A ‘Hybrid’ Radiotherapy Regimen Designed for Immunomodulation: Combining High-Dose Radiotherapy with Low-Dose Radiotherapy


Abstract
:Simple Summary
Abstract
1. Introduction
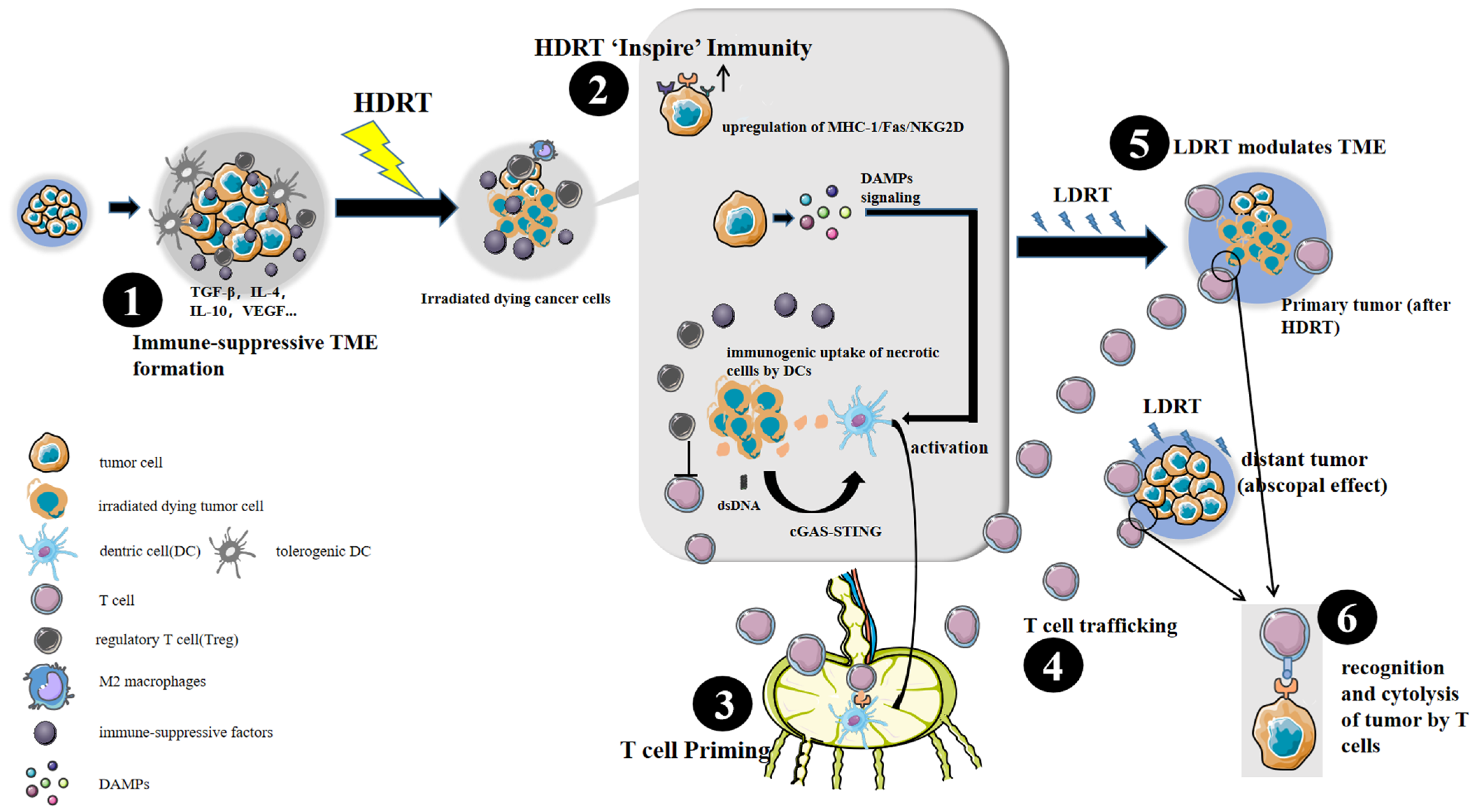
2. HDRT + LDRT in Stimulating Anti-Tumor Immunity: A Preclinical Snapshot
3. Immunobiology Mechanisms of HDRT + LDRT
3.1. HDRT: A Powerful Tool to Inspire Anti-Tumor Immunity
3.2. LDRT: An Immune Modulator
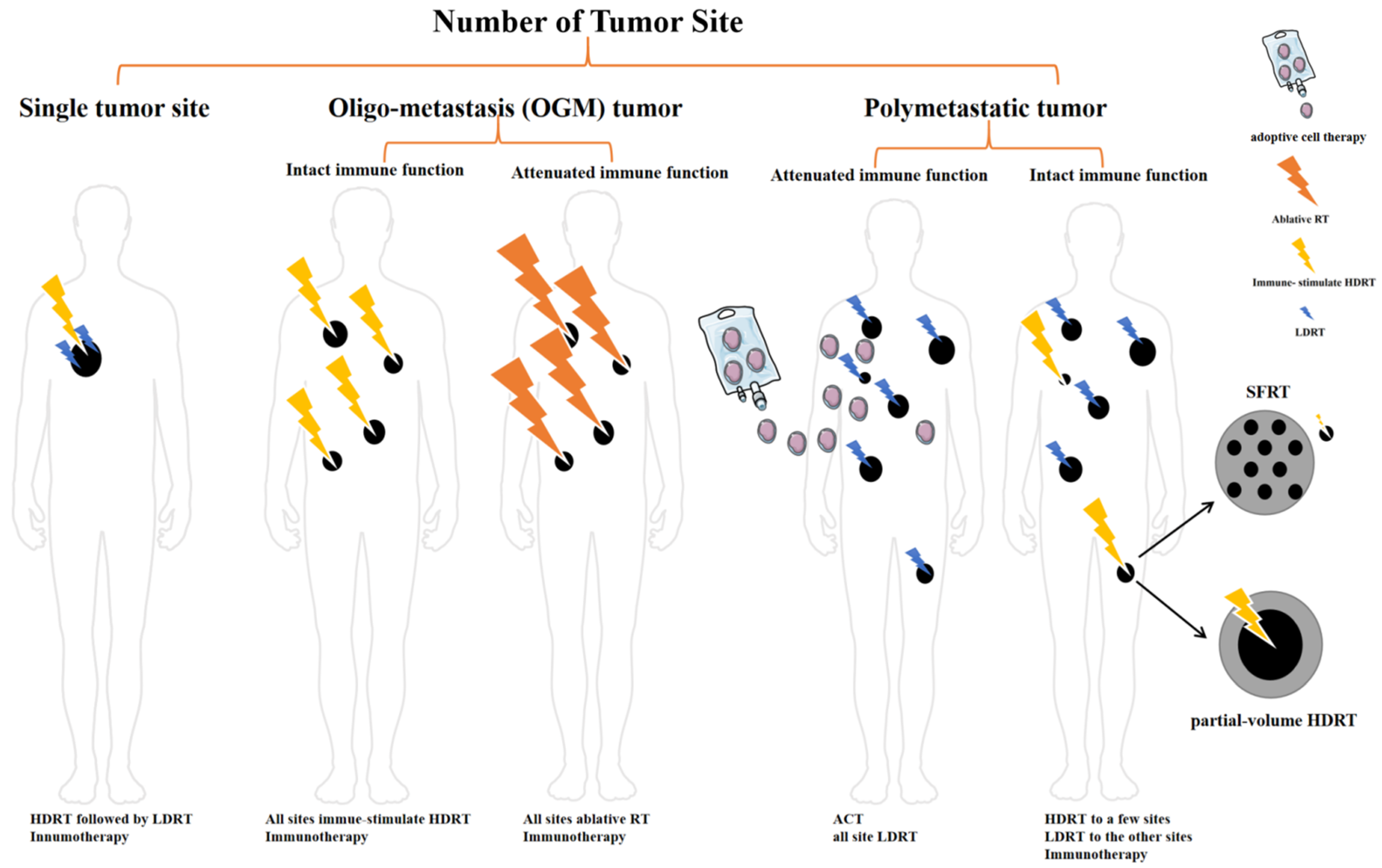
4. Clinical Challenges: How Will We Bridge the Gap between Theory and Practice?
4.1. How to Decide the Number of HDRT Site
4.2. Target Volume of HDRT
4.3. The Impact of Patients’ Immune Function
5. Clinical Decision Suggestions
5.1. Single Tumor Site
5.2. Oligo-Metastasis (OGM) Tumor
5.3. Polymetastatic Disease/High Tumor Burden
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donlon, N.; Power, R.; Hayes, C.; Reynolds, J.; Lysaght, J. Radiotherapy, immunotherapy, and the tumour microenvironment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021, 502, 84–96. [Google Scholar] [CrossRef]
- Yu, S.; Wang, Y.; He, P.; Shao, B.; Liu, F.; Xiang, Z.; Yang, T.; Zeng, Y.; He, T.; Ma, J.; et al. Effective Combinations of Immunotherapy and Radiotherapy for Cancer Treatment. Front. Oncol. 2022, 12, 809304. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Guha, C.; Schoenfeld, J.; Morris, Z.; Monjazeb, A.; Sikora, A.; Crittenden, M.; Shiao, S.; Khleif, S.; Gupta, S.; et al. Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 2021, 9, e002038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niedermann, G. Abscopal Effects with Hypofractionated Schedules Extending into the Effector Phase of the Tumor-Specific T-Cell Response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [Green Version]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.-A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gao, M.; Huang, Z.; Yu, J.; Meng, X. SBRT combined with PD-1/PD-L1 inhibitors in NSCLC treatment: A focus on the mechanisms, advances, and future challenges. J. Hematol. Oncol. 2020, 13, 105. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; De Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; DeMaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- De Olza, M.O.; Bourhis, J.; Irving, M.; Coukos, G.; Herrera, F.G. High versus low dose irradiation for tumor immune reprogramming. Curr. Opin. Biotechnol. 