
Mutational Signature and Integrative Genomic Analysis of Human Papillomavirus-Associated Penile Squamous Cell Carcinomas from Latin American Patients

 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. HPV Genotyping
2.3. Whole-Exome Sequencing
2.4. Data Processing
2.5. Variant Calling
2.6. Variant Filtering
2.7. Copy Number Variation by Sequencing (CNV-seq)
2.8. UTR-Perturbing Variants
2.9. Integrated Genomic and Molecular Characterization of HPV-Associated PSCC
2.10. Pathway Analysis
3. Results
3.1. High Frequency of HPV Infection in PSCC
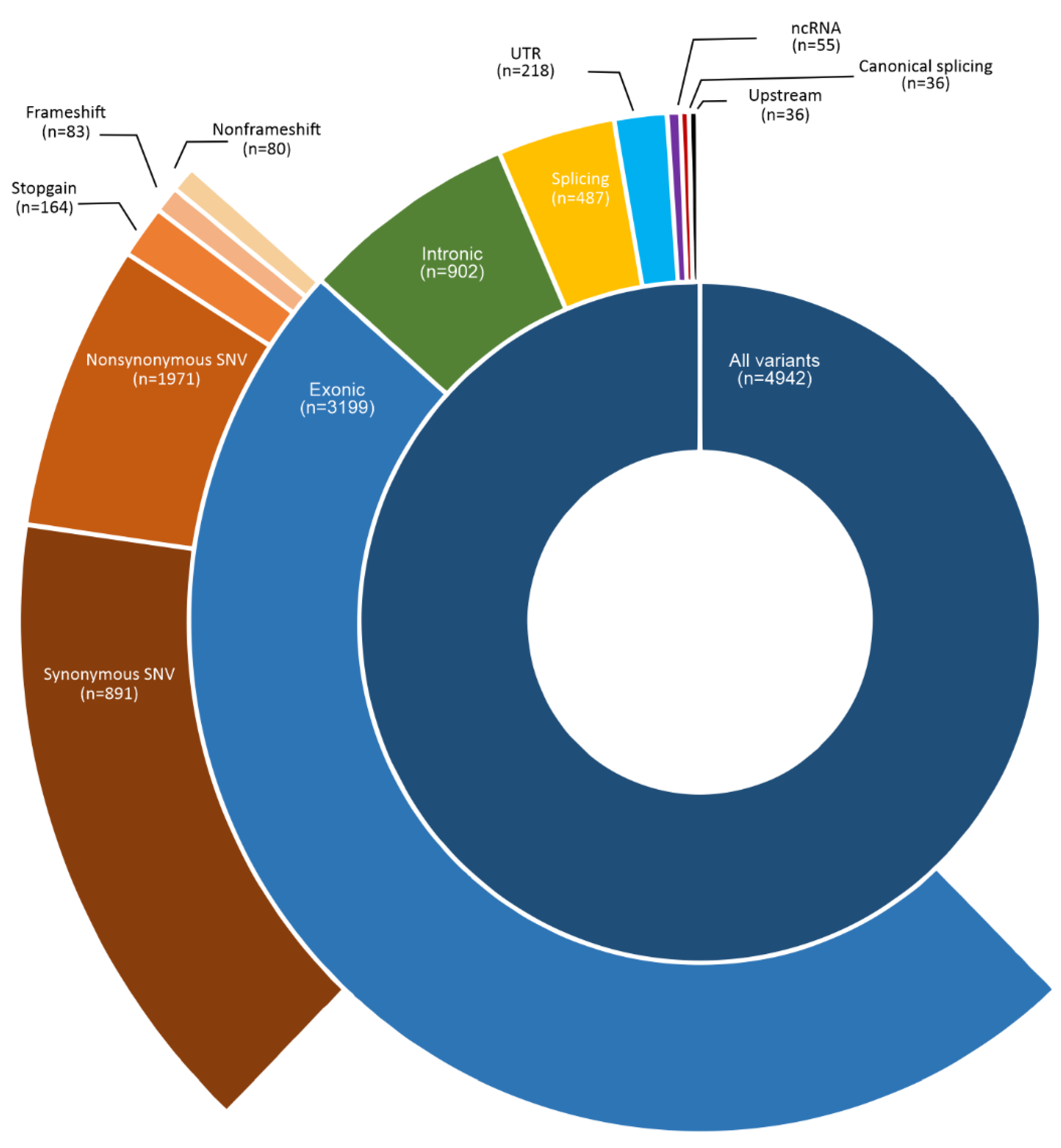
3.2. Somatic Variants in HPV-Associated PSCC in Latin Americans
3.3. Novel Genes Altered Exclusively in PSCC from Latin Americans
3.4. Copy Number Variation in HPV-Associated PSCC
3.5. UTRs Variants and Their Biological Consequences in miRNA-mRNA Interactions
3.6. Spectrum of Somatic Variants in HPV-Associated PSCC
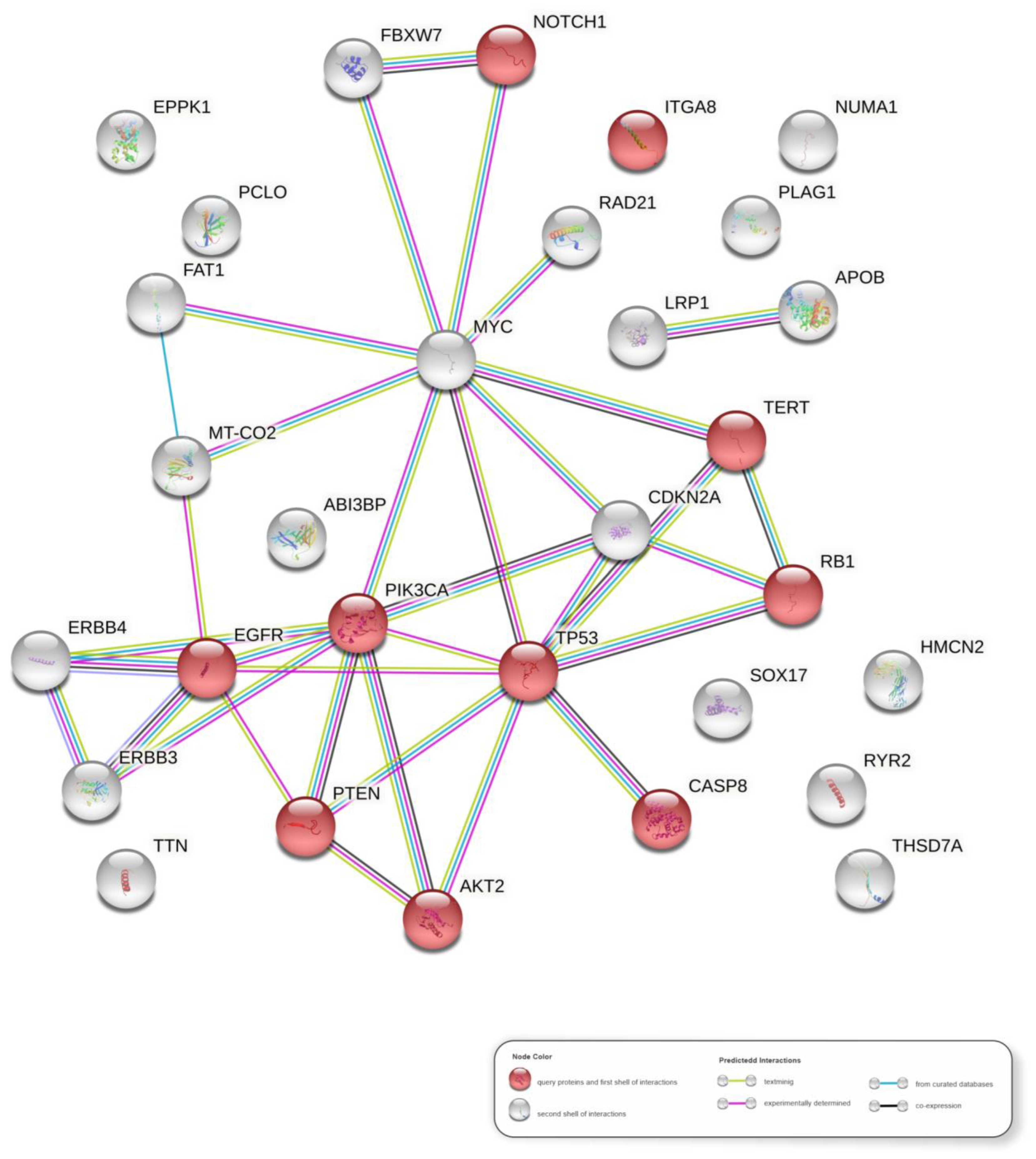
3.7. Molecular Pathways in HPV-Associated PSCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Necchi, A.; Muneer, A.; Tobias-Machado, M.; Tran, A.T.H.; Van Rompuy, A.-S.; Spiess, P.E.; Albersen, M. Penile Cancer. Nat. Rev. Dis. Primers 2021, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Koifman, L.; Vides, A.J.; Koifman, N.; Carvalho, J.P.; Ornellas, A.A. Epidemiological Aspects of Penile Cancer in Rio de Janeiro: Evaluation of 230 Cases. Int. Braz J. Urol. 2011, 37, 231–243. [Google Scholar] [CrossRef][Green Version]
- Coelho, R.W.P.; Pinho, J.D.; Moreno, J.S.; Garbis, D.V.E.O.; do Nascimento, A.M.T.; Larges, J.S.; Calixto, J.R.R.; Ramalho, L.N.Z.; da Silva, A.A.M.; Nogueira, L.R.; et al. Penile Cancer in Maranhão, Northeast Brazil: The Highest Incidence Globally? BMC Urol. 2018, 18, 50. [Google Scholar] [CrossRef]
- Douglawi, A.; Masterson, T.A. Penile Cancer Epidemiology and Risk Factors: A Contemporary Review. Curr. Opin. Urol. 2019, 29, 145–149. [Google Scholar] [CrossRef]
- Kidd, L.C.; Chaing, S.; Chipollini, J.; Giuliano, A.R.; Spiess, P.E.; Sharma, P. Relationship between Human Papillomavirus and Penile Cancer—Implications for Prevention and Treatment. Transl. Androl. Urol. 2017, 6, 791–802. [Google Scholar] [CrossRef]
- Hansen, B.T.; Orumaa, M.; Lie, A.K.; Brennhovd, B.; Nygård, M. Trends in Incidence, Mortality and Survival of Penile Squamous Cell Carcinoma in Norway 1956-2015: Trends in Penile Squamous Cell Carcinoma. Int. J. Cancer 2018, 142, 1586–1593. [Google Scholar] [CrossRef]
- D’Hauwers, K.W.M.; Depuydt, C.E.; Bogers, J.J.; Noel, J.C.; Delvenne, P.; Marbaix, E.; Donders, A.R.T.; Tjalma, W.A.A. Human Papillomavirus, Lichen Sclerosus and Penile Cancer: A Study in Belgium. Vaccine 2012, 30, 6573–6577. [Google Scholar] [CrossRef]
