
Pten and p53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
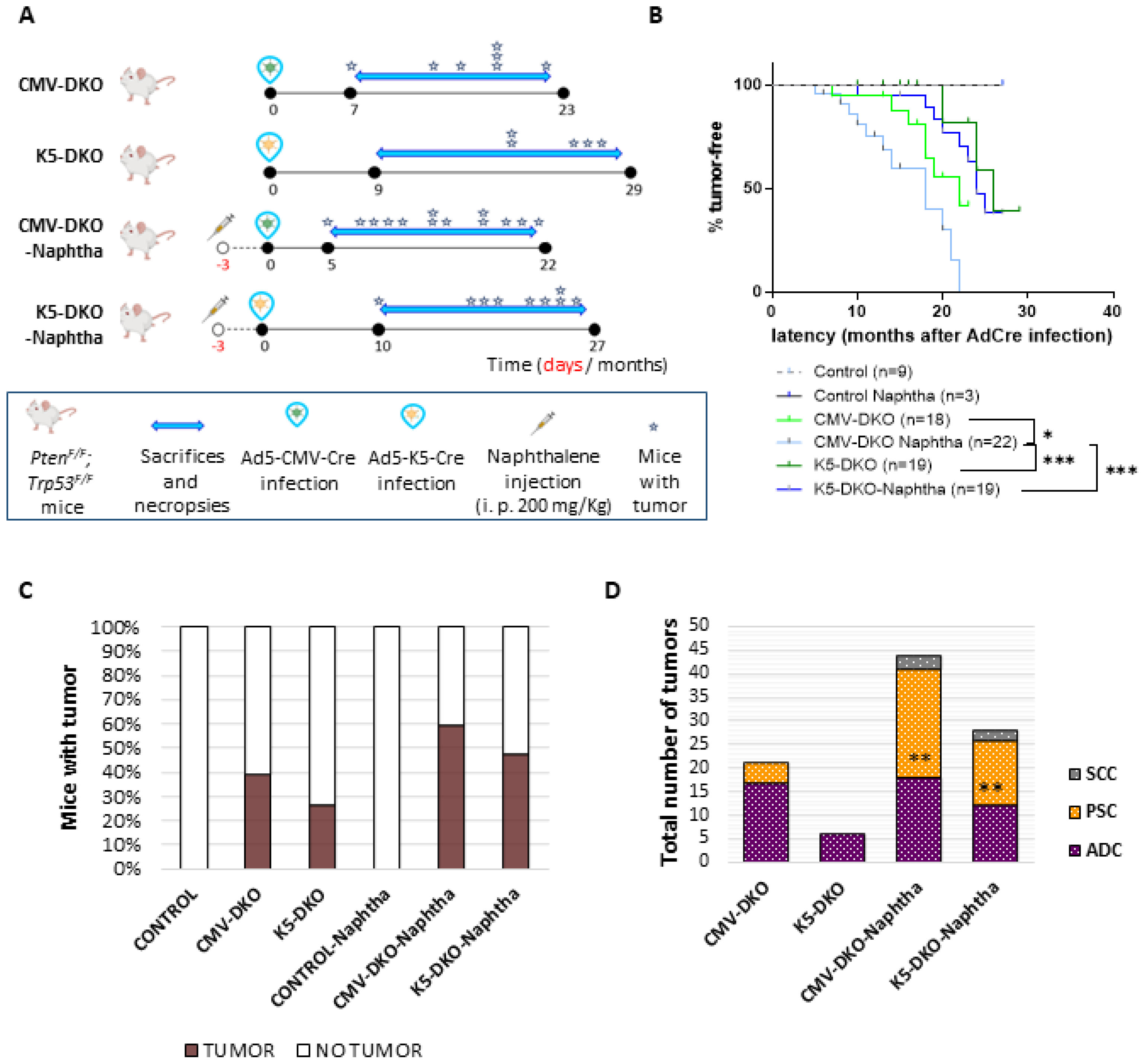
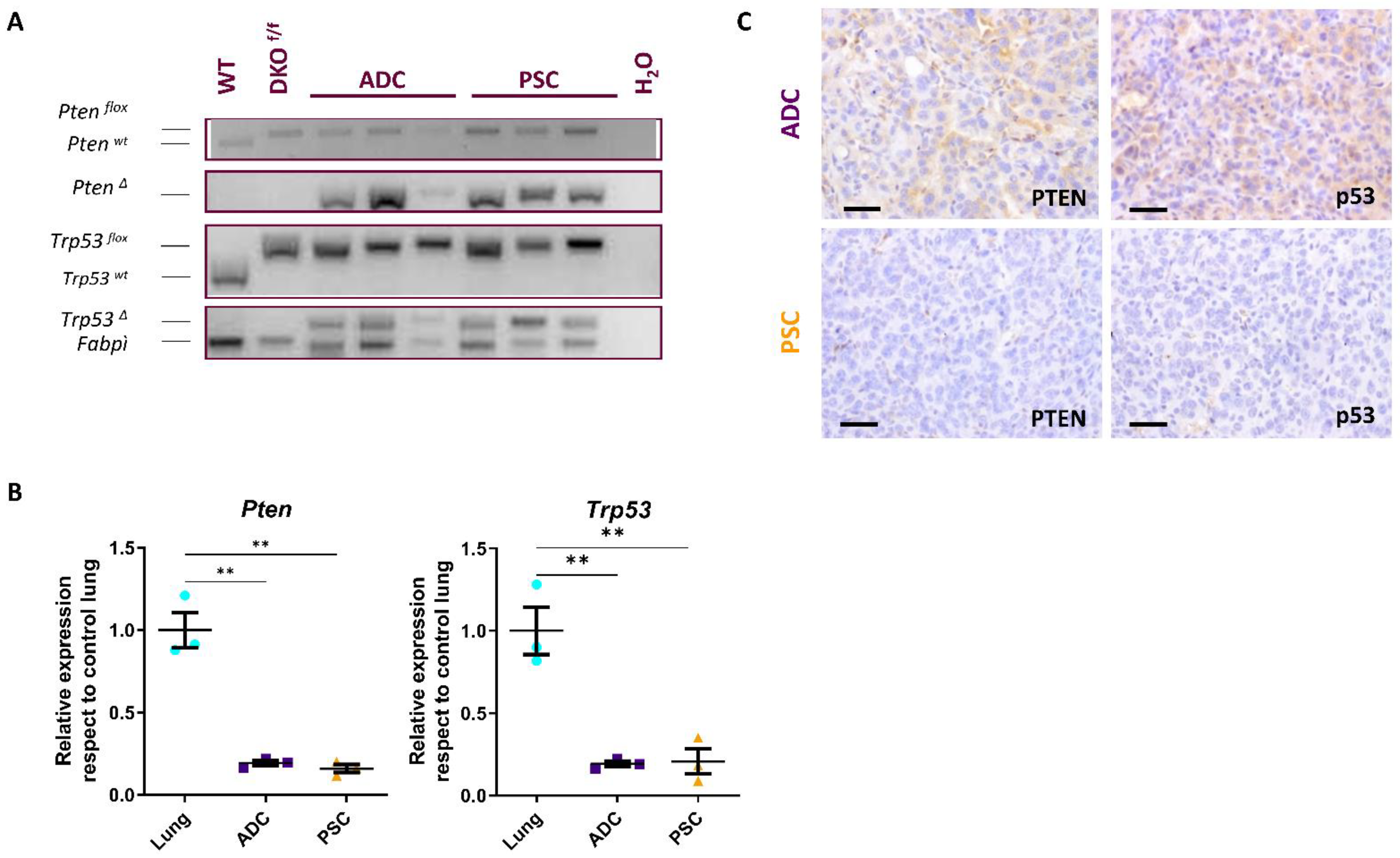
2.1. Combined Deletion of Pten and p53 in Epithelial Cells of Adult Lungs
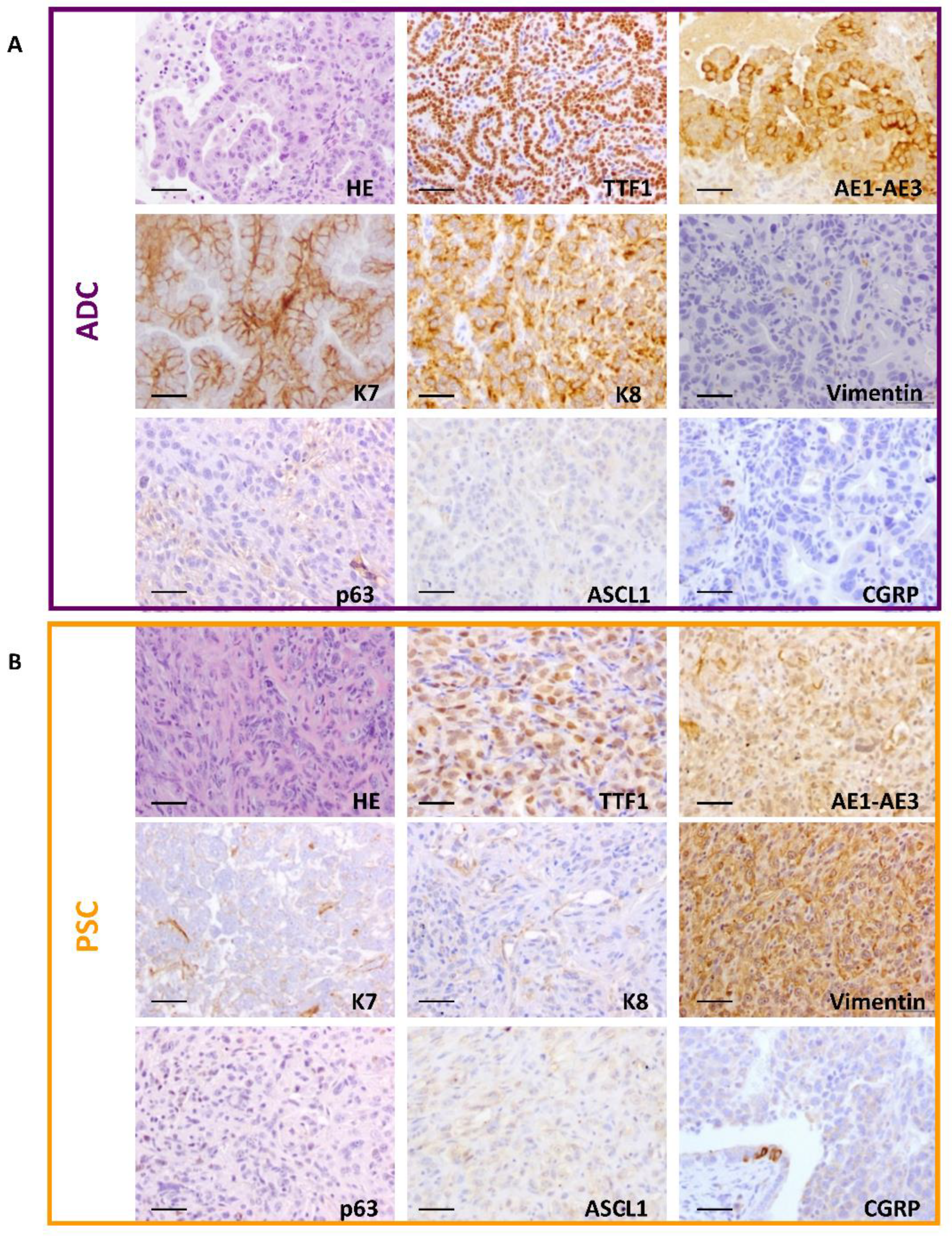
2.2. Development of Adenocarcinoma and Pulmonary Sarcomatoid Carcinoma in Double-Deficient Lungs
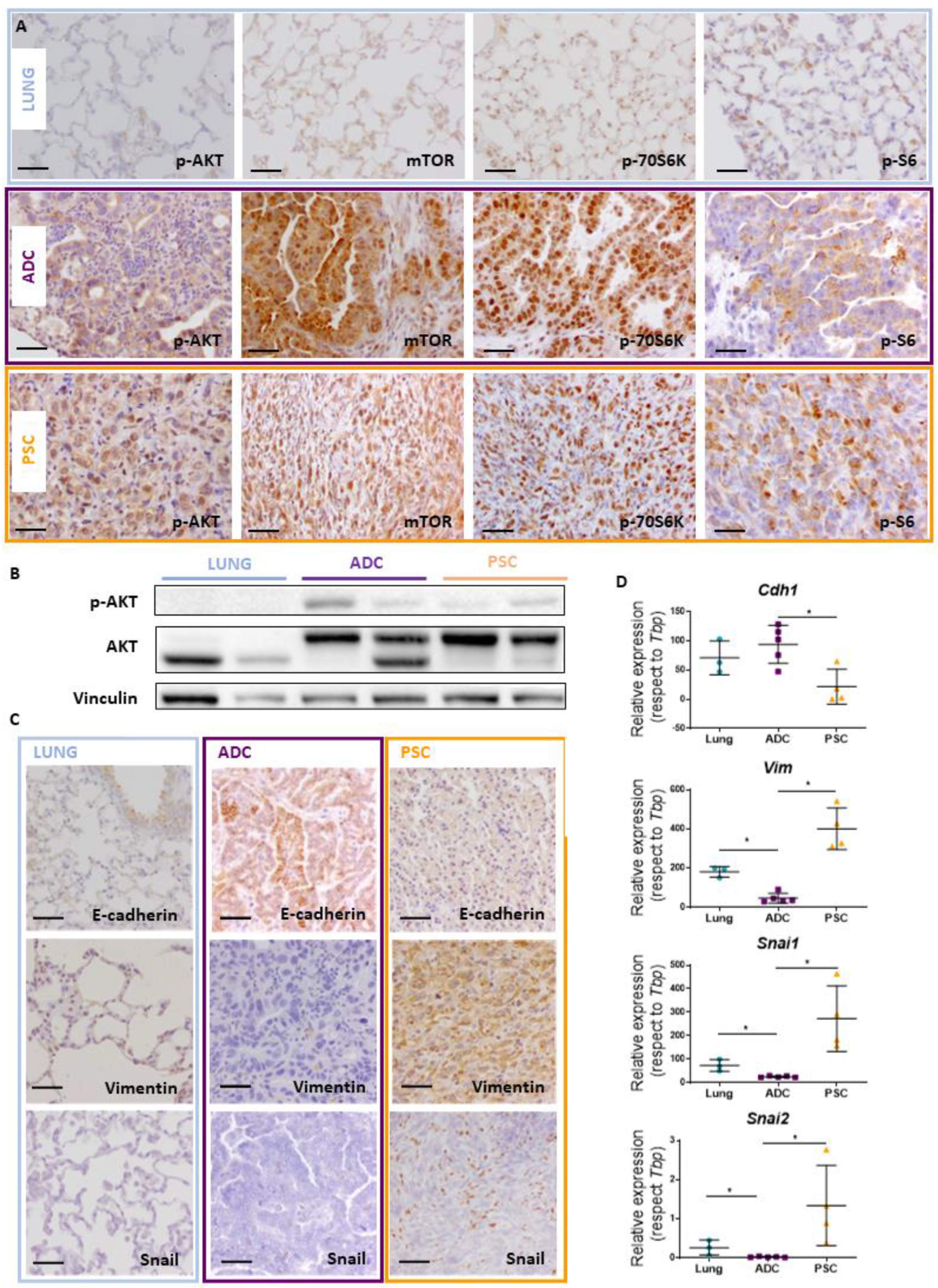
2.3. Akt Pathway Activation and Epithelial–Mesenchymal Transition (EMT) Occur in Pten and Trp53 Double-Deficient Mouse Tumors
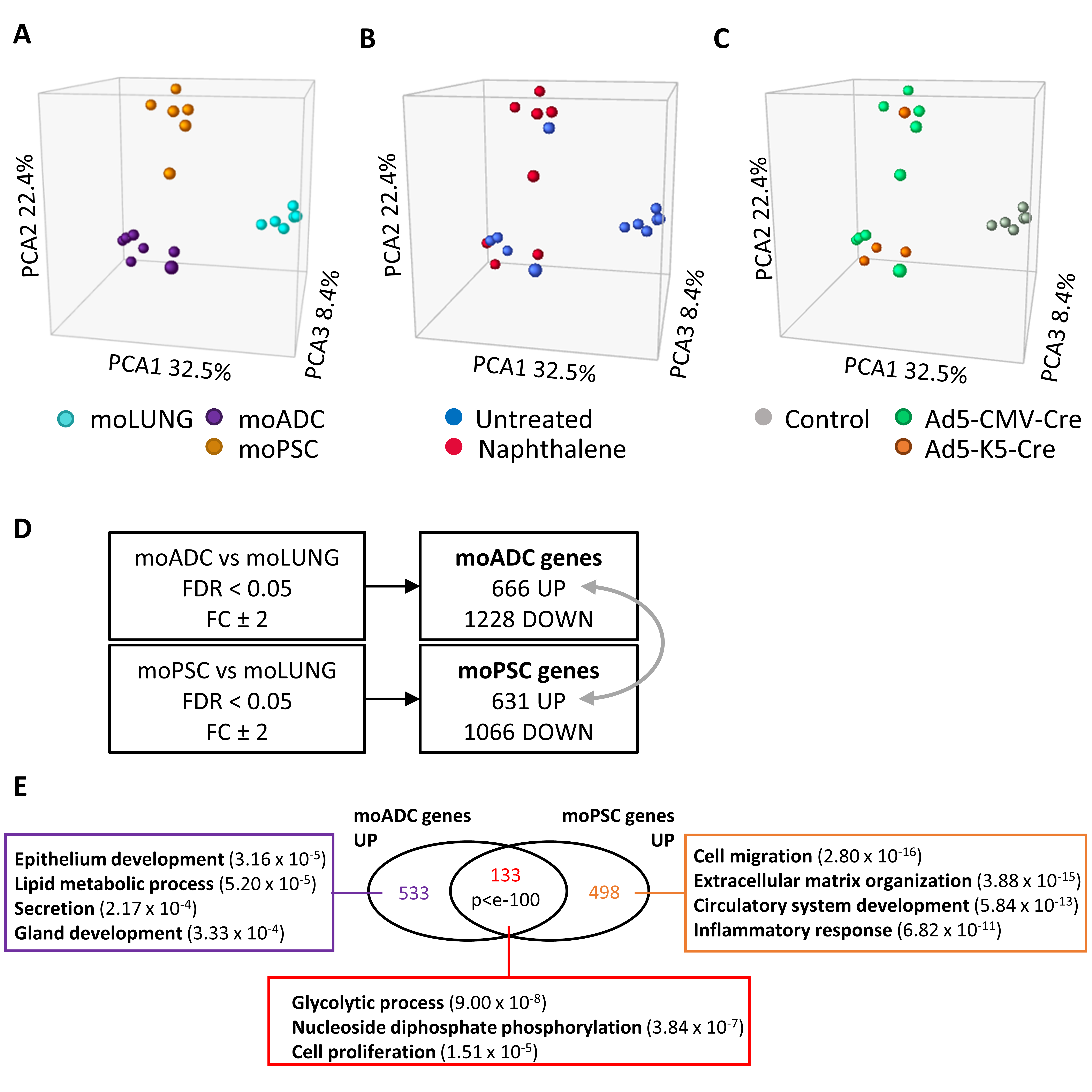
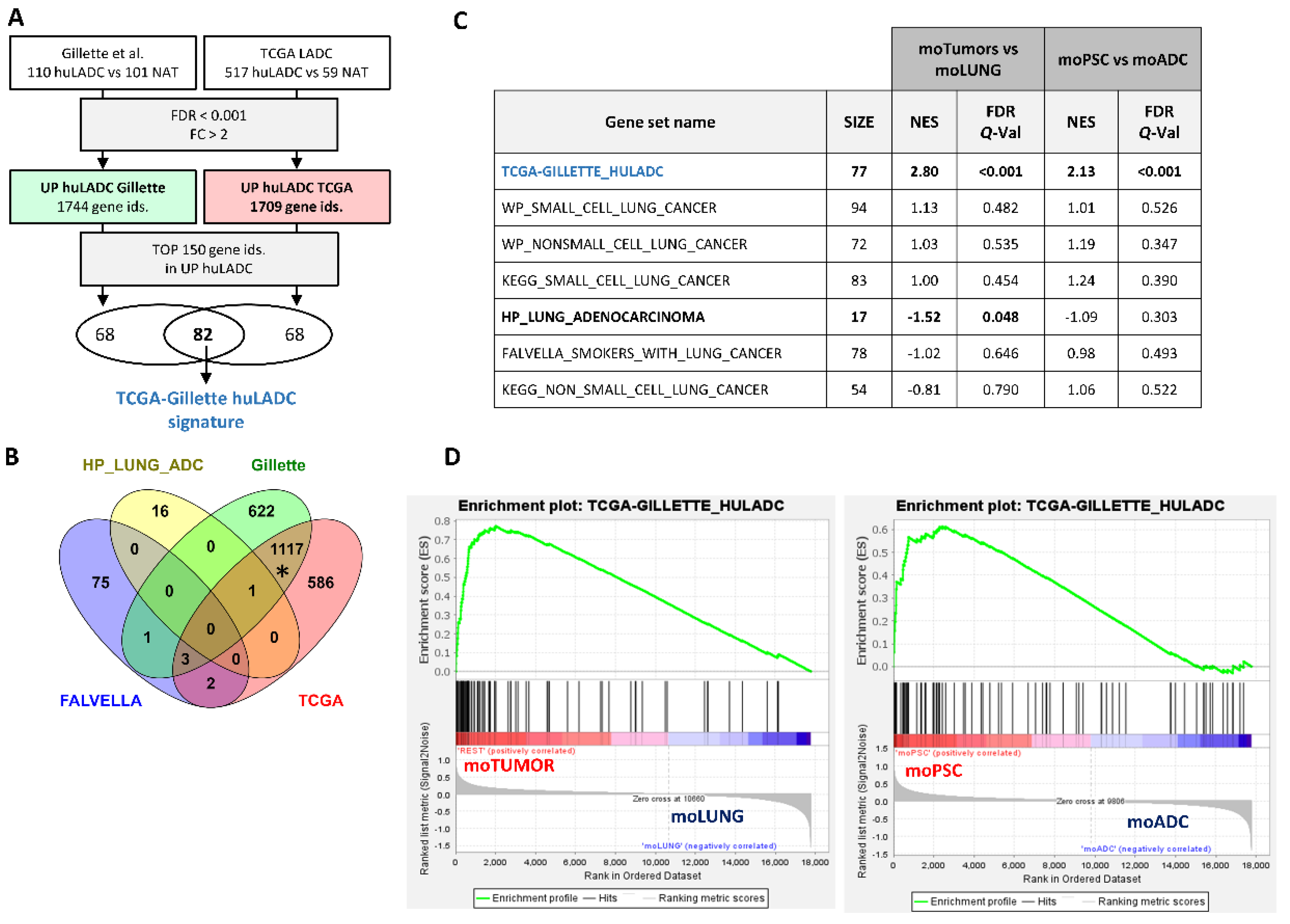
2.4. Transcriptomic Analysis of Double-Deficient Pten and Trp53 Mouse Lung Tumors
3. Discussion
4. Materials and Methods
4.1. Mice and Adenoviral Infections
4.2. Naphthalene Treatment
4.3. Genotyping
4.4. Histology and Immunostaining/Immunohistochemistry
4.5. RNA Extraction and RT-qPCR
4.6. Western Blot Analysis
4.7. Transcriptome Analyses
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Meza, R.; Meernik, C.; Jeon, J.; Cote, M.L. Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS ONE 2015, 10, e0121323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Damotte, D.; Alifano, M. New Therapeutic Strategies for Lung Cancer. Cancers 2021, 13, 1937. [Google Scholar] [CrossRef] [PubMed]
- Weissferdt, A. Pulmonary Sarcomatoid Carcinomas: A Review. Adv. Anat. Pathol. 2018, 25, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.; Girard, N.; Ung, M.; Monnet, I.; Cazes, A.; Bonnette, P.; Duruisseaux, M.; Mazieres, J.; Antoine, M.; Cadranel, J.; et al. Efficacy of first-line chemotherapy in patients with advanced lung sarcomatoid carcinoma. J. Thorac. Oncol. 2013, 8, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Maneenil, K.; Xue, Z.; Liu, M.; Boland, J.; Wu, F.; Stoddard, S.M.; Molina, J.; Yang, P. Sarcomatoid Carcinoma of the Lung: The Mayo Clinic Experience in 127 Patients. Clin. Lung Cancer 2018, 19, e323–e333. [Google Scholar] [CrossRef]