2020, 65, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Kane, N.; Kobayashi, N.; Kono, E.A.; Yamashiro, J.M.; Nickols, N.G.; Reiter, R.E. High-dose per Fraction Radiotherapy Induces Both Antitumor Immunity and Immunosuppressive Responses in Prostate Tumors. Clin. Cancer Res. 2021, 27, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, N.; Romero, T.; Diaz-Perez, S.; Rettig, M.B.; Steinberg, M.L.; Kishan, A.U.; Schaue, D.; Reiter, R.E.; Knudsen, B.S.; Nickols, N.G. Significant changes in macrophage and CD8 T cell densities in primary prostate tumors 2 weeks after SBRT. Prostate Cancer Prostatic Dis. online ahead of print. 2022. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin. Cancer Res. 2018, 24, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhou, J.; Wu, M.; Hu, C.; Yang, J.; Li, D.; Wu, P.; Chen, Y.; Chen, P.; Lin, S.; et al. Low-Dose Total Body Irradiation Can Enhance Systemic Immune Related Response Induced by Hypo-Fractionated Radiation. Front. Immunol. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Herrera, F.G.; Ronet, C.; de Olza, M.O.; Barras, D.; Crespo, I.; Andreatta, M.; Corria-Osorio, J.; Spill, A.; Benedetti, F.; Genolet, R.; et al. Low-Dose Radiotherapy Reverses Tumor Immune Desertification and Resistance to Immunotherapy. Cancer Discov. 2021, 12, 108–133. [Google Scholar] [CrossRef]
- He, K.; Barsoumian, H.B.; Bertolet, G.; Verma, V.; Leuschner, C.; Koay, E.J.; Ludmir, E.B.; Hsu, E.; Pisipati, E.; Voss, T.A.; et al. Novel Use of Low-Dose Radiotherapy to Modulate the Tumor Microenvironment of Liver Metastases. Front. Immunol. 2021, 12, 812210. [Google Scholar] [CrossRef]
- Barsoumian, H.B.; Ramapriyan, R.; Younes, A.I.; Caetano, M.S.; Menon, H.; Comeaux, N.I.; Cushman, T.R.; Schoenhals, J.E.; Cadena, A.P.; Reilly, T.P.; et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J. Immunother. Cancer 2020, 8, e000537. [Google Scholar] [CrossRef]
- Barsoumian, H.B.; Sezen, D.; Menon, H.; Younes, A.I.; Hu, Y.; He, K.; Puebla-Osorio, N.; Wasley, M.; Hsu, E.; Patel, R.R.; et al. High Plus Low Dose Radiation Strategy in Combination with TIGIT and PD1 Blockade to Promote Systemic Antitumor Responses. Cancers 2022, 14, 221. [Google Scholar] [CrossRef]
- Patel, R.R.; He, K.; Barsoumian, H.B.; Chang, J.Y.; Tang, C.; Verma, V.; Comeaux, N.; Chun, S.G.; Gandhi, S.; Truong, M.T.; et al. High-dose irradiation in combination with non-ablative low-dose radiation to treat metastatic disease after progression on immunotherapy: Results of a phase II trial. Radiother. Oncol. 2021, 162, 60–67. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos-Hoff, M.H. The Radiobiology of TGFβ. Semin. Cancer Biol. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.A.