- Macedo, J.; Silva, E.; Nogueira, L.; Coelho, R.; Silva, J.; Santos, A.; Teixeira-Júnior, A.A.; Belfort, M.; Silva, G.; Khayat, A.; et al. Genomic Profiling Reveals the Pivotal Role of HrHPV Driving Copy Number and Gene Expression Alterations, Including MRNA Downregulation of TP53 and RB1 in Penile Cancer. Mol. Carcinog. 2020, 59, 604–617. [Google Scholar] [CrossRef]
- Silva, J.D.; Nogueira, L.; Coelho, R.; Deus, A.; Khayat, A.; Marchi, R.; Oliveira, E.D.; Santos, A.P.D.; Cavalli, L.; Pereira, S. HPV-Associated Penile Cancer: Impact of Copy Number Alterations in MiRNA/MRNA Interactions and Potential Druggable Targets. CBM 2021, 32, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Costa, C.C.; Farias Ramos, I.; Laus, A.C.; Sussuchi, L.; Reis, R.M.; Khayat, A.S.; Cavalli, L.R.; Pereira, S.R. Upregulated MiRNAs on the TP53 and RB1 Binding Seedless Regions in High-Risk HPV-Associated Penile Cancer. Front. Genet. 2022. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Feber, A.; Worth, D.C.; Chakravarthy, A.; de Winter, P.; Shah, K.; Arya, M.; Saqib, M.; Nigam, R.; Malone, P.R.; Tan, W.S.; et al. CSN1 Somatic Mutations in Penile Squamous Cell Carcinoma. Cancer Res. 2016, 76, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, K.; Chen, Y.; Zhou, J.; Liang, Y.; Yang, X.; Li, X.; Cao, Y.; Wang, D.; Luo, L.; et al. Mutational Landscape of Penile Squamous Cell Carcinoma in a Chinese Population. Int. J. Cancer 2019, 145, 1280–1289. [Google Scholar] [CrossRef]
- Chahoud, J.; Gleber-Netto, F.O.; McCormick, B.Z.; Rao, P.; Lu, X.; Guo, M.; Morgan, M.B.; Chu, R.A.; Martinez-Ferrer, M.; Eterovic, A.K.; et al. Whole-Exome Sequencing in Penile Squamous Cell Carcinoma Uncovers Novel Prognostic Categorization and Drug Targets Similar to Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 2560–2570. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cleary, J.G.; Braithwaite, R.; Gaastra, K.; Hilbush, B.S.; Inglis, S.; Irvine, S.A.; Jackson, A.; Littin, R.; Rathod, M.; Ware, D.; et al. Comparing Variant Call Files for Performance Benchmarking of Next-Generation Sequencing Variant Calling Pipelines. bioRxiv 2015. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids. Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Liu, J.; Yan, J.; Mao, R.; Ren, G.; Liu, X.; Zhang, Y.; Wang, J.; Wang, Y.; Li, M.; Qiu, Q.; et al. Exome Sequencing Identified Six Copy Number Variations as a Prediction Model for Recurrence of Primary Prostate Cancers with Distinctive Prognosis. Transl. Cancer Res. 2020, 9, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Quillet, A.; Saad, C.; Ferry, G.; Anouar, Y.; Vergne, N.; Lecroq, T.; Dubessy, C. Improving Bioinformatics Prediction of MicroRNA Targets by Ranks Aggregation. Front. Genet. 2020, 10, 1330. [Google Scholar] [CrossRef] [PubMed]
