
DNA Damage Regulates the Functions of the RNA Binding Protein Sam68 through ATM-Dependent Phosphorylation

 , , , and
, , , and 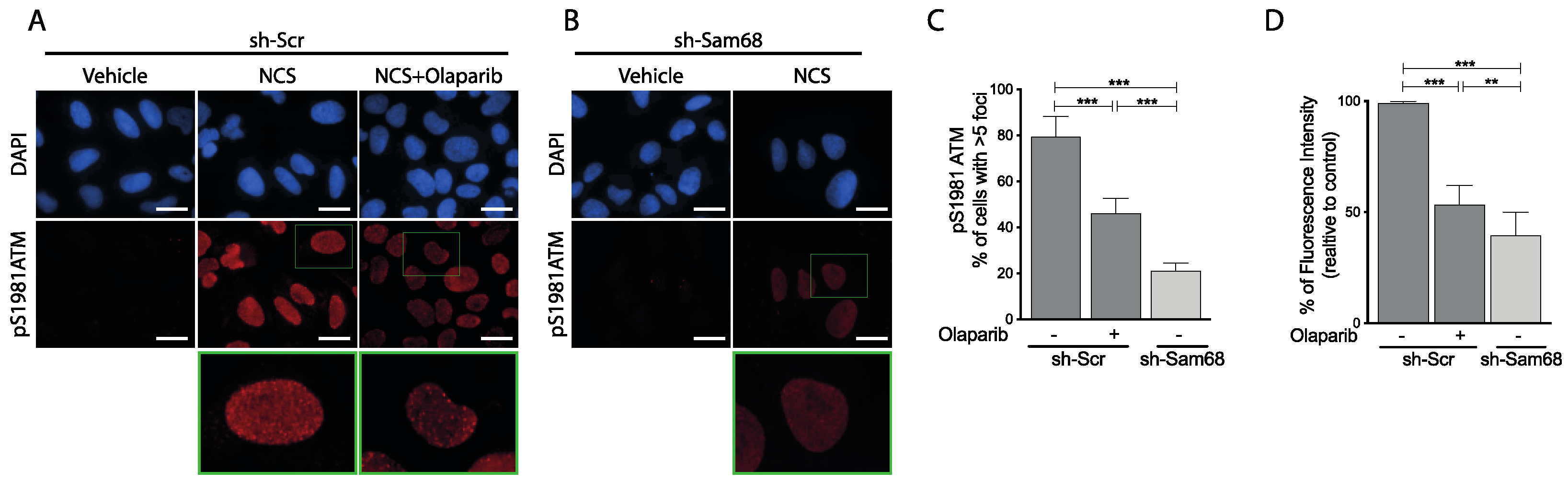{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Antibodies and Other Reagents
2.3. Cell Culture and Transfections
2.4. Immunoblotting and Immunoprecipitation
2.5. PolyA Pulldown Assay
2.6. Survival Assay
2.7. RT-PCR and qPCR Analyses
2.8. Immunofluorescence Analysis
2.9. Quantification and Statistical Analysis
3. Results
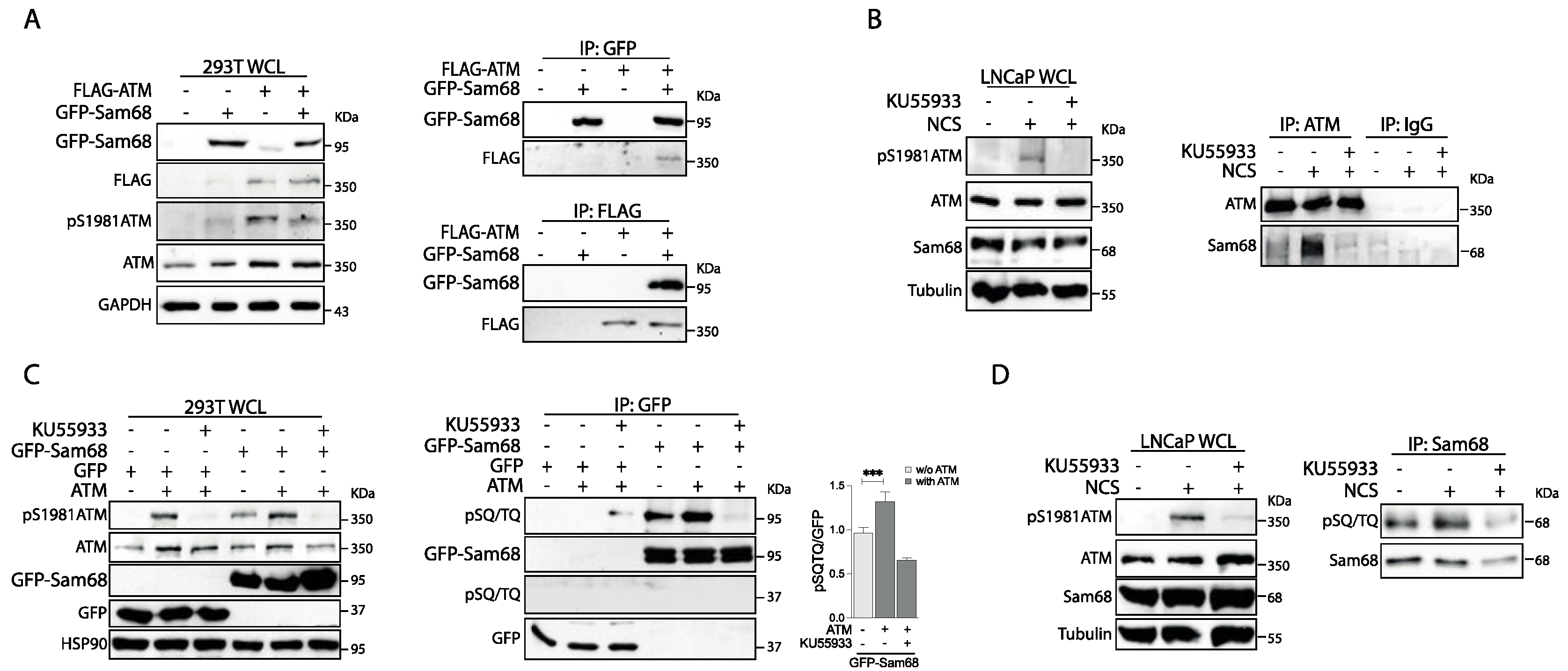
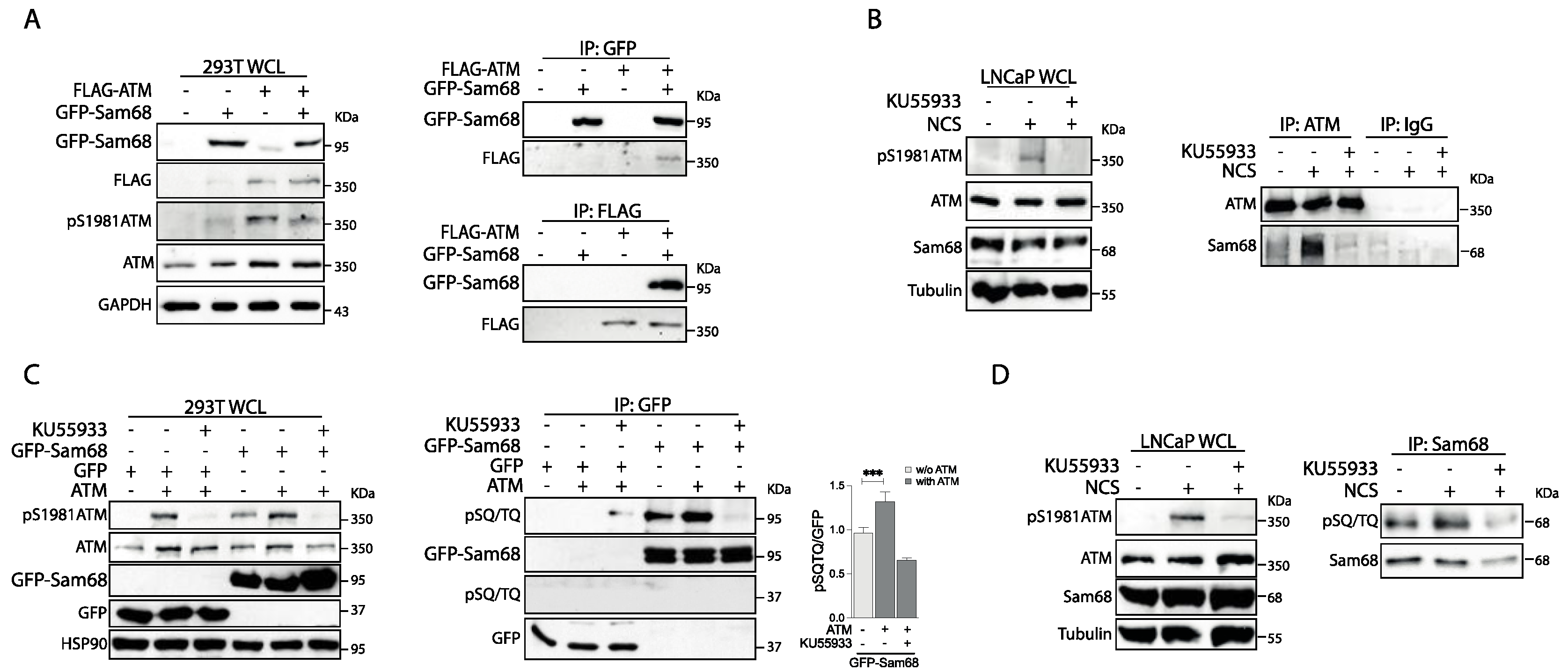
3.1. Sam68 Is a Substrate of the ATM Kinase in Prostate Cancer Cells
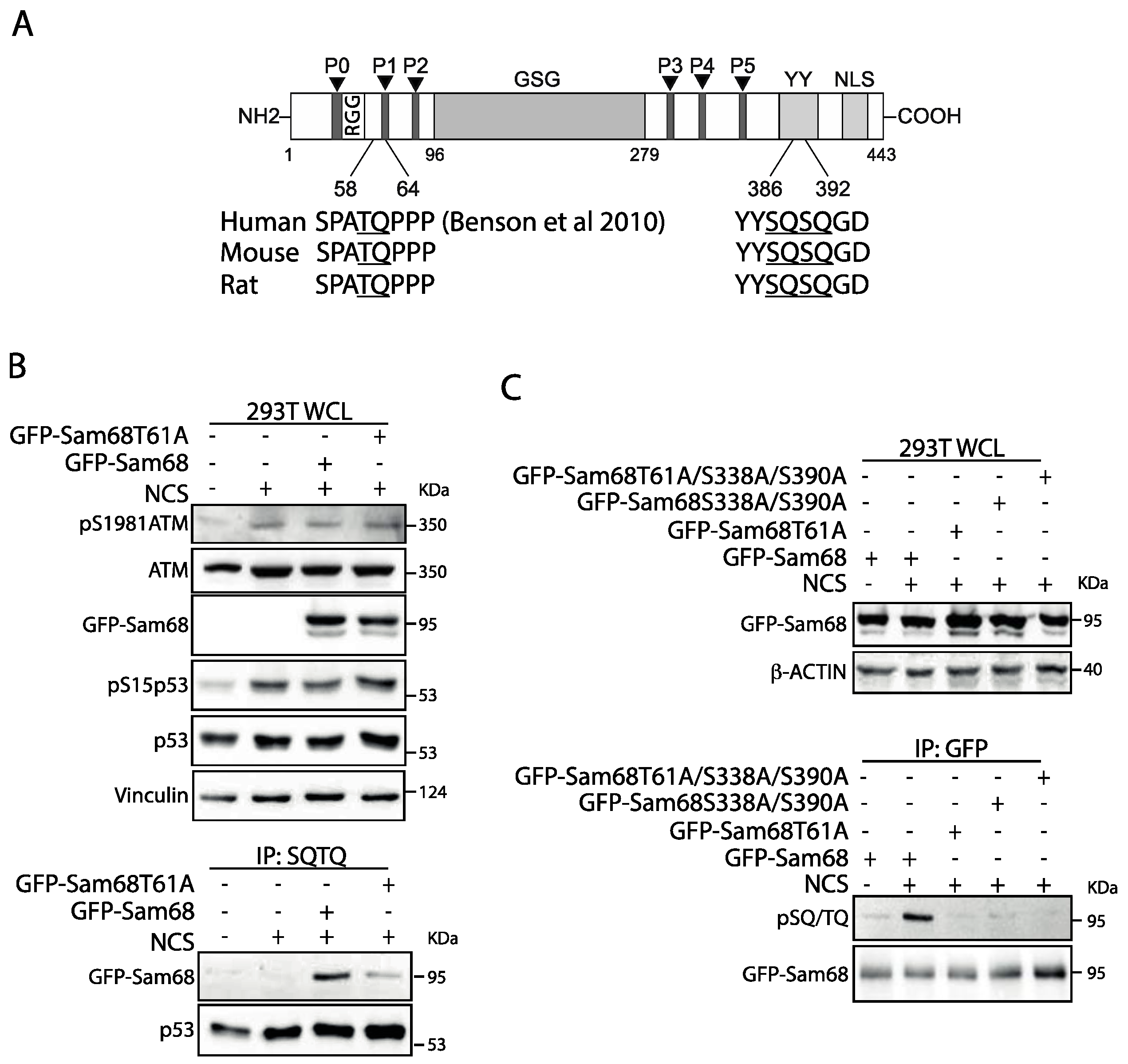
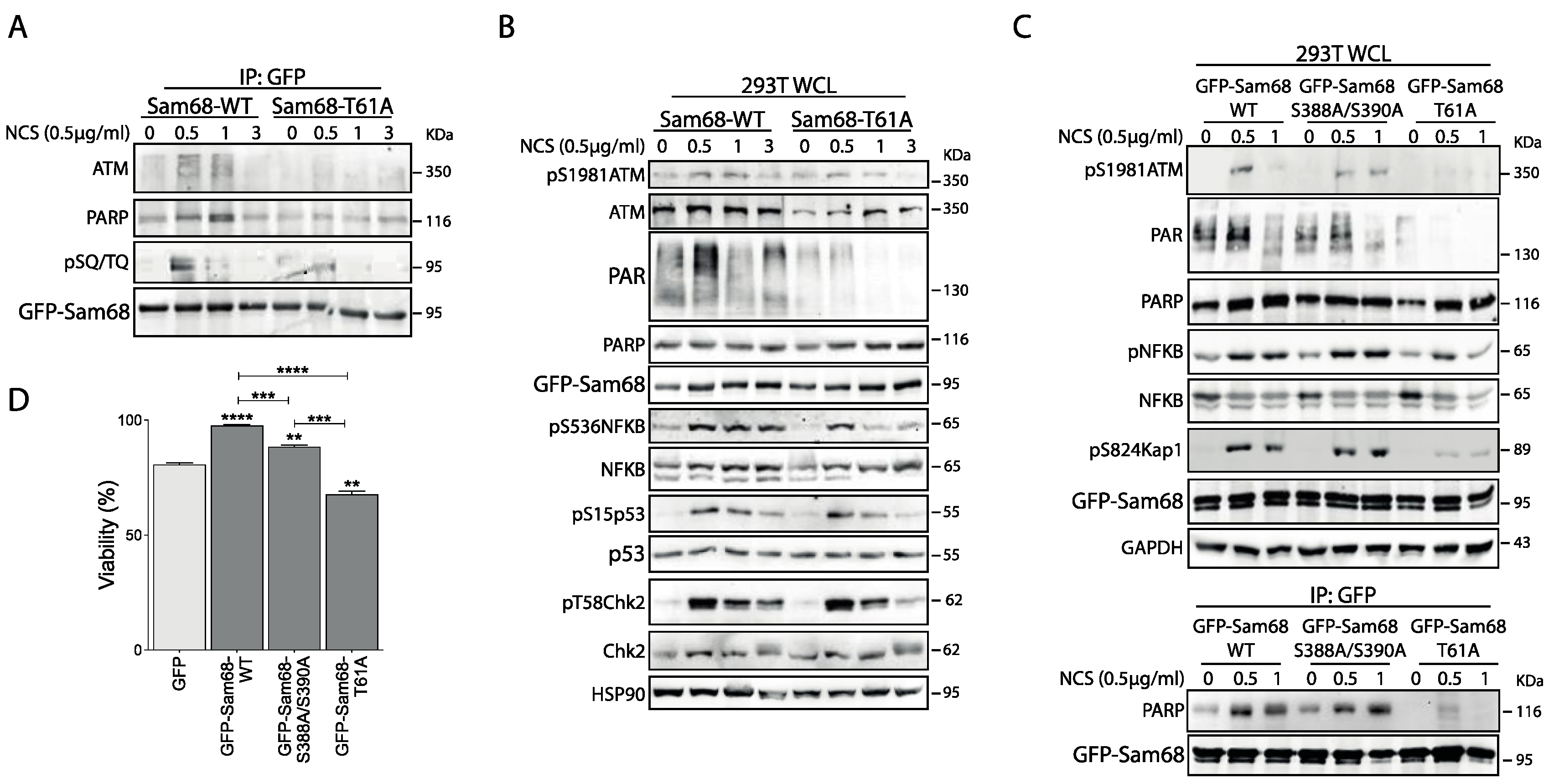
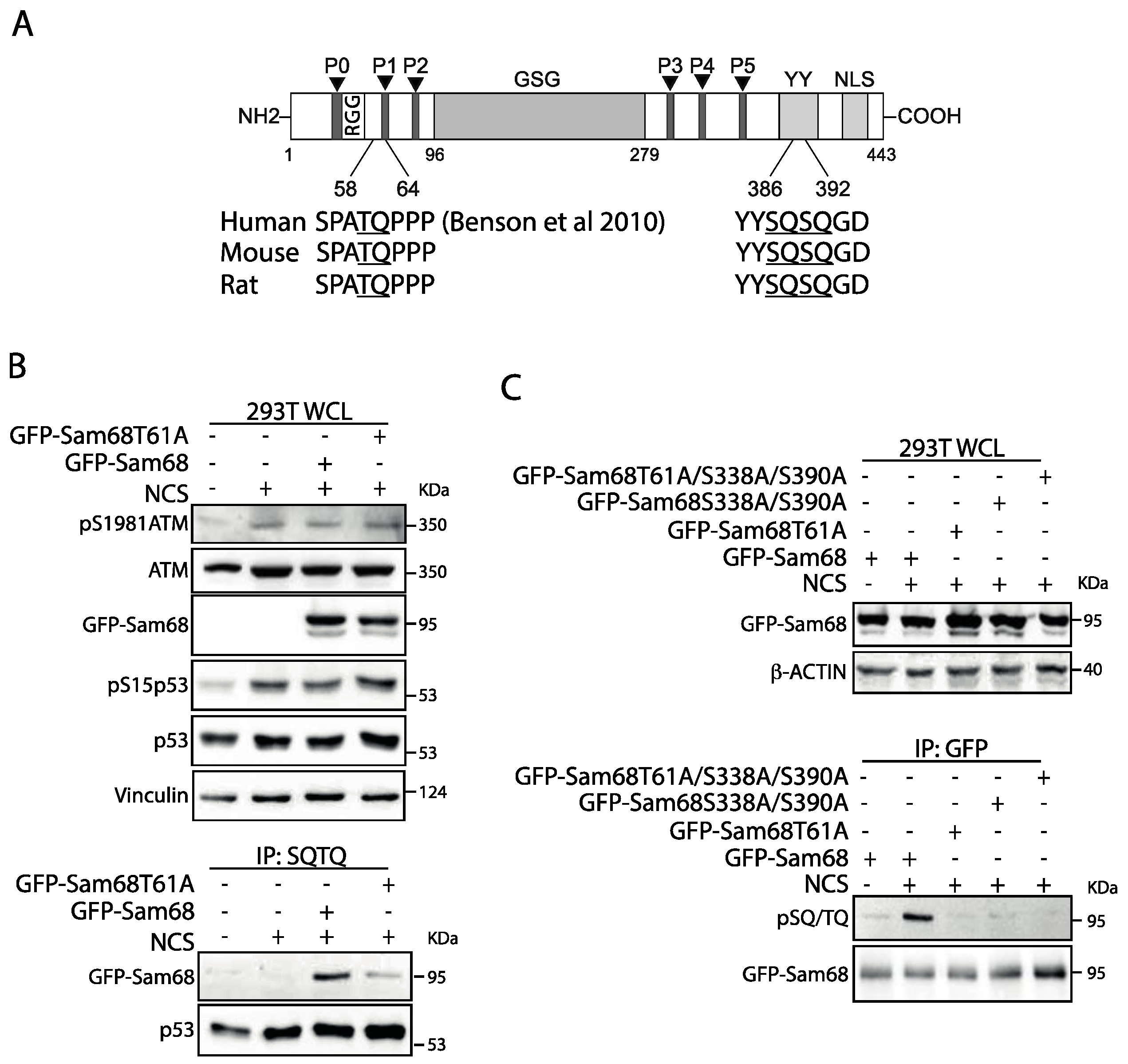
3.2. Threonine 61 in Sam68 Is a Specific Target of ATM Kinase Activity
3.3. ATM Promotes the Interaction of Sam68 with PARP1 through Threonine 61 Phosphorylation
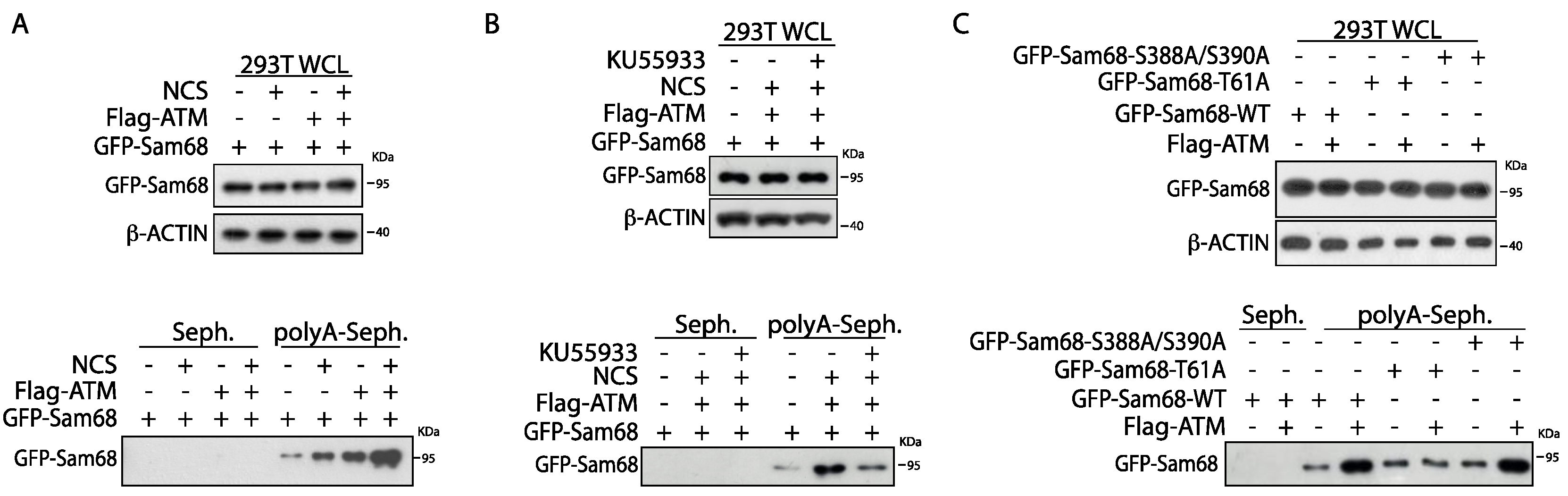
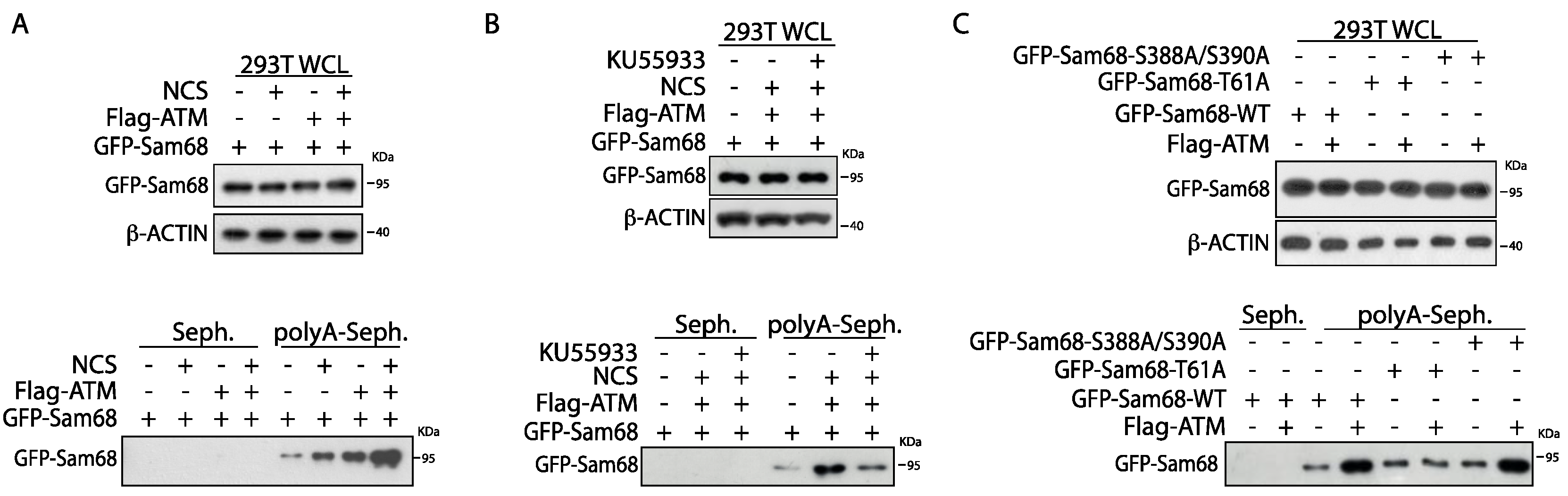
3.4. ATM Modulates the RNA Binding Affinity of Sam68 upon DDR Induction
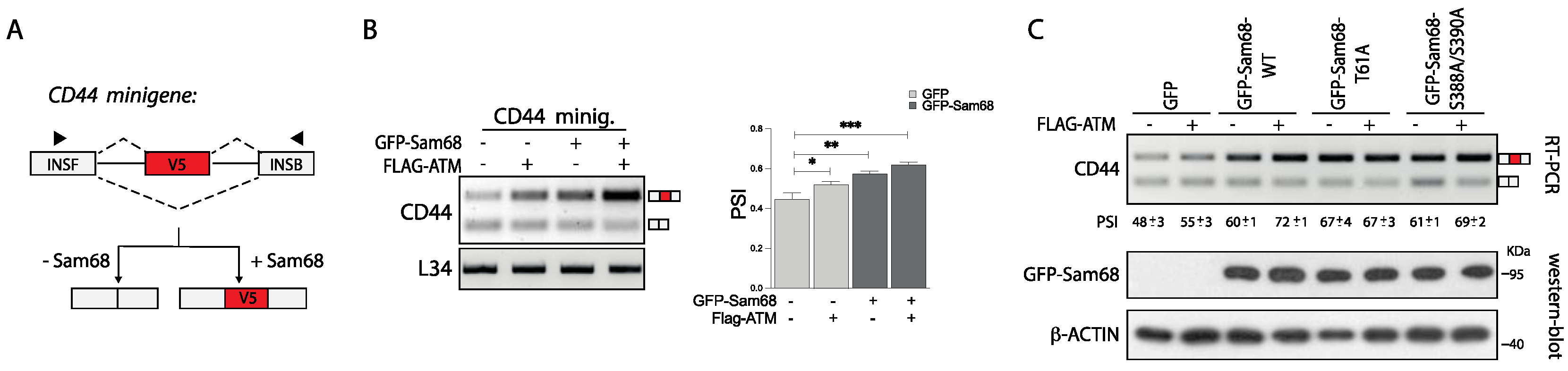
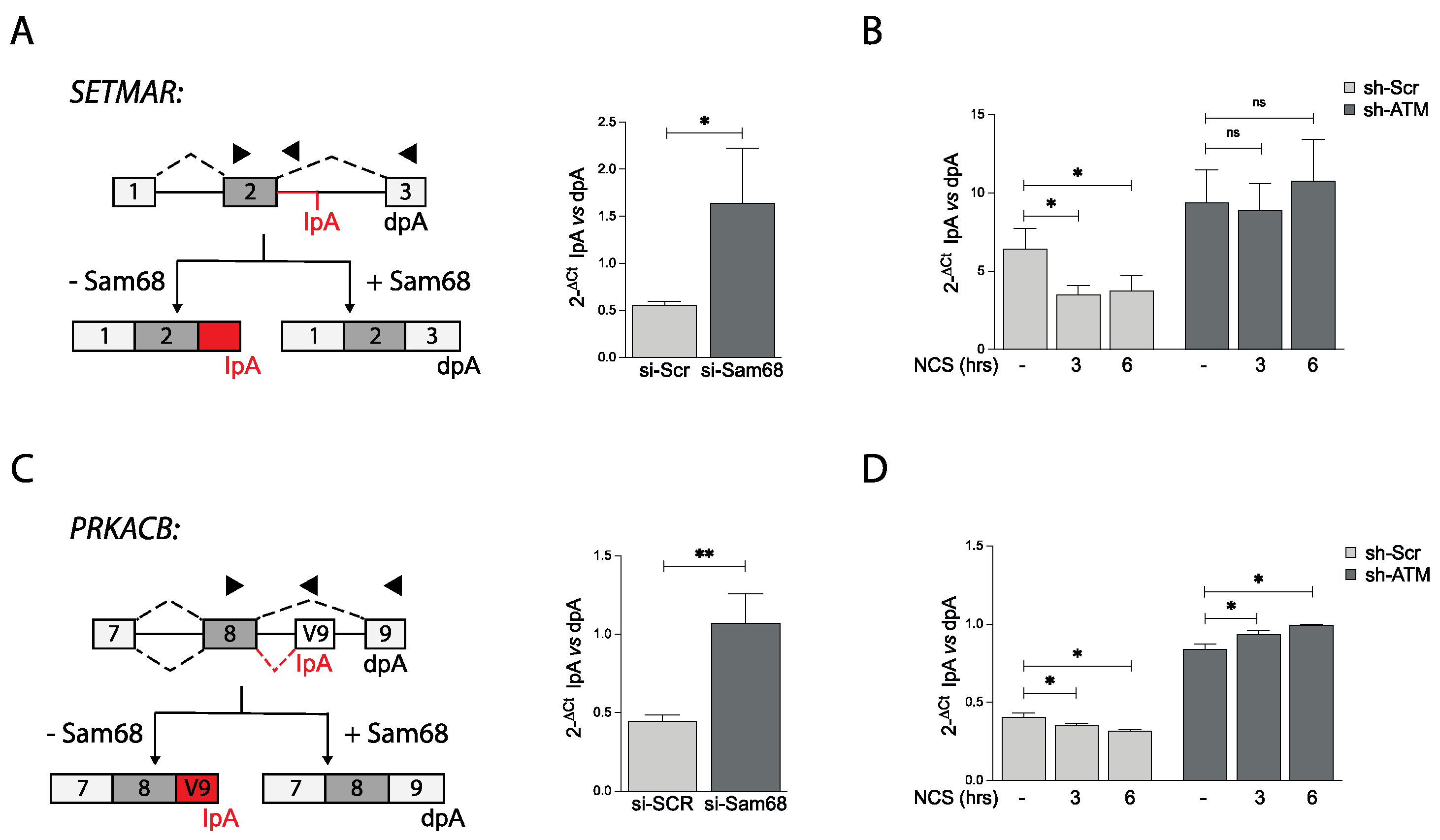
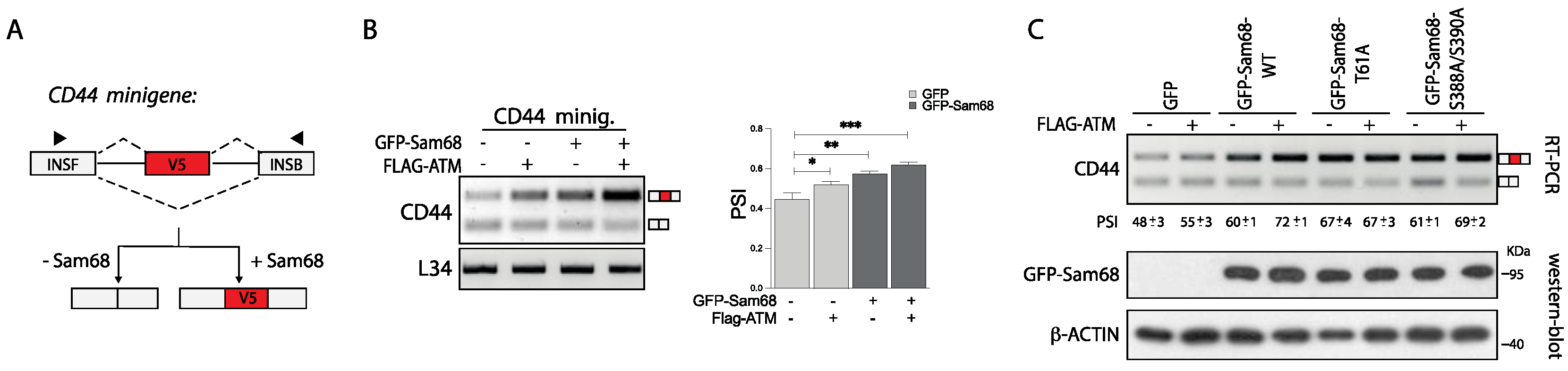
3.5. ATM Promotes the RNA Processing Activity of Sam68
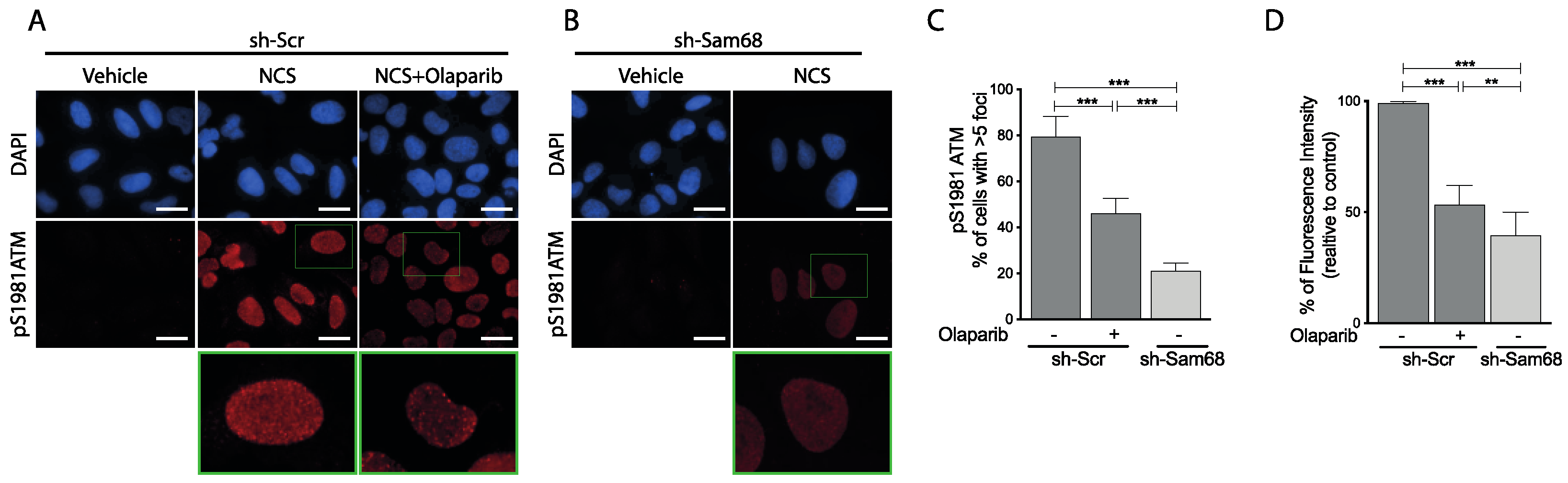
3.6. ATM Modulates Sam68 Function upon DDR Induction in Prostate Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Front. Cell. Dev. Biol. 2020, 8, 599048. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Biamonti, G. Pre-mRNA processing factors meet the DNA damage response. Front. Genet. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: The unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, M.; Sfaxi, R.; Vagner, S. Reciprocal Links between Pre-messenger RNA 3′-End Processing and Genome Stability. Trends Biochem. Sci. 2021, 46, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, R.J.; Marengo, M.S.; Wassarman, D.A. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol. Cell Biol. 2006, 26, 9256–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.J.; Pérez Santangelo, M.S.; Paronetto, M.P.; de la Mata, M.; Pelisch, F.; Boireau, S.; Glover-Cutter, K.; Ben-Dov, C.; Blaustein, M.; Lozano, J.J.; et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009, 137, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, I.; Chakraborty, A.; Saulnier, O.; Benoit-Pilven, C.; Vacher, S.; Labiod, D.; Lam, E.W.F.; Bièche, I.; Delattre, O.; Pouzoulet, F.; et al. ZRANB2 and SYF2-mediated splicing programs converging on ECT2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020, 48, 2676–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, Y.W.; Cattoglio, C.; Tjian, R. The intertwined roles of transcription and repair proteins. Mol. Cell 2013, 52, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Nickless, A.; Cheruiyot, A.; Flanagan, K.C.; Piwnica-Worms, D.; Stewart, S.A.; You, Z. p38 MAPK inhibits nonsense-mediated RNA decay in response to persistent DNA damage in noncycling cells. J. Biol. Chem. 2017, 292, 15266–15276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busa, R.; Geremia, R.; Sette, C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010, 