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Paris, S.; Barsoumian, H.; Abana, C.O.; He, K.; Sezen, D.; Wasley, M.; Masrorpour, F.; Chen, D.; Yang, L.; et al. A radioenhancing nanoparticle mediated immunoradiation improves survival and generates long-term antitumor immune memory in an anti-PD1-resistant murine lung cancer model. J. Nanobiotechnol. 2021, 19, 416. [Google Scholar] [CrossRef]
- Savage, T.; Pandey, S.; Guha, C. Postablation Modulation after Single High-Dose Radiation Therapy Improves Tumor Control via Enhanced Immunomodulation. Clin. Cancer Res. 2020, 26, 910–921. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, 602. [Google Scholar] [CrossRef]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Caetano, M.S.; Wang, X.; Menon, H.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Cortez, M.A.; et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int. J. Radiat. Oncol. 2019, 104, 903–912. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.; Harrington, K. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Menon, H.; Ramapriyan, R.; Cushman, T.R.; Verma, V.; Kim, H.H.; Schoenhals, J.E.; Atalar, C.; Selek, U.; Chun, S.G.; Chang, J.Y.; et al. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front. Immunol. 2019, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, C.; Zelefsky, M.J.; Lovelock, M.; Fuks, Z.; Hunt, M.; Rosenzweig, K.; Zatcky, J.; Kim, B.; Yamada, Y. Predictors of Local Control After Single-Dose Stereotactic Image-Guided Intensity-Modulated Radiotherapy for Extracranial Metastases. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. OncoImmunology 2014, 3, e28518. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Kho, V.M.; Mekers, V.E.; Span, P.N.; Bussink, J.; Adema, G.J. Radiotherapy and cGAS/STING signaling: Impact on MDSCs in the tumor microenvironment. Cell. Immunol. 2021, 362, 104298. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Chen, Z.J. Old dogs, new trick: Classic cancer therapies activate cGAS. Cell Res. 2020, 30, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Mole, R. Whole body irradiation: Radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, Z.; Ma, S.; Liu, X. Radiotherapy and Cytokine Storm: Risk and Mechanism. Front. Oncol. 2021, 11, 670464. [Google Scholar] [CrossRef]