- Rennie, W.; Liu, C.; Carmack, C.S.; Wolenc, A.; Kanoria, S.; Lu, J.; Long, D.; Ding, Y. STarMir: A Web Server for Prediction of MicroRNA Binding Sites. Nucleic Acids Res. 2014, 42, W114–W118. [Google Scholar] [CrossRef]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold Web Server for Statistical Folding and Rational Design of Nucleic Acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and Effective Prediction of MicroRNA/Target Duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Long, D.; Lee, R.; Williams, P.; Chan, C.Y.; Ambros, V.; Ding, Y. Potent Effect of Target Structure on MicroRNA Function. Nat. Struct. Mol. Biol. 2007, 14, 287–294. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-Protein Interaction Networks, with Increased Coverage and Integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Kumar Gupta, A.; Kumar, M. HPVbase—A Knowledgebase of Viral Integrations, Methylation Patterns and MicroRNAs Aberrant Expression: As Potential Biomarkers for Human Papillomaviruses Mediated Carcinomas. Sci. Rep. 2015, 5, 12522. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A Manually Curated Database of Viral Integration Sites in the Human Genome. Nucleic Acids Res. 2020, 48, D633–D641. [Google Scholar] [CrossRef] [PubMed]
- Anna Szymonowicz, K.; Chen, J. Biological and Clinical Aspects of HPV-Related Cancers. Cancer Biol. Med. 2020, 17, 864–878. [Google Scholar] [CrossRef]
- Ribera-Cortada, I.; Guerrero-Pineda, J.; Trias, I.; Veloza, L.; Garcia, A.; Marimon, L.; Diaz-Mercedes, S.; Alamo, J.R.; Rodrigo-Calvo, M.T.; Vega, N.; et al. Pathogenesis of Penile Squamous Cell Carcinoma: Molecular Update and Systematic Review. Int. J. Mol. Sci. 2021, 23, 251. [Google Scholar] [CrossRef]
- Schwaederle, M.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Squamousness: Next-Generation Sequencing Reveals Shared Molecular Features across Squamous Tumor Types. Cell Cycle 2015, 14, 2355–2361. [Google Scholar] [CrossRef]
- Cubilla, A.L.; Lloveras, B.; Alejo, M.; Clavero, O.; Chaux, A.; Kasamatsu, E.; Monfulleda, N.; Tous, S.; Alemany, L.; Klaustermeier, J.; et al. Value of P16INK4a in the Pathology of Invasive Penile Squamous Cell Carcinomas: A Report of 202 Cases. Am. J. Surg. Pathol. 2011, 35, 253–261. [Google Scholar] [CrossRef]