38, 3005–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Miñana, B.; Valcárcel, J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.J.; Nieto Moreno, N.; Giono, L.E.; Cambindo Botto, A.E.; Dujardin, G.; Bastianello, G.; Lavore, S.; Torres-Méndez, A.; Menck, C.F.M.; Blencowe, B.J.; et al. Major Roles for Pyrimidine Dimers, Nucleotide Excision Repair, and ATR in the Alternative Splicing Response to UV Irradiation. Cell Rep. 2017, 18, 2868–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Shao, Y.; Wang, Q.; Zhai, Y.; Li, X. Genotoxic stress causes the accumulation of DNA-dependent protein kinase catalytic subunit phosphorylated at serine 2056 at nuclear speckles and alters pre-mRNA alternative splicing. FEBS Open Bio 2019, 9, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Bielli, P.; Sette, C. Oncogenic dysregulation of pre-mRNA processing by protein kinases: Challenges and therapeutic opportunities. FEBS J. 2021, 288, 6250–6272. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubbury, S.J.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Wickramasinghe, V.O.; Savill, J.M.; Chavali, S.; Jonsdottir, A.B.; Rajendra, E.; Grüner, T.; Laskey, R.A.; Babu, M.M.; Venkitaraman, A.R. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol. Cell 2013, 51, 737–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfano, L.; Caporaso, A.; Altieri, A.; Dell’Aquila, M.; Landi, C.; Bini, L.; Pentimalli, F.; Giordano, A. Depletion of the RNA binding protein HNRNPD impairs homologous recombination by inhibiting DNA-end resection and inducing R-loop accumulation. Nucleic Acids Res. 2019, 47, 4068–4085. [Google Scholar] [CrossRef] [Green Version]