- Liu, X.; Jia, Y.; Stoopler, M.B.; Shen, Y.; Cheng, H.; Chen, J.; Mansukhani, M.; Koul, S.; Halmos, B.; Borczuk, A.C. Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J. Clin. Oncol. 2016, 34, 794–802. [Google Scholar] [CrossRef]
- Schrock, A.B.; Li, S.D.; Frampton, G.M.; Suh, J.; Braun, E.; Mehra, R.; Buck, S.C.; Bufill, J.A.; Peled, N.; Karim, N.A.; et al. Pulmonary Sarcomatoid Carcinomas Commonly Harbor Either Potentially Targetable Genomic Alterations or High Tumor Mutational Burden as Observed by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2017, 12, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Velcheti, V.; Rimm, D.L.; Schalper, K.A. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1). J. Thorac. Oncol. 2013, 8, 803–805. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Xu, J.; Li, L.; Li, R.; Wang, Y.; Tian, Y.; Guo, W.; Wang, Z.; Tan, F.; Ying, J.; et al. Integrated molecular characterization reveals potential therapeutic strategies for pulmonary sarcomatoid carcinoma. Nat. Commun. 2020, 11, 4878. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, M.; Roy, S.; LaFramboise, W.A.; Petrosko, P.; Miller, C.; Incharoen, P.; Dacic, S. KRAS mutation is predictive of outcome in patients with pulmonary sarcomatoid carcinoma. Histopathology 2018, 73, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Aisner, D.L.; Sholl, L.M.; Berry, L.D.; Rossi, M.R.; Chen, H.; Fujimoto, J.; Moreira, A.L.; Ramalingam, S.S.; Villaruz, L.C.; Otterson, G.A.; et al. The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations-The Lung Cancer Mutation Consortium (LCMC2). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1038–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Spoerke, J.M.; O’Brien, C.; Huw, L.; Koeppen, H.; Fridlyand, J.; Brachmann, R.K.; Haverty, P.M.; Pandita, A.; Mohan, S.; Sampath, D.; et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6771–6783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Lorz, C.; Oteo, M.; Santos, M. Neuroendocrine Lung Cancer Mouse Models: An Overview. Cancers 2020, 13, 14. [Google Scholar] [CrossRef]
- Santos, M. New models of large-cell neuroendocrine carcinoma and small-cell lung carcinoma. Mol. Cell. Oncol. 2020, 7, 1702413. [Google Scholar] [CrossRef]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef]
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol. Oncol. 2013, 7, 165–177. [Google Scholar] [CrossRef]
- Hynds, R.E.; Frese, K.K.; Pearce, D.R.; Gronroos, E.; Dive, C.; Swanton, C. Progress towards non-small-cell lung cancer models that represent clinical evolutionary trajectories. Open Biol. 2021, 11, 200247. [Google Scholar] [CrossRef]
- Lazaro, S.; Perez-Crespo, M.; Enguita, A.B.; Hernandez, P.; Martinez-Palacio, J.; Oteo, M.; Sage, J.; Paramio, J.M.; Santos, M. Ablating all three retinoblastoma family members in mouse lung leads to neuroendocrine tumor formation. Oncotarget 2017, 8, 4373–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro, S.; Perez-Crespo, M.; Lorz, C.; Bernardini, A.; Oteo, M.; Enguita, A.B.; Romero, E.; Hernandez, P.; Tomas, L.; Morcillo, M.A.; et al. Differential development of large-cell neuroendocrine or small-cell lung carcinoma upon inactivation of 4 tumor suppressor genes. Proc. Natl. Acad. Sci. USA 2019, 116, 22300–22306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, T.P.; McBride, J.T.; Peake, J.L.; Pinkerton, K.E.; Stripp, B.R. Cell proliferation contributes to PNEC hyperplasia after acute airway injury. Am. J. Physiol. 