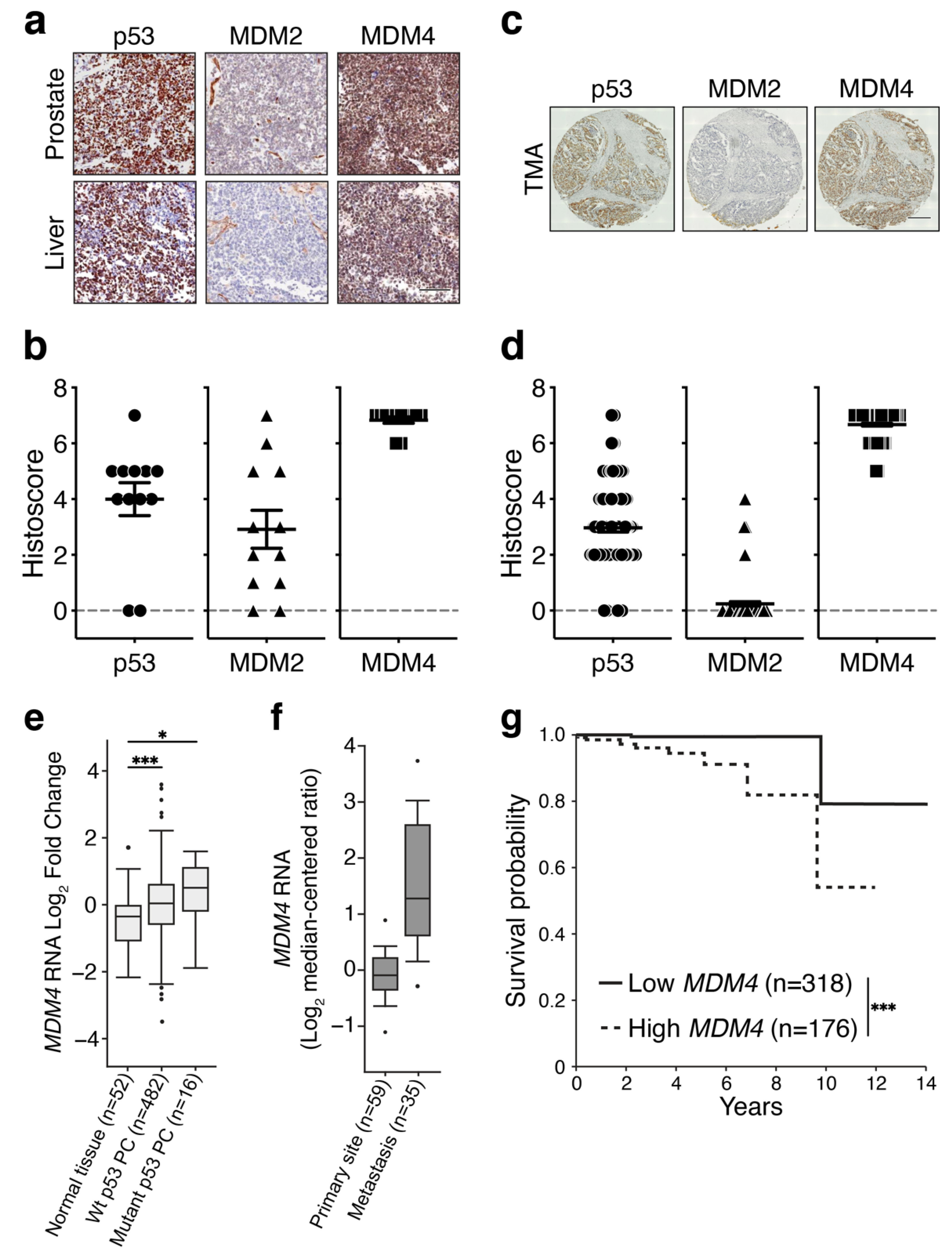
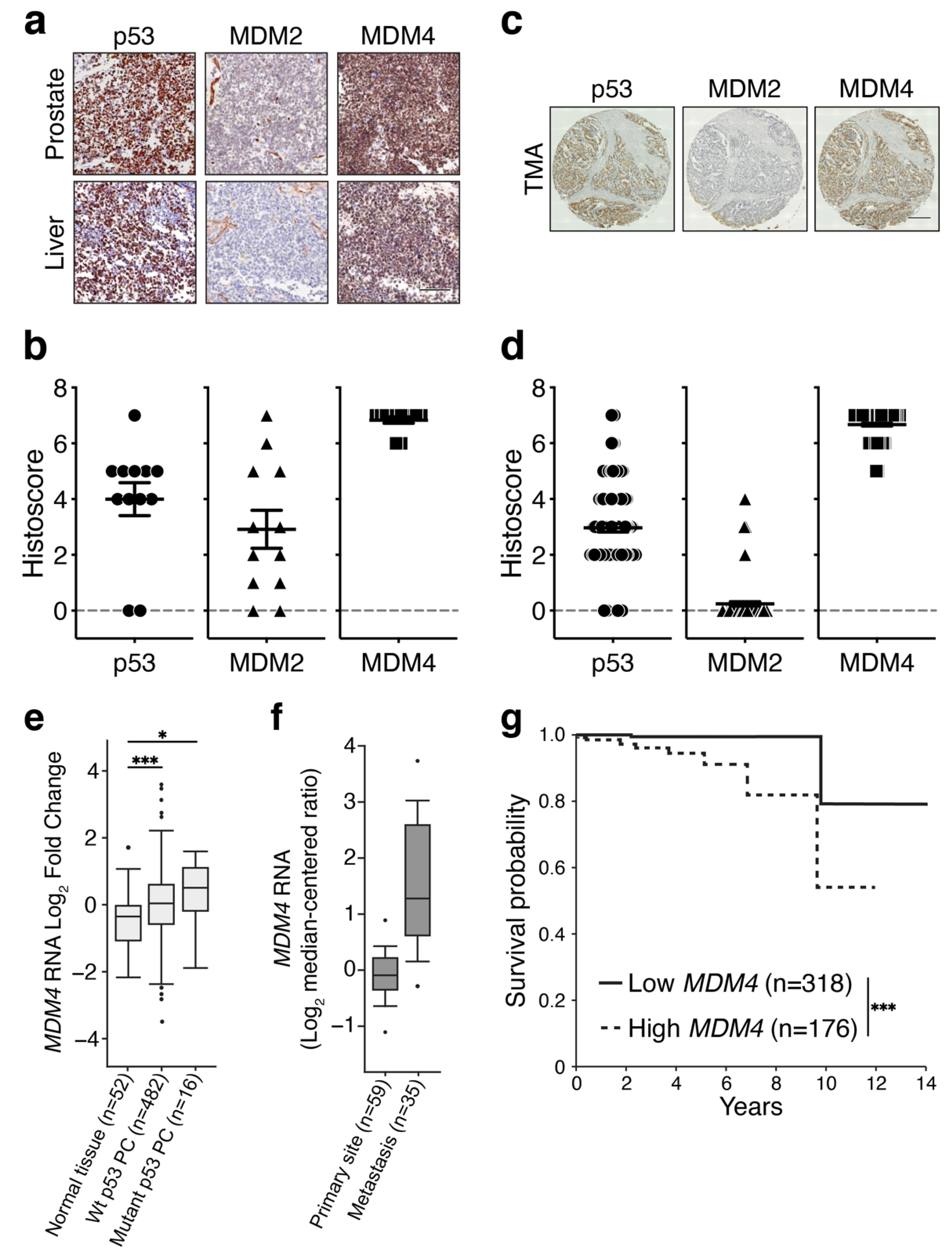
3.1. MDM4 Levels Are High in Primary and Metastatic PC
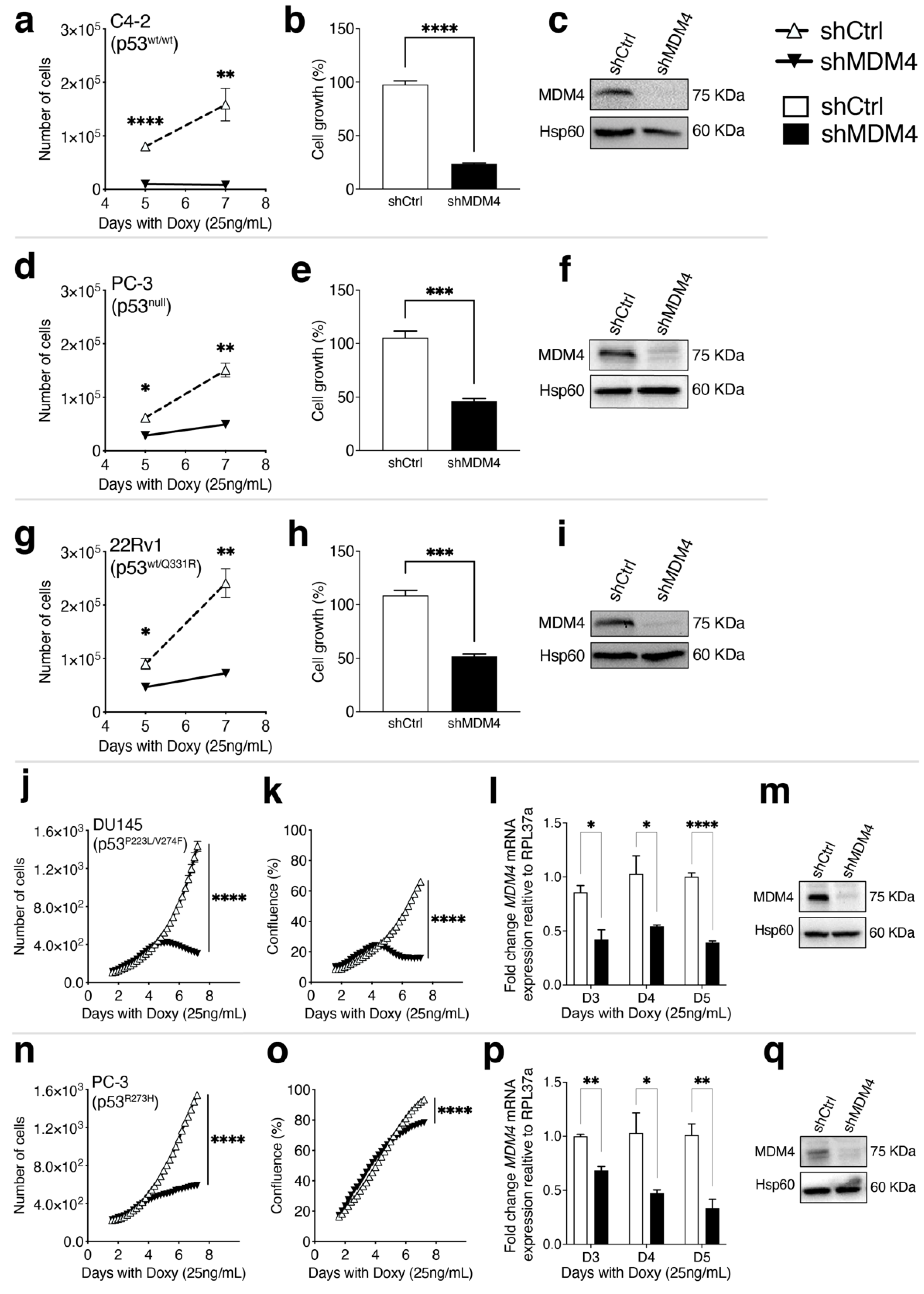
3.2. MDM4 Expression Is Required for the Growth of PC Cells In Vitro
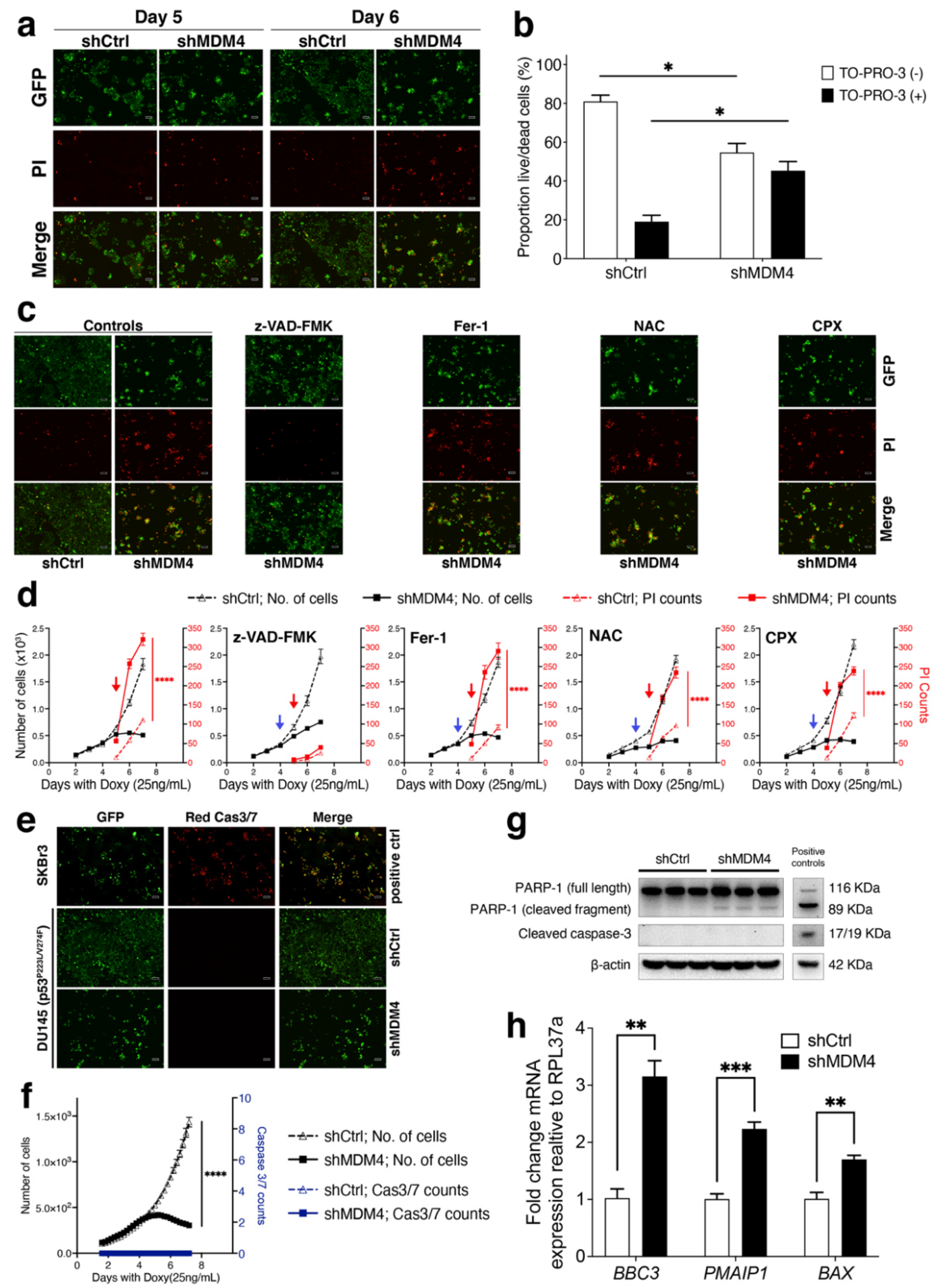
3.3. MDM4 Knockdown Causes Apoptotic Death In Vitro in PC Cells with Endogenous Mutant p53
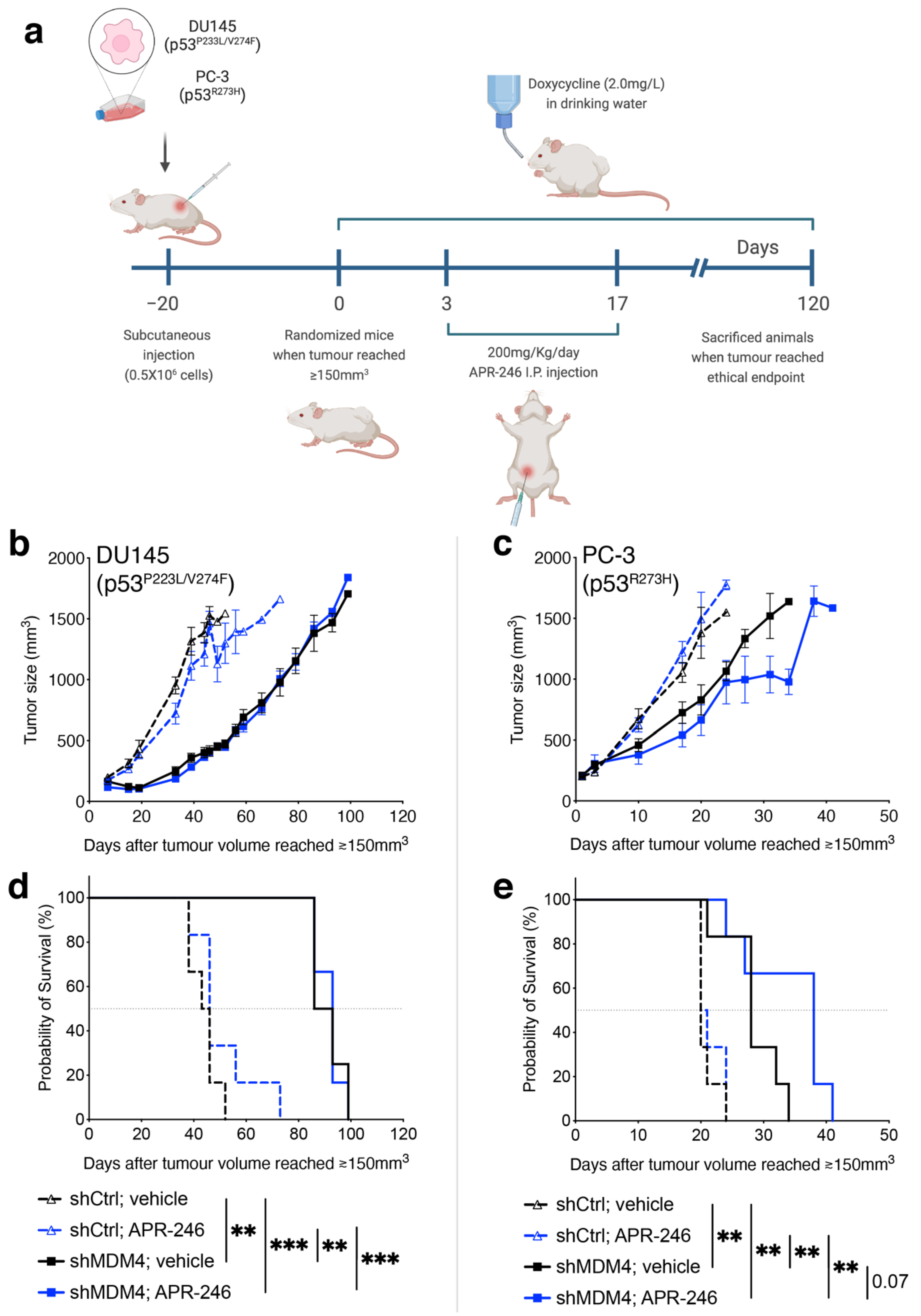
3.4. Downregulation of MDM4 Attenuates the Tumour Growth of DU145 Xenotransplants In Vivo
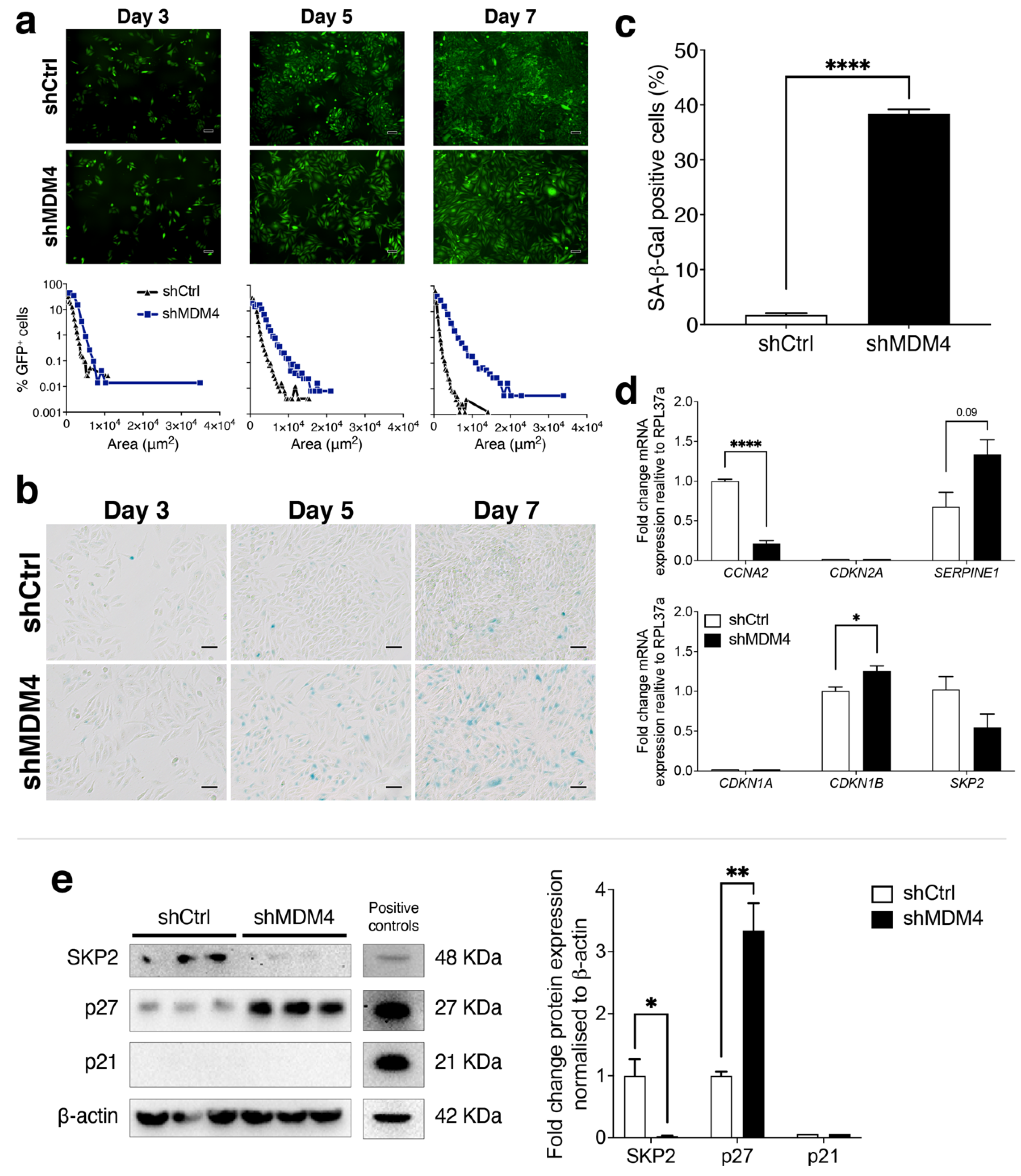
3.5. A Cellular Senescence Response Is Induced in PC-3 (p53R273H) Following MDM4 KD
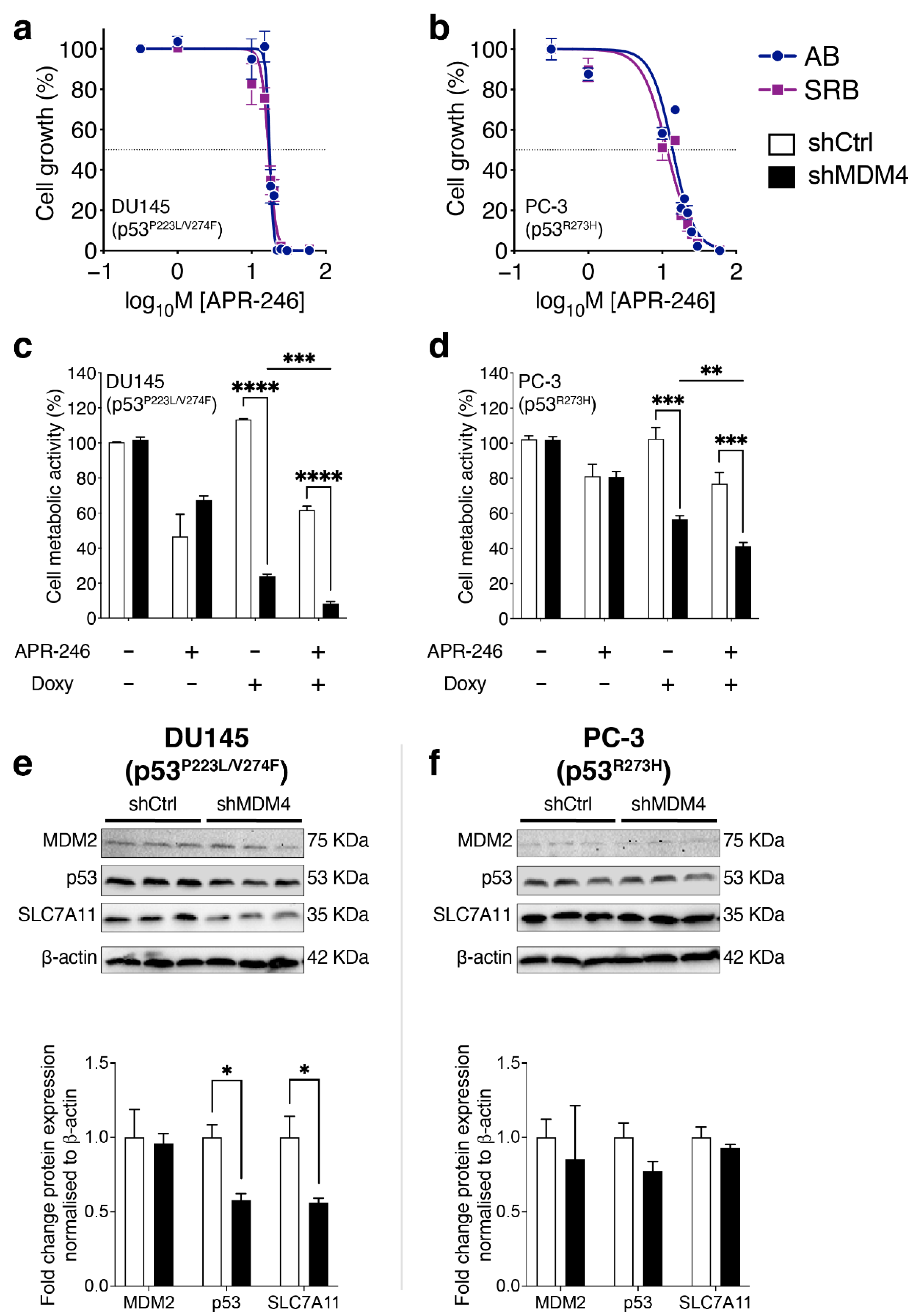
3.6. Treatment of PC Cells by Targeting MDM4 Alone or in Combination with APR-246
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris JPt Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; Aust, D.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar]
- Hong, M.K.; Macintyre, G.; Wedge, D.C.; Van Loo, P.; Patel, K.; Lunke, S.; Alexandrov, L.B.; Sloggett, C.; Cmero, M.; Marass, F.; et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 2015, 6, 6605. [Google Scholar] [CrossRef]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Gesztes, W.; Schafer, C.; Young, D.; Fox, J.; Jiang, J.; Chen, Y.; Kuo, H.C.; Mwamukonda, K.B.; Dobi, A.; Burke, A.P.; et al. Focal p53 protein expression and lymphovascular invasion in primary prostate tumors predict metastatic progression. Sci. Rep. 2022, 12, 5404. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.I.; Stricker, P.D.; Kench, J.G.; Grogan, J.; Haynes, A.M.; Henshall, S.M.; Grygiel, J.J.; Delprado, W.; Turner, J.J.; Horvath, L.G.; et al. p53 nuclear accumulation as an early indicator of lethal prostate cancer. Br. J. Cancer 2019, 121, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Teroerde, M.; Nientiedt, C.; Duensing, A.; Hohenfellner, M.; Stenzinger, A.; Duensing, S. Revisiting the Role of p53 in Prostate Cancer. In Prostate Cancer; Bott, S.R.J., Ng, K.L., Eds.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Haupt, S.; Mejia-Hernandez, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Haupt, S.; Buckley, D.; Pang, J.-M.B.; Panimaya, J.; Paul, P.J.; Gamel, C.; Takano, E.A.; Lee, Y.-Y.; Hidding, S.; Rogers, T.-M.; et al. Targeting Mdmx to treat Breast Cancers with wild type p53. Cell Death Dis. 2015, 6, e1821. [Google Scholar] [CrossRef]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef]
- Garcia, D.; Warr, M.R.; Martins, C.P.; Brown Swigart, L.; Passegue, E.; Evan, G.I. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011, 25, 1746–1757. [Google Scholar] [CrossRef]
