
Molecular Profiles of Serum-Derived Extracellular Vesicles in High-Grade Serous Ovarian Cancer

 , , ,
, , ,
Abstract
Simple Summary
Abstract
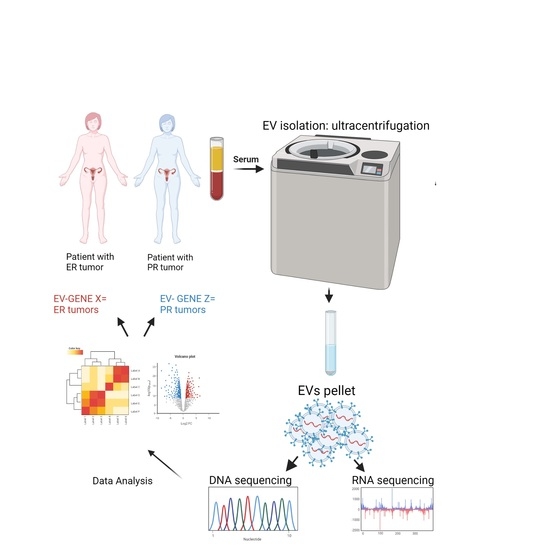
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Sample Collection
2.2. Medium and Large EV Isolation from Serum
2.3. Medium and Large EV Characterization and Quantification
2.4. Nucleic Acid Isolation from m/lEVs
2.5. Whole-Genome Sequencing
2.6. Mutation Calling and CNV Identification
2.7. RNA Sequencing
2.8. Statistical Analysis
3. Results
3.1. Patient Groups and Sequencing Data
3.2. Somatic Alterations in EV-DNA
3.3. Comparison between EV-DNA and Tumor DNA
3.4. Transcriptomic Expression Profiles in EV-RNA
3.5. Differential Expression and Enriched Pathways between the R0 and NACT Groups
3.6. Differential Expression and Enriched Pathways between the NACT-ER and NACT-PR
Groups
3.7. Comparison between EV-RNA and Tumor RNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 925. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.D.; Nick, A.M.; Coleman, R.L.; Westin, S.N.; Ramirez, P.T.; Soliman, P.T.; Fellman, B.; Meyer, L.A.; Schmeler, K.M.; Lu, K.H.; et al. Laparoscopic Surgical Algorithm to Triage the Timing of Tumor Reductive Surgery in Advanced Ovarian Cancer. Obstet. Gynecol. 2018, 132, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Nick, A.M.; Coleman, R.L.; Ramirez, P.T.; Sood, A.K. A framework for a personalized surgical approach to ovarian cancer. Na.t Rev. Clin. Oncol. 2015, 12, 239–245. [Google Scholar] [CrossRef]
- Lee, S.; Zhao, L.; Little, L.D.; Westin, S.N.; Jazarei, A.A.; Fleming, N.D.; Zhang, J.; Futreal, P.A.; Sood, A.K. Distinct T cell receptor repertoire diversity of clinically defined high-grade serous ovarian cancer treatment subgroups. iScience 2021, 24, 102053. [Google Scholar] [CrossRef]
- Lee, S.; Zhao, L.; Rojas, C.; Bateman, N.W.; Yao, H.; Lara, O.D.; Celestino, J.; Morgan, M.B.; Nguyen, T.V.; Conrads, K.A.; et al. Molecular Analysis of Clinically Defined Subsets of High-Grade Serous Ovarian Cancer. Cell Rep. 2020, 31, 107502. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Abbosh, C.; Swanton, C.; Birkbak, N.J. Clonal haematopoiesis: A source of biological noise in cell-free DNA analyses. Ann. Oncol. 2019, 30, 358–359. [Google Scholar] [CrossRef]
- Ma, S.; McGuire, M.H.; Mangala, L.S.; Lee, S.; Stur, E.; Hu, W.; Bayraktar, E.; Villar-Prados, A.; Ivan, C.; Wu, S.Y.; et al. Gain-of-function p53 protein transferred via small extracellular vesicles promotes conversion of fibroblasts to a cancer-associated phenotype. Cell Rep. 2021, 34, 108726. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [CrossRef]
- Genomes Project, C.; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Lai, D.; Ha, G.; Shah, S. HMMcopy: Copy Number Prediction With correction for GC and Mappability Bias for HTS Data. R package Version 1.38.0. 2022. Available online: https://rdrr.io/bioc/HMMcopy/ (accessed on 18 November 2021).