- Gadhikar, M.A.; Zhang, J.; Shen, L.; Rao, X.; Wang, J.; Zhao, M.; Kalu, N.N.; Johnson, F.M.; Byers, L.A.; Heymach, J.; et al. CDKN2A/P16 Deletion in Head and Neck Cancer Cells Is Associated with CDK2 Activation, Replication Stress, and Vulnerability to CHK1 Inhibition. Cancer Res. 2018, 78, 781–797. [Google Scholar] [CrossRef]
- Sievers, P.; Hielscher, T.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Berghoff, A.S.; Neidert, M.C.; Wirsching, H.-G.; Mawrin, C.; Ketter, R.; et al. CDKN2A/B Homozygous Deletion Is Associated with Early Recurrence in Meningiomas. Acta Neuropathol. 2020, 140, 409–413. [Google Scholar] [CrossRef]
- da Mata, S.; Ferreira, J.; Nicolás, I.; Esteves, S.; Esteves, G.; Lérias, S.; Silva, F.; Saco, A.; Cochicho, D.; Cunha, M.; et al. P16 and HPV Genotype Significance in HPV-Associated Cervical Cancer—A Large Cohort of Two Tertiary Referral Centers. Int. J. Mol. Sci. 2021, 22, 2294. [Google Scholar] [CrossRef]
- Zito Marino, F.; Sabetta, R.; Pagliuca, F.; Brunelli, M.; Aquino, G.; Perdonà, S.; Botti, G.; Facchini, G.; Fiorentino, F.; Di Lauro, G.; et al. Discrepancy of P16 Immunohistochemical Expression and HPV RNA in Penile Cancer. A Multiplex in Situ Hybridization/Immunohistochemistry Approach Study. Infect. Agents Cancer 2021, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhang, J.; Chen, Y.; Hu, Q.; Shen, H.; Huang, R.; Liu, Q.; Kaur, J.; Long, M.; Battaglia, S.; et al. Whole-exome Sequencing of Ovarian Cancer Families Uncovers Putative Predisposition Genes. Int. J. Cancer 2020, 146, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Meyerson, W.U.; Dursi, L.J.; Wang, L.-B.; Dong, G.; Liang, W.-W.; Weerasinghe, A.; Li, S.; Li, Y.; Kelso, S.; et al. Retrospective Evaluation of Whole Exome and Genome Mutation Calls in 746 Cancer Samples. Nat. Commun. 2020, 11, 4748. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Deng, C.; Li, Z.; Chen, J.; Yao, K.; Huang, K.; Liu, T.; Liu, Z.; Qin, Z.; Zhou, F.; et al. Molecular Characterization and Integrative Genomic Analysis of a Panel of Newly Established Penile Cancer Cell Lines. Cell Death Dis. 2018, 9, 684. [Google Scholar] [CrossRef]
- Stecca, C.E.; Alt, M.; Jiang, D.M.; Chung, P.; Crook, J.M.; Kulkarni, G.S.; Sridhar, S.S. Recent Advances in the Management of Penile Cancer: A Contemporary Review of the Literature. Oncol. Ther. 2021, 9, 21–39. [Google Scholar] [CrossRef]