- De Lange, J.; Teunisse, A.F.; Vries, M.V.; Lodder, K.; Lam, S.; Luyten, G.P.; Bernal, F.; Jager, M.J.; Jochemsen, A.G. High levels of Hdmx promote cell growth in a subset of uveal melanomas. Am. J. Cancer Res. 2012, 2, 492–507. [Google Scholar] [PubMed]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Xu, Z.Y.; Mo, S.; Yuan, L.; Cheng, X.D.; Qin, J.J. Targeting MDMX for Cancer Therapy: Rationale, Strategies, and Challenges. Front. Oncol. 2020, 10, 1389. [Google Scholar] [CrossRef] [PubMed]
- Miranda, P.J.; Buckley, D.; Raghu, D.; Pang, J.B.; Takano, E.A.; Vijayakumaran, R.; Teunisse, A.F.; Posner, A.; Procter, T.; Herold, M.J.; et al. MDM4 is a rational target for treating breast cancers with mutant p53. J. Pathol. 2017, 241, 661–670. [Google Scholar] [CrossRef]
- Herold, M.J.; van den Brandt, J.; Seibler, J.; Reichardt, H.M. Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc. Natl. Acad. Sci. USA 2008, 105, 18507–18512. [Google Scholar] [CrossRef]
- Wang, H.; Ma, X.; Ren, S.; Buolamwini, J.K.; Yan, C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 2011, 10, 69–79. [Google Scholar] [CrossRef]
- Czekanska, E.M. Assessment of cell proliferation with resazurin-based fluorescent dye. Methods Mol. Biol. 2011, 740, 27–32. [Google Scholar]
- Page, B.; Page, M.; Noel, C. A new fluorometric assay for cytotoxicity measurements in-vitro. Int. J. Oncol. 1993, 3, 473–476. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Louria-Hayon, I.; Grossman, T.; Sionov, R.V.; Alsheich, O.; Pandolfi, P.P.; Haupt, Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J. Biol. Chem. 2003, 278, 33134–33141. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.J.; Raghu, D.; Chan, A.L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Haupt, S.; Vijayakumaran, R.; Panimaya, J.; Burgess, A.; Lim, E.; Haupt, Y. The role of MDM2 and MDM4 in breast cancer development and prevention. J. Mol. Cell Biol. 2017, 9, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Thorne, H.; Sandhu, S.; Hamilton, A.; Mintoff, C.; Christie, E.; Spruyt, O.; Williams, S.; McNally, O.; Mileshkin, L.; et al. A community-based model of rapid autopsy in end-stage cancer patients. Nat. Biotechnol. 2016, 34, 1010–1014. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Stegeman, S.; Moya, L.; Selth, L.A.; Spurdle, A.B.; Clements, J.A.; Batra, J. A genetic variant of MDM4 influences regulation by multiple microRNAs in prostate cancer. Endocr. Relat. Cancer 2015, 22, 265–276. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- McCann, J.J.; Vasilevskaya, I.A.; McNair, C.; Gallagher, P.; Neupane, N.P.; de Leeuw, R.; Shafi, A.A.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; et al. Mutant p53 elicits context-dependent pro-tumorigenic phenotypes. Oncogene 2022, 41, 444–458. [Google Scholar] [CrossRef]
- Roh, J.L.; Park, J.Y.; Kim, E.H. XI-011 enhances cisplatin-induced apoptosis by functional restoration of p53 in head and neck cancer. Apoptosis 2014, 19, 1594–1602. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef] [PubMed]
- Molnar, T.; Pallagi, P.; Tel, B.; Kiraly, R.; Csoma, E.; Jenei, V.; Varga, Z.; Gogolak, P.; Odile Hueber, A.; Mate, Z.; et al. Caspase-9 acts as a regulator of necroptotic cell death. FEBS J. 2021, 288, 6476–6491. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.R.; Kelley, K.; Tuttle, R.; Berberich, S.J. HdmX overexpression inhibits oncogene induced cellular senescence. Cell Cycle 2010, 9, 3376–3382. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Xu, S.; Wu, W.; Huang, H.; Huang, R.; Xie, L.; Su, A.; Liu, S.; Zheng, R.; Yuan, Y.; Zheng, H.L.; et al. The p53/miRNAs/Ccna2 pathway serves as a novel regulator of cellular senescence: Complement of the canonical p53/p21 pathway. Aging Cell 2019, 18, e12918. [Google Scholar] [CrossRef]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar] [CrossRef]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022, 107, 403–416. [Google Scholar] [CrossRef]
- Fujihara, K.M.; Zhang, B.; Jackson, T.D.; Nijiagel, B.; Ang, C.; Nikolic, I.; Sutton, V.; Trapani, J.; Simpson, K.J. Stojanovski, D.; et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase and synergizes with serine and glycine dietary restriction. Sci. Adv. 2022, in press. [Google Scholar]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Fujihara, K.M.; Corrales Benitez, M.