- Kulzer, L.; Rubner, Y.; Deloch, L.; Allgäuer, A.; Frey, B.; Fietkau, R.; Dörrie, J.; Schaft, N.; Gaipl, U. Norm- and hypo-fractionated radiotherapy is capable of activating human dendritic cells. J. Immunotoxicol. 2014, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Tait, S.W. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Vaes, R.; Hendriks, L.; Vooijs, M.; De Ruysscher, D. Biomarkers of Radiotherapy-Induced Immunogenic Cell Death. Cells 2021, 10, 930. [Google Scholar] [CrossRef]
- Zanoni, M.; Sarti, A.; Zamagni, A.; Cortesi, M.; Pignatta, S.; Arienti, C.; Tebaldi, M.; Sarnelli, A.; Romeo, A.; Bartolini, D.; et al. Irradiation causes senescence, ATP release, and P2 × 7 receptor isoform switch in glioblastoma. Cell Death Dis. 2022, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 109, 199. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.; Schneider, R.; Formenti, S.C.; Dustin, M.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. 2015, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar] [PubMed]
- Formenti, S.C.; Demaria, S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. JNCI J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, C.T.; Palena, C.; Chakarborty, M.; Tsang, K.-Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of Tumor Cells Up-Regulates Fas and Enhances CTL Lytic Activity and CTL Adoptive Immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External Beam Radiation of Tumors Alters Phenotype of Tumor Cells to Render Them Susceptible to Vaccine-Mediated T-Cell Killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef] [Green Version]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Yang, M.; Zhang, J.; Yin, Y.; Fan, X.; Zhang, Y.; Qin, S.; Zhang, H.; Yu, F. Immunogenic Cell Death Induction by Ionizing Radiation. Front. Immunol. 2021, 12, 705361. [Google Scholar] [CrossRef]
- Martinez-Zubiaurre, I.; Chalmers, A.J.; Hellevik, T. Radiation-Induced Transformation of Immunoregulatory Networks in the Tumor Stroma. Front. Immunol. 2018, 9, 1679. [Google Scholar] [CrossRef]
- Gorchs, L.; Hellevik, T.; Bruun, J.-A.; Camilio, K.-A.; Al-Saad, S.; Stuge, T.-B.; Martinez-Zubiaurre, I. Cancer-Associated Fibroblasts from Lung Tumors Maintain Their Immunosuppressive Abilities after High-Dose Irradiation. Front. Oncol. 2015, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Crittenden, M.R.; Savage, T.; Cottam, B.; Baird, J.; Rodriguez, P.C.; Newell, P.; Young, K.; Jackson, A.M.; Gough, M.J. Expression of Arginase I in Myeloid Cells Limits Control of Residual Disease after Radiation Therapy of Tumors in Mice. Radiat. Res. 2014, 182, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, Y.; Nirschl, T.R.; Kochel, C.M.; Lopez-Bujanda, Z.; Theodros, D.; Mao, W.; Carrera-Haro, M.A.; Ghasemzadeh, A.; Marciscano, A.E.; Velarde, E.; et al. Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2017, 5, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Pilones, K.; García-Martínez, E.; Rudqvist, N.; Formenti, S.; Demaria, S. Barriers to Radiation-Induced Tumor Vaccination. Front. Immunol. 2017, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Li, J.-F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.-N.; Pollard, J.W. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Xiong, S.; Zhang, L.; Chu, Y. Enhancement of antitumor immunity by low-dose total body irradiationis associated with selectively decreasing the proportion and number of T regulatory cells. Cell. Mol. Immunol. 2010, 7, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Monjazeb, A.M.; Giobbie-Hurder, A.; Lako, A.; Thrash, E.M.; Brennick, R.C.; Kao, K.Z.; Manuszak, C.; Gentzler, R.D.; Tesfaye, A.; Jabbour, S.K.; et al. A Randomized Trial of Combined PD-L1 and CTLA-4 Inhibition with Targeted Low-Dose or Hypofractionated Radiation for Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 2470–2480. [Google Scholar] [CrossRef]
- Song, C.W.; Payne, J.T.; Levitt, S.H. Vascularity and Blood Flow in X-Irradiated Walker Carcinoma 256 of Rats. Radiology 1972, 104, 693–697. [Google Scholar] [CrossRef]
- Song, C.W.; Levitt, S.H. Vascular Changes in Walker 256 Carcinoma of Rats Following X Irradiation. Radiology 1971, 100, 397–407. [Google Scholar] [CrossRef]
- Wong, H.H.; Song, C.W.; Levitt, S.H. Early Changes in the Functional Vasculature of Walker Carcinoma 256 Following Irradiation. Radiology 1973, 108, 429–434. [Google Scholar] [CrossRef]
- Chen, L.; Endler, A.; Shibasaki, F. Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Exp. Mol. Med. 2009, 41, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezen, D.; Patel, R.R.; Tang, C.; Onstad, M.; Nagarajan, P.; Patel, S.P.; Welsh, J.W.; Lin, L.L. Immunotherapy combined with high- and low-dose radiation to all sites leads to complete clearance of disease in a patient with metastatic vaginal melanoma. Gynecol. Oncol. 2021, 161, 645–652. [Google Scholar] [CrossRef]
- Brooks, E.D.; Chang, J.Y. Time to abandon single-site irradiation for inducing abscopal effects. Nat. Rev. Clin. Oncol. 2018, 16, 123–135. [Google Scholar] [CrossRef]