1997, 272, L486–L493. [Google Scholar] [CrossRef]
- Hong, K.U.; Reynolds, S.D.; Watkins, S.; Fuchs, E.; Stripp, B.R. In Vivo differentiation potential of tracheal basal cells: Evidence for multipotent and unipotent subpopulations. Am. J. Physiol. 2004, 286, L643–L649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- Weissferdt, A.; Kalhor, N.; Correa, A.M.; Moran, C.A. “Sarcomatoid” carcinomas of the lung: A clinicopathological study of 86 cases with a new perspective on tumor classification. Hum. Pathol. 2017, 63, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Terra, S.B.S.P.; Jang, J.S.; Bi, L.; Kipp, B.R.; Jen, J.; Yi, E.S.; Boland, J.M. Molecular characterization of pulmonary sarcomatoid carcinoma: Analysis of 33 cases. Mod. Pathol. 2016, 29, 824–831. [Google Scholar] [CrossRef]
- Baldovini, C.; Rossi, G.; Ciarrocchi, A. Approaches to Tumor Classification in Pulmonary Sarcomatoid Carcinoma. Lung Cancer 2019, 10, 131–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, D.; Liu, H.; Chen, J. Pulmonary sarcomatoid carcinoma: Progress, treatment and expectations. Ther. Adv. Med. Oncol. 2020, 12, 1758835920950207. [Google Scholar] [CrossRef]
- Thomas, V.T.; Hinson, S.; Konduri, K. Epithelial-mesenchymal transition in pulmonary carcinosarcoma: Case report and literature review. Ther. Adv. Med. Oncol. 2012, 4, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Manzotti, G.; Torricelli, F.; Benedetta, D.; Lococo, F.; Sancisi, V.; Rossi, G.; Piana, S.; Ciarrocchi, A. An Epithelial-to-Mesenchymal Transcriptional Switch Triggers Evolution of Pulmonary Sarcomatoid Carcinoma (PSC) and Identifies Dasatinib as New Therapeutic Option. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2348–2360. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, Y.; Jiang, J.; Liu, Z.; Mu, C. Transformation of Two Cases of Lung Adenocarcinoma into Pulmonary Sarcomatoid Carcinoma following Treatment. Can. Respir. J. 2021, 2021, 6661772. [Google Scholar] [CrossRef]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225.e35. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists With Venn’s Diagrams, 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 27 May 2021).
- Frullanti, E.; Colombo, F.; Falvella, F.S.; Galvan, A.; Noci, S.; De Cecco, L.; Incarbone, M.; Alloisio, M.; Santambrogio, L.; Nosotti, M.; et al. Association of lung adenocarcinoma clinical stage with gene expression pattern in noninvolved lung tissue. Int. J. Cancer 2012, 131, E643–E648. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Song, J.Y.; Kwon, M.C.; Proost, N.; Zevenhoven, J.; Berns, A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 4952–4957. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, F.; Xu, C.; Chen, X.; Hou, X.; Li, Q.; Li, P.; Xie, Z.; Liu, Y.; Chang, L.; et al. Genomic origin and intratumor heterogeneity revealed by sequencing on carcinomatous and sarcomatous components of pulmonary sarcomatoid carcinoma. Oncogene 2021, 40, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Kasai, T.; Hashimoto, H.; Iwata, Y.; Manabe, H. Sarcomatoid carcinoma of the lung: A clinicopathologic study of 37 cases. Cancer 1999, 86, 608–616. [Google Scholar] [CrossRef]