- Bielli, P.; Busa, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisone, P.; Pradella, D.; Di Matteo, A.; Belloni, E.; Ghigna, C.; Paronetto, M.P. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. Biomed. Res. Int. 2015, 2015, 528954. [Google Scholar] [CrossRef] [Green Version]
- Najib, S.; Martín-Romero, C.; González-Yanes, C.; Sánchez-Margalet, V. Role of Sam68 as an adaptor protein in signal transduction. Cell. Mol. Life Sci. 2005, 62, 36–43. [Google Scholar] [CrossRef]
- La Rosa, P.; Bielli, P.; Compagnucci, C.; Cesari, E.; Volpe, E.; FarioliVecchioli, S.; Sette, C. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating. Elife 2016, 5, e20750. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Messina, V.; Bianchi, E.; Barchi, M.; Vogel, G.; Moretti, C.; Palombi, F.; Stefanini, M.; Geremia, R.; Richard, S.; et al. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J. Cell. Biol. 2009, 185, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Messina, V.; Barchi, M.; Geremia, R.; Richard, S.; Sette, C. Sam68 marks the transcriptionally active stages of spermatogenesis and modulates alternative splicing in male germ cells. Nucleic Acids Res. 2011, 39, 4961–4974. [Google Scholar] [CrossRef] [Green Version]
- Huot, M.; Vogel, G.; Zabarauskas, A.; Ngo, C.T.; Coulombe-Huntington, J.; Majewski, J.; Richard, S. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol. Cell 2012, 46, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Richard, S. Sam68 Regulates S6K1 Alternative Splicing during Adipogenesis. Mol. Cell. Biol. 2015, 35, 1926–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Wang, L.; Li, Y.; Xiong, H.; Wu, J.; Li, J.; Li, M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. J. Pathol. 2010, 222, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.; Gaughan, L.; Dalgliesh, C.; El-Sherif, A.; Robson, C.N.; Leung, H.Y.; Elliott, D.J. The RNA-binding and adaptor protein Sam68 modulates signal-dependent splicing and transcriptional activity of the androgen receptor. J. Pathol. 2008, 215, 67–77. [Google Scholar] [CrossRef]
- Li, N.; Richard, S. Sam68 functions as a transcriptional coactivator of the p53 tumor suppressor. Nucleic Acids Res. 2016, 44, 8726–8741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, Y.D.; Mitchell, R.R.; Risueño, R.M.; Orlando, L.; Tanasijevic, B.; Boyd, A.L.; Aslostovar, L.; Salci, K.R.; Shapovalova, Z.; Russell, J.; et al. Sam68 Allows Selective Targeting of Human Cancer Stem Cells. Cell Chem. Biol. 2017, 24, 833–844.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palombo, R.; Frisone, P.; Fidaleo, M.; Mercatelli, N.; Sette, C.; Paronetto, M.P. The Promoter-Associated Noncoding RNA. Cancer Res. 2019, 79, 3570–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Cappellari, M.; Busa, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010, 70, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Chen, W.; Wang, J.; Qi, L.; Pan, H.; Feng, Z.; Tian, D. SAM68 promotes tumorigenesis in lung adenocarcinoma by regulating metabolic conversion via PKM alternative splicing. Theranostics 2021, 11, 3359–3375. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Fu, K.; Hodgson, A.; Wier, E.M.; Wen, M.G.; Kamenyeva, O.; Xia, X.; Koo, L.Y.; Wan, F. Sam68 Is Required for DNA Damage Responses via Regulating Poly(ADP-ribosyl)ation. PLoS Biol. 2016, 14, e1002543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, K.; Sun, X.; Wier, E.M.; Hodgson, A.; Liu, Y.; Sears, C.L.; Wan, F. Sam68/KHDRBS1 is critical for colon tumorigenesis by regulating genotoxic stress-induced NF-κB activation. eLife 2016, 5, e15018. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Sun, X.; Wier, E.M.; Hodgson, A.; Hobbs, R.P.; Wan, F. Sam68/KHDRBS1-dependent NF-κB activation confers radioprotection to the colon epithelium in γ-irradiated mice. eLife 2016, 5, e21957. [Google Scholar] [CrossRef]
- Busa, R.; Paronetto, M.P.; Farini, D.; Pierantozzi, E.; Botti, F.; Angelini, D.F.; Attisani, F.; Vespasiani, G.; Sette, C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 2007, 26, 4372–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellari, M.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Saarikettu, J.; Silvennoinen, O.; Sette, C. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 2014, 33, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.; Markert, E.; Zhou, Y.; Robson, C.N.; Elliott, D.J.; Lindberg, J.; Leung, H.Y.; Rajan, P. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci. Rep. 2015, 5, 13426. [Google Scholar] [CrossRef]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Richard, S. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 2003, 1653, 73–86. [Google Scholar] [CrossRef]