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Cetintas, V.B.; Batada, N.N. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J. Transl. Med. 2020, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, A.M.; Bouska, A.; Arrate, M.P.; Eischen, C.M. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene 2015, 34, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Wohlberedt, K.; Klusmann, I.; Derevyanko, P.K.; Henningsen, K.; Choo, J.; Manzini, V.; Magerhans, A.; Giansanti, C.; Eischen, C.M.; Jochemsen, A.G.; et al. Mdm4 supports DNA replication in a p53-independent fashion. Oncogene 2020, 39, 4828–4843. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Piersigilli, A.; Gou, J.; Ogunwobi, O.; Bargonetti, J. Context-dependent roles of MDMX (MDM4) and MDM2 in breast cancer proliferation and circulating tumor cells. Breast Cancer Res. 2019, 21, 5. [Google Scholar] [CrossRef]
- Xiong, S.; Pant, V.; Zhang, Y.; Aryal, N.K.; You, M.J.; Kusewitt, D.; Lozano, G. The p53 inhibitor Mdm4 cooperates with multiple genetic lesions in tumourigenesis. J. Pathol. 2017, 241, 501–510. [Google Scholar] [CrossRef]
- Uchida, C.; Miwa, S.; Isobe, T.; Kitagawa, K.; Hattori, T.; Oda, T.; Yasuda, H.; Kitagawa, M. Effects of MdmX on Mdm2-mediated downregulation of pRB. FEBS Lett. 2006, 580, 1753–1758. [Google Scholar] [CrossRef]