- Blaukovitsch, M.; Halbwedl, I.; Kothmaier, H.; Gogg-Kammerer, M.; Popper, H.H. Sarcomatoid carcinomas of the lung--are these histogenetically heterogeneous tumors? Virchows Arch. 2006, 449, 455–461. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Dooley, A.L.; Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 2009, 4, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Ferone, G.; Song, J.Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.P.; Proost, N.; Gargiulo, G.; Berns, A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 2016, 30, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Semenova, E.A.; Nagel, R.; Berns, A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015, 29, 1447–1462. [Google Scholar] [CrossRef] [Green Version]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Iwanaga, K.; Yang, Y.; Raso, M.G.; Ma, L.; Hanna, A.E.; Thilaganathan, N.; Moghaddam, S.; Evans, C.M.; Li, H.; Cai, W.W.; et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 2008, 68, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.L.; Olive, K.P.; Tuveson, D.A.; Bronson, R.; Crowley, D.; Brown, M.; Jacks, T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005, 65, 10280–10288. [Google Scholar] [CrossRef] [Green Version]
- Vieira, T.; Antoine, M.; Ruppert, A.M.; Fallet, V.; Duruisseaux, M.; Giroux Leprieur, E.; Poulot, V.; Rabbe, N.; Sclick, L.; Beau-Faller, M.; et al. Blood vessel invasion is a major feature and a factor of poor prognosis in sarcomatoid carcinoma of the lung. Lung Cancer 2014, 85, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN is a potent suppressor of small cell lung cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Liu, Y.N.; Pierce, R.; Abou-Kheir, W.; Casey, O.; Seng, V.; Camacho, D.; Simpson, R.M.; Kelly, K. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am. J. Pathol. 