- Sette, C. Post-translational regulation of star proteins and effects on their biological functions. Adv. Exp. Med. Biol. 2010, 693, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Larocque, D.; Tyner, A.L.; Richard, S. Tyrosine phosphorylation of sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. J. Biol. Chem. 2005, 280, 38639–38647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, K.; Sun, X.; Xia, X.; Hobbs, R.P.; Guo, Y.; Coulombe, P.A.; Wan, F. Sam68 is required for the growth and survival of nonmelanoma skin cancer. Cancer Med. 2019, 8, 6106–6113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Venables, J.P.; Elliott, D.J.; Geremia, R.; Rossi, P.; Sette, C. Tr-kit promotes the formation of a multimolecular complex composed by Fyn, PLCgamma1 and Sam68. Oncogene 2003, 22, 8707–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell. Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Pellegrini, L.; Jolly, A.; Farini, D.; Cesari, E.; Bielli, P.; de la Grange, P.; Sette, C. Functional Interaction between U1snRNP and Sam68 Insures Proper 3′ End Pre-mRNA Processing during Germ Cell Differentiation. Cell Rep. 2019, 26, 2929–2941.e5. [Google Scholar] [CrossRef] [Green Version]
- Tresini, M.; Marteijn, J.A.; Vermeulen, W. Bidirectional coupling of splicing and ATM signaling in response to transcription-blocking DNA damage. RNA Biol. 2016, 13, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.J.; Wang, Y.; Luo, H.; Leng, M.; Zhang, J.; Yang, T.; Besusso, D.; Jung, S.Y.; Qin, J. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 2007, 282, 17330–17334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krietsch, J.; Caron, M.C.; Gagné, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Williamson, E.A.; Nickoloff, J.A.; Hromas, R.A.; Lee, S.H. Metnase Mediates Loading of Exonuclease 1 onto Single Strand Overhang DNA for End Resection at Stalled Replication Forks. J. Biol. Chem. 2017, 292, 1414–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stagni, V.; Orecchia, S.; Mignini, L.; Beji, S.; Antonioni, A.; Caggiano, C.; Barilà, D.; Bielli, P.; Sette, C. DNA Damage Regulates the Functions of the RNA Binding Protein Sam68 through ATM-Dependent Phosphorylation. Cancers 2022, 14, 3847. https://doi.org/10.3390/cancers14163847
Stagni V, Orecchia S, Mignini L, Beji S, Antonioni A, Caggiano C, Barilà D, Bielli P, Sette C. DNA Damage Regulates the Functions of the RNA Binding Protein Sam68 through ATM-Dependent Phosphorylation. Cancers. 2022; 14(16):3847. https://doi.org/10.3390/cancers14163847
Chicago/Turabian StyleStagni, Venturina, Silvia Orecchia, Luca Mignini, Sara Beji, Ambra Antonioni, Cinzia Caggiano, Daniela Barilà, Pamela Bielli, and Claudio Sette. 2022. "DNA Damage Regulates the Functions of the RNA Binding Protein Sam68 through ATM-Dependent Phosphorylation" Cancers 14, no. 16: 3847. https://doi.org/10.3390/cancers14163847
APA StyleStagni, V., Orecchia, S., Mignini, L., Beji, S., Antonioni, A., Caggiano, C., Barilà, D., Bielli, P., & Sette, C. (2022). DNA Damage Regulates the Functions of the RNA Binding Protein Sam68 through ATM-Dependent Phosphorylation. Cancers, 14(16), 3847. https://doi.org/10.3390/cancers14163847