- Huang, K.-B.; Guo, S.-J.; Li, Y.-H.; Zhang, X.-K.; Chen, D.; Spiess, P.E.; Li, Z.-S.; Deng, C.-Z.; Chen, J.-P.; Zhou, Q.-H.; et al. Genome-Wide Profiling Reveals HPV Integration Pattern and Activated Carcinogenic Pathways in Penile Squamous Cell Carcinoma. Cancers 2021, 13, 6104. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with Open Crowdsource Efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Januario, T.; Ye, X.; Bainer, R.; Alicke, B.; Smith, T.; Haley, B.; Modrusan, Z.; Gould, S.; Yauch, R.L. PRC2-Mediated Repression of SMARCA2 Predicts EZH2 Inhibitor Activity in SWI/SNF Mutant Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 12249–12254. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: An Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Izutsu, K.; Ando, K.; Nishikori, M.; Shibayama, H.; Teshima, T.; Kuroda, J.; Kato, K.; Imaizumi, Y.; Nosaka, K.; Sakai, R.; et al. Phase II Study of Tazemetostat for Relapsed or Refractory B-cell Non-Hodgkin Lymphoma with EZH2 Mutation in Japan. Cancer Sci. 2021, 112, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma and Advanced Solid Tumours: A First-in-Human, Open-Label, Phase 1 Study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.-W.; Jahan, T.; et al. Tazemetostat in Advanced Epithelioid Sarcoma with Loss of INI1/SMARCB1: An International, Open-Label, Phase 2 Basket Study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Pyziak, K.; Sroka-Porada, A.; Rzymski, T.; Dulak, J.; Łoboda, A. Potential of Enhancer of Zeste Homolog 2 Inhibitors for the Treatment of SWI/SNF Mutant Cancers and Tumor Microenvironment Modulation. Drug Dev. Res. 2021, 82, 730–753. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Macdonald, E.; Venneti, S.; Wang, X.Q.D.; Witkowski, L.; Jelinic, P.; Kong, T.; Martinez, D.; Morin, G.; et al. CDK4/6 Inhibitors Target SMARCA4-Determined Cyclin D1 Deficiency in Hypercalcemic Small Cell Carcinoma of the Ovary. Nat. Commun. 2019, 10, 558. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic Analysis of Telomere Length and Somatic Alterations in 31 Cancer Types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, J.-H.; Han, J.H.; Cho, N.H.; Kim, S.J.; Kim, S.I.; Choo, S.H.; Kim, J.S.; Park, B.; Kwon, J.E. TERT Promoter Mutations in Penile Squamous Cell Carcinoma: High Frequency in Non-HPV-Related Type and Association with Favorable Clinicopathologic Features. J. Cancer Res. Clin. Oncol. 2021, 147, 1125–1135. [Google Scholar] [CrossRef]
- Bell, R.J.A.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The Transcription Factor GABP Selectively Binds and Activates the Mutant TERT Promoter in Cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11, 589929. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of P53: MicroRNAs and P53. J. Cell Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Secker, G.A.; Harvey, N.L. Regulation of VEGFR Signalling in Lymphatic Vascular Development and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 7760. [Google Scholar] [CrossRef]