2011, 179, 422–435. [Google Scholar] [CrossRef]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M.; Laddha, S.V.; Castillo-Martin, M.; Sun, Y.; Cordon-Cardo, C.; Chan, C.S.; Levine, A.J. Involvement of tumor suppressors PTEN and p53 in the formation of multiple subtypes of liposarcoma. Cell Death Differ. 2015, 22, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Dudgeon, C.; Chan, C.; Kang, W.; Sun, Y.; Emerson, R.; Robins, H.; Levine, A.J. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev. 2014, 28, 2613–2620. [Google Scholar] [CrossRef] [Green Version]
- Puzio-Kuter, A.M.; Castillo-Martin, M.; Kinkade, C.W.; Wang, X.; Shen, T.H.; Matos, T.; Shen, M.M.; Cordon-Cardo, C.; Abate-Shen, C. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009, 23, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Guijarro, M.V.; Dahiya, S.; Danielson, L.S.; Segura, M.F.; Vales-Lara, F.M.; Menendez, S.; Popiolek, D.; Mittal, K.; Wei, J.J.; Zavadil, J.; et al. Dual Pten/Tp53 suppression promotes sarcoma progression by activating Notch signaling. Am. J. Pathol. 2013, 182, 2015–2027. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Rong, L.; Owczarek, T.B.; Bernardo, M.D.; Shoulson, R.L.; Chua, C.W.; Kim, J.Y.; Lankarani, A.; Chakrapani, P.; Syed, T.; et al. Novel Mouse Models of Bladder Cancer Identify a Prognostic Signature Associated with Risk of Disease Progression. Cancer Res. 2021, 81, 5161–5175. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.A.; Hudson, A.L.; Clarke, S.J.; Molloy, M.P.; Howell, V.M. From mice to men: GEMMs as trial patients for new NSCLC therapies. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 27, pp. 118–127. [Google Scholar] [CrossRef]