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad.Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [PubMed]
- Allenson, K.; Castillo, J.; San Lucas, F.A.S.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 2017, 28, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romero, N.; Carrion-Navarro, J.; Esteban-Rubio, S.; Lazaro-Ibanez, E.; Peris-Celda, M.; Alonso, M.M.; Guzman-De-Villoria, J.; Fernandez-Carballal, C.; de Mendivil, A.O.; Garcia-Duque, S.; et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget 2017, 8, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108. [Google Scholar] [CrossRef]
- Del Re, M.; Marconcini, R.; Pasquini, G.; Rofi, E.; Vivaldi, C.; Bloise, F.; Restante, G.; Arrigoni, E.; Caparello, C.; Bianco, M.G.; et al. PD-L1 mRNA expression in plasma-derived exosomes is associated with response to anti-PD-1 antibodies in melanoma and NSCLC. Brit. J. Cancer 2018, 118, 820–824. [Google Scholar] [CrossRef]
- Shao, H.; Chung, J.; Balaj, L.; Charest, A.; Bigner, D.D.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Weissleder, R.; Lee, H. Protein Typing of Circulating Microvesicles Allows Real-time Monitoring of Glioblastoma Therapy. Eur. J. Cancer 2012, 48, 137–138. [Google Scholar] [CrossRef][Green Version]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Stucci, L.S.; Ascierto, P.A.; Capone, M.; Madonna, G.; Lopalco, P.; Silvestris, F. Serum exosomes as predictors of clinical response to ipilimumab in metastatic melanoma. Oncoimmunology 2018, 7, e1387706. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; Mcgee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Bioph. Res. Co. 2014, 451, 295–301. [Google Scholar] [CrossRef]
- Harada, T.; Yamamoto, H.; Kishida, S.; Kishida, M.; Awada, C.; Takao, T.; Kikuchi, A. Wnt5b-associated exosomes promote cancer cell migration and proliferation. Cancer Sci. 2017, 108, 42–52. [Google Scholar] [CrossRef]
- McBride, J.D.; Rodriguez-Menocal, L.; Guzman, W.; Candanedo, A.; Garcia-Contreras, M.; Badiavas, E.V. Bone Marrow Mesenchymal Stem Cell-Derived CD63(+) Exosomes Transport Wnt3a Exteriorly and Enhance Dermal Fibroblast Proliferation, Migration, and Angiogenesis In Vitro. Stem. Cells Dev. 2017, 26, 1384–1398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, X.D.; Zhang, X.; Sun, Y.X.; Yan, Y.M.; Shi, H.; Zhu, Y.H.; Wu, L.J.; Pan, Z.J.; Zhu, W.; et al. Human Umbilical Cord Mesenchymal Stem Cell Exosomes Enhance Angiogenesis Through the Wnt4/beta-Catenin Pathway. Stem. Cell Transl. Med. 2015, 4, 513–522. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Corvigno, S.; Ma, S.; Celestino, J.; Fleming, N.D.; Hajek, R.A.; Lankenau Ahumada, A.; Jennings, N.B.; Thompson, E.J.; Tang, H.; et al. Molecular Profiles of Serum-Derived Extracellular Vesicles in High-Grade Serous Ovarian Cancer. Cancers 2022, 14, 3589. https://doi.org/10.3390/cancers14153589
Zhao L, Corvigno S, Ma S, Celestino J, Fleming ND, Hajek RA, Lankenau Ahumada A, Jennings NB, Thompson EJ, Tang H, et al. Molecular Profiles of Serum-Derived Extracellular Vesicles in High-Grade Serous Ovarian Cancer. Cancers. 2022; 14(15):3589. https://doi.org/10.3390/cancers14153589
Chicago/Turabian StyleZhao, Li, Sara Corvigno, Shaolin Ma, Joseph Celestino, Nicole D. Fleming, Richard A. Hajek, Adrian Lankenau Ahumada, Nicholas B. Jennings, Erika J. Thompson, Hongli Tang, and et al. 2022. "Molecular Profiles of Serum-Derived Extracellular Vesicles in High-Grade Serous Ovarian Cancer" Cancers 14, no. 15: 3589. https://doi.org/10.3390/cancers14153589
APA StyleZhao, L., Corvigno, S., Ma, S., Celestino, J., Fleming, N. D., Hajek, R. A., Lankenau Ahumada, A., Jennings, N. B., Thompson, E. J., Tang, H., Westin, S. N., Jazaeri, A. A., Zhang, J., Futreal, P. A., Sood, A. K., & Lee, S. (2022). Molecular Profiles of Serum-Derived Extracellular Vesicles in High-Grade Serous Ovarian Cancer. Cancers, 14(15), 3589. https://doi.org/10.3390/cancers14153589