- Bai, M.; Li, Z.-G.; Ba, Y. Influence of KDR Genetic Variation on the Efficacy and Safety of Patients with Chemotherapy Refractory Metastatic CRC Who Received Apatinib Treatment. Int. J. Mol. Sci. 2021, 14, 1041–1055. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Seeholzer, T.; Kurz, S.; Schlauderer, F.; Woods, S.; Gehring, T.; Widmann, S.; Lammens, K.; Krappmann, D. BCL10-CARD11 Fusion Mimics an Active CARD11 Seed That Triggers Constitutive BCL10 Oligomerization and Lymphocyte Activation. Front. Immunol. 2018, 9, 2695. [Google Scholar] [CrossRef]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef]
- Qiu, Z.; Lin, A.; Li, K.; Lin, W.; Wang, Q.; Wei, T.; Zhu, W.; Luo, P.; Zhang, J. A Novel Mutation Panel for Predicting Etoposide Resistance in Small-Cell Lung Cancer. Drug Des. Dev. Ther. 2019, 13, 2021–2041. [Google Scholar] [CrossRef]
- Renou, L.; Boelle, P.-Y.; Deswarte, C.; Spicuglia, S.; Benyoucef, A.; Calvo, J.; Uzan, B.; Belhocine, M.; Cieslak, A.; Landman-Parker, J.; et al. Homeobox Protein TLX3 Activates MiR-125b Expression to Promote T-Cell Acute Lymphoblastic Leukemia. Blood Adv. 2017, 1, 733–747. [Google Scholar] [CrossRef]
- Kawahara, Y. Human Diseases Caused by Germline and Somatic Abnormalities in MicroRNA and MicroRNA-Related Genes: MicroRNAs and Human Genetic Diseases. Congenit. Anom. 2014, 54, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chhichholiya, Y.; Suryan, A.K.; Suman, P.; Munshi, A.; Singh, S. SNPs in MiRNAs and Target Sequences: Role in Cancer and Diabetes. Front. Genet. 2021, 12, 793523. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D. Single Nucleotide Alterations in MicroRNAs and Human Cancer-A Not Fully Explored Field. Non-Coding RNA Res. 2020, 5, 27–31. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | WES and qPCR (%) | aCGH (%) | Parameters | WES and qPCR (%) | aCGH (%) |
|---|---|---|---|---|---|
| Histologic site | Histologic type | ||||
| Glans | 10 (33.3) | 5 (27.8) | Usual | 12 (40.0) | 7 (38.8) |
| Glans + foreskin | 8 (26.7) | 8 (44.4) | Condylomatous | 14 (46.6) | 9 (50.0) |
| Foreskin | 2 (6.7) | 2 (11.1) | Mixed | 2 (6.7) | 1 (5.6) |
| Glans and other areas | 10 (33.3) | 3 (16.7) | Basaloid | 2 (6.7) | 1 (5.6) |
| Predominant lesion | Surgical procedure: | ||||
| Ulcerated Vegetative Ulcer/vegetative Verrucous Nodular ulcerated No information | 11 (36.7) 10 (33.3) 3 (10.0) 2 (6.7) 3 (10.0) 1 (3.3) | 6 (33.3) 6 (33.3) 2 (11.1) 2 (11.1) 2 (11.1) 0 | Preserved penis Amputation (partial) Amputation (total) Glandectomy (total) No information | 1 (3.3) 17 (56.7) 9 (30.0) 2 (6.7) 1 (3.3) | 1 (5.6) 12 (66.6) 5 (27.8) 0 (0.0) 0 (0.0) |