- Segovia, C.; San Jose-Eneriz, E.; Munera-Maravilla, E.; Martinez-Fernandez, M.; Garate, L.; Miranda, E.; Vilas-Zornoza, A.; Lodewijk, I.; Rubio, C.; Segrelles, C.; et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat. Med. 2019, 25, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cruz, A.B.; Santos, M.; Garcia-Escudero, R.; Moral, M.; Segrelles, C.; Lorz, C.; Saiz, C.; Buitrago-Perez, A.; Costa, C.; Paramio, J.M. Spontaneous tumor formation in Trp53-deficient epidermis mediated by chromosomal instability and inflammation. Anticancer Res. 2009, 29, 3035–3042. [Google Scholar]
- Bornachea, O.; Santos, M.; Martinez-Cruz, A.B.; Garcia-Escudero, R.; Duenas, M.; Costa, C.; Segrelles, C.; Lorz, C.; Buitrago, A.; Saiz-Ladera, C.; et al. EMT and induction of miR-21 mediate metastasis development in Trp53-deficient tumours. Sci. Rep. 2012, 2, 434. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lázaro, S.; Lorz, C.; Enguita, A.B.; Seller, I.; Paramio, J.M.; Santos, M. Pten and p53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma. Cancers 2022, 14, 3671. https://doi.org/10.3390/cancers14153671
Lázaro S, Lorz C, Enguita AB, Seller I, Paramio JM, Santos M. Pten and p53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma. Cancers. 2022; 14(15):3671. https://doi.org/10.3390/cancers14153671
Chicago/Turabian StyleLázaro, Sara, Corina Lorz, Ana Belén Enguita, Iván Seller, Jesús M. Paramio, and Mirentxu Santos. 2022. "Pten and p53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma" Cancers 14, no. 15: 3671. https://doi.org/10.3390/cancers14153671
APA StyleLázaro, S., Lorz, C., Enguita, A. B., Seller, I., Paramio, J. M., & Santos, M. (2022). Pten and p53 Loss in the Mouse Lung Causes Adenocarcinoma and Sarcomatoid Carcinoma. Cancers, 14(15), 3671. https://doi.org/10.3390/cancers14153671