- Simeckova, S.; Kahounova, Z.; Fedr, R.; Remsik, J.; Slabakova, E.; Suchankova, T.; Prochazkova, J.; Bouchal, J.; Kharaishvili, G.; Kral, M.; et al. High Skp2 expression is associated with a mesenchymal phenotype and increased tumorigenic potential of prostate cancer cells. Sci. Rep. 2019, 9, 5695. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, D.D.; Wu, Y.P.; Su, D.; Zhou, T.Y.; Gai, R.H.; Fu, Y.Y.; Zheng, L.; He, Q.J.; Zhu, H.; et al. MDM2 promotes epithelial-mesenchymal transition and metastasis of ovarian cancer SKOV3 cells. Br. J. Cancer 2017, 117, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Wu, J.; Lu, G.; Wang, X. MDM4 alternative splicing and implication in MDM4 targeted cancer therapies. Am. J. Cancer Res. 2021, 11, 5864–5880. [Google Scholar]
- Pardieu, B.; Pasanisi, J.; Ling, F.; Dal Bello, R.; Penneroux, J.; Su, A.; Joudinaud, R.; Chat, L.; Wu, H.C.; Duchmann, M.; et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 2022, 36, 1585–1595. [Google Scholar] [CrossRef]
- Ceder, S.; Eriksson, S.E.; Cheteh, E.H.; Dawar, S.; Corrales Benitez, M.; Bykov, V.J.N.; Fujihara, K.M.; Grandin, M.; Li, X.; Ramm, S.; et al. A thiol-bound drug reservoir enhances APR-246-induced mutant p53 tumor cell death. EMBO Mol. Med. 2021, 13, e10852. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejía-Hernández, J.O.; Raghu, D.; Caramia, F.; Clemons, N.; Fujihara, K.; Riseborough, T.; Teunisse, A.; Jochemsen, A.G.; Abrahmsén, L.; Blandino, G.; et al. Targeting MDM4 as a Novel Therapeutic Approach in Prostate Cancer Independent of p53 Status. Cancers 2022, 14, 3947. https://doi.org/10.3390/cancers14163947
Mejía-Hernández JO, Raghu D, Caramia F, Clemons N, Fujihara K, Riseborough T, Teunisse A, Jochemsen AG, Abrahmsén L, Blandino G, et al. Targeting MDM4 as a Novel Therapeutic Approach in Prostate Cancer Independent of p53 Status. Cancers. 2022; 14(16):3947. https://doi.org/10.3390/cancers14163947
Chicago/Turabian StyleMejía-Hernández, Javier Octavio, Dinesh Raghu, Franco Caramia, Nicholas Clemons, Kenji Fujihara, Thomas Riseborough, Amina Teunisse, Aart G. Jochemsen, Lars Abrahmsén, Giovanni Blandino, and et al. 2022. "Targeting MDM4 as a Novel Therapeutic Approach in Prostate Cancer Independent of p53 Status" Cancers 14, no. 16: 3947. https://doi.org/10.3390/cancers14163947
APA StyleMejía-Hernández, J. O., Raghu, D., Caramia, F., Clemons, N., Fujihara, K., Riseborough, T., Teunisse, A., Jochemsen, A. G., Abrahmsén, L., Blandino, G., Russo, A., Gamell, C., Fox, S. B., Mitchell, C., Takano, E. A., Byrne, D., Miranda, P. J., Saleh, R., Thorne, H., ... Haupt, S. (2022). Targeting MDM4 as a Novel Therapeutic Approach in Prostate Cancer Independent of p53 Status. Cancers, 14(16), 3947. https://doi.org/10.3390/cancers14163947