| Lymphatic invasion | Perineural invasion | ||||
| Yes | 5 (16.7) | 3 (16.7) | Yes | 9 (30.0) | 5 (27.8) |
| No | 22 (73.3) | 13 (72.2) | No | 18 (60.0) | 11 (61.1) |
| No information | 3 (10.0) | 2 (11.1) | No information | 3 (10.0) | 2 (11.1) |
| Tumor Grade: | Tumor size (mm): | ||||
| G1 | 2 (6.7) | 1 (5.6) | 0.6–2.0 | 2 (6.7) | 0 |
| G2 | 20 (66.7) | 12 (66.6) | 2.1–5.0 | 17 (56.6) | 13 (72.2) |
| G3 | 8 (26.6) | 5 (27.8) | 5.1–10.0 | 11 (36.7) | 5 (27.8) |
| Primary tumor (T) | Infection HPV | ||||
| pT1 pT2 pT3 No information | 5 (16.7) 11 (36.7) 13 (43.3) 1 (3.33) | 3 (16.7) 7 (38.9) 8 (44.4) 0 | Positive Negative | 30 (100.0) 0 | 18 (100.0) 0 |
| Phimosis | Marital status | ||||
| Yes | 15 (50.0) | 7 (38.9) | Married | 20 (66.7) | 15 (83.3) |
| No | 9 (30.0) | 6 (33.3) | Single | 7 (23.3) | 3 (16.7) |
| No information | 6 (20.0) | 5 (27.8) | No information | 3 (10.0) | 0 |
| Gene | Cytoband | Integration Sites (HPV Subtypes) * | Variants | |||
|---|---|---|---|---|---|---|
| KMT2C | 7q36.1 | yes (16, 18) | c.11261C > T | c.4844G > A | c.162-17T > G | c.9059C > G |
| KMT2D | 12q13.12 | yes (16, 18) | c.511C > T c.9614G > C | c.9232G > C c.9186G > C | c.8914G > A c.12133C > T | |
| SMARCA4 | 19p13.2 | yes (16, 18) | c.3575G > A | c.1249C > T | c.3694G > A | chr19:g.11003471C > T |
| CIC | 19q13.2 | yes (16, 18) | c.1264C > T | chr19:g.42274618G > A | chr19:g.42287248CC > TT:p.? | |
| PTPRB | 12q15 | yes (18) | c.2886G > C | c.979G > C | c.2357C > G | chr12:g.70609388C > T |
| NBEA | 13q13.3 | yes (16, 26) ** | c.6226C > T c.1572T > G | c.4644 + 5G > A | c.6060G > A | c.5463C > A |
| FAM135B | 8q24.23 | yes (16, 18, 45) | c.2974T > A | chr8:g.138132825A > C | ||
| GTF2I | 7q11.23 | yes (16) | c.178G > C | c.20C > G | c.233A > G | chr7:g.74589997C > A |
| AJUBA | 14q11.2 | yes (16, 18) | c.1006G > C | chr14:g. 22978448GAGAA > -:p.? | c.762_ 763insGA | c.445dupG |
| CR1 | 1q32.2 | yes (16, 18) | c.4067G > C | c.68G > A | c.5383G > T | chr1:g.207588649A > T |
| Gene | Start | End | Cytoband | HPV Integration Site (Subtypes) * |
|---|---|---|---|---|
| NOTCH1 ☨ | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 136407694 | 136518542 | 9q34.3–9q34.3 | yes (16, 18) | |
| PTCH1 | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 84866993 | 98578322 | 9q21.33–9q22.31 | yes (16, 18) | |
| TSC1 | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 131060785 | 133209449 | 9q34.12–9q34.2 | yes (16, 18) | |
| NUP214 | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 131060785 | 133209449 | 9q34.12–9q34.2 | yes (16, 18) | |
| SET | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 128220134 | 128822687 | 9q34.11–9q34.11 | yes (16, 18) | |
| ABL1 | 129873232 | 131057397 | 9q34.11–9q34.12 | yes (16, 18) |
| 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) | |
| TNC ☨ | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 114792157 | 120990030 | 9q32–9q33.1 | yes (16, 18) | |
| FANCC ☨ | 89311043 | 138219684 | 9q22.2–9q34.2 | yes (16, 18) |
| 84866993 | 98578322 | 9q21.33–9q22.31 | yes (16, 18) | |
| PLAG1 ☨ | 54136261 | 89971540 | 8q21.12–8q13.2 | yes (16, 18, 68) |
| 56072840 | 143934540 | 8q21.12–8q24.22 | yes (16, 18, 68) | |
| MYC ☨ | 117528401 | 133257113 | 8q24.12–8q24.22 | yes (16, 18) |
| 56072840 | 143934540 | 8q21.12–8q24.22 | yes (16, 18, 68) | |
| RAD21 | 109399826 | 117521303 | 8q23.2–8q24.11 | yes (16, 18) |
| 56072840 | 143934540 | 8q21.12–8q24.22 | yes (16, 18, 68) | |
| NUMA1 | 70078357 | 73083990 | 11q13.3–11q13.4 | yes (16) |
| 71463803 | 72023397 | 11q13.4–11q13.4 | yes (16) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canto, L.M.; da Silva, J.M.; Castelo-Branco, P.V.; da Silva, I.M.; Nogueira, L.; Fonseca-Alves, C.E.; Khayat, A.; Birbrair, A.; Pereira, S.R. Mutational Signature and Integrative Genomic Analysis of Human Papillomavirus-Associated Penile Squamous Cell Carcinomas from Latin American Patients. Cancers 2022, 14, 3514. https://doi.org/10.3390/cancers14143514
Canto LM, da Silva JM, Castelo-Branco PV, da Silva IM, Nogueira L, Fonseca-Alves CE, Khayat A, Birbrair A, Pereira SR. Mutational Signature and Integrative Genomic Analysis of Human Papillomavirus-Associated Penile Squamous Cell Carcinomas from Latin American Patients. Cancers. 2022; 14(14):3514. https://doi.org/10.3390/cancers14143514
Chicago/Turabian StyleCanto, Luisa Matos, Jenilson Mota da Silva, Patrícia Valèria Castelo-Branco, Ingrid Monteiro da Silva, Leudivan Nogueira, Carlos Eduardo Fonseca-Alves, André Khayat, Alexander Birbrair, and Silma Regina Pereira. 2022. "Mutational Signature and Integrative Genomic Analysis of Human Papillomavirus-Associated Penile Squamous Cell Carcinomas from Latin American Patients" Cancers 14, no. 14: 3514. https://doi.org/10.3390/cancers14143514
APA StyleCanto, L. M., da Silva, J. M., Castelo-Branco, P. V., da Silva, I. M., Nogueira, L., Fonseca-Alves, C. E., Khayat, A., Birbrair, A., & Pereira, S. R. (2022). Mutational Signature and Integrative Genomic Analysis of Human Papillomavirus-Associated Penile Squamous Cell Carcinomas from Latin American Patients. Cancers, 14(14), 3514. https://doi.org/10.3390/cancers14143514