- Tang, C.; Welsh, J.W.; de Groot, P.; Massarelli, E.; Chang, J.Y.; Hess, K.R.; Basu, S.; Curran, M.A.; Cabanillas, M.E.; Subbiah, V.; et al. Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin. Cancer Res. 2017, 23, 1388–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.R.; Verma, V.; Barsoumian, H.B.; Ning, M.S.; Chun, S.G.; Tang, C.; Chang, J.Y.; Lee, P.P.; Gandhi, S.; Balter, P.; et al. Use of Multi-Site Radiation Therapy for Systemic Disease Control. Int. J. Radiat. Oncol. Biol. Phys. 2020, 109, 352–364. [Google Scholar] [CrossRef]
- Iyengar, P.; Wardak, Z.; Gerber, D.; Tumati, V.; Ahn, C.; Hughes, R.; Dowell, J.; Cheedella, N.; Nedzi, L.; Westover, K.; et al. Consolidative Radiotherapy for Limited Metastatic Non-Small-Cell Lung Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2018, 4, e173501. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Lemons, J.M.; Karrison, T.G.; Pitroda, S.P.; Melotek, J.M.; Zha, Y.; Al-Hallaq, H.A.; Arina, A.; Khodarev, N.N.; Janisch, L.; et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Onderdonk, B.E.; Bhave, S.R.; Karrison, T.; Lemons, J.M.; Chang, P.; Zha, Y.; Carll, T.; Krausz, T.; Huang, L.; et al. Improved Survival Associated with Local Tumor Response Following Multisite Radiotherapy and Pembrolizumab: Secondary Analysis of a Phase I Trial. Clin. Cancer Res. 2020, 26, 6437–6444. [Google Scholar] [CrossRef] [PubMed]
- Milano, M.T.; Mihai, A.; Kang, J.; Singh, D.P.; Verma, V.; Qiu, H.; Chen, Y.; Kong, F.-M. Stereotactic body radiotherapy in patients with multiple lung tumors: A focus on lung dosimetric constraints. Expert Rev. Anticancer Ther. 2019, 19, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.W.; Tang, C.; de Groot, P.; Naing, A.; Hess, K.R.; Heymach, J.V.; Papadimitrakopoulou, V.A.; Cushman, T.R.; Subbiah, V.; Chang, J.Y.; et al. Phase II Trial of Ipilimumab with Stereotactic Radiation Therapy for Metastatic Disease: Outcomes, Toxicities, and Low-Dose Radiation–Related Abscopal Responses. Cancer Immunol. Res. 2019, 7, 1903–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, H.; Chen, D.; Ramapriyan, R.; Verma, V.; Barsoumian, H.B.; Cushman, T.R.; Younes, A.; Cortez, M.A.; Erasmus, J.J.; De Groot, P.; et al. Influence of low-dose radiation on abscopal responses in patients receiving high-dose radiation and immunotherapy. J. Immunother. Cancer 2019, 7, 237. [Google Scholar] [CrossRef] [Green Version]
- Schoenhals, J.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor–Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170. [Google Scholar] [CrossRef] [Green Version]
- Asperud, J.; Arous, D.; Edin, N.F.J.; Malinen, E. Spatially fractionated radiotherapy: Tumor response modelling including immunomodulation. Phys. Med. Biol. 2021, 66, 175012. [Google Scholar] [CrossRef]
- Markovsky, E.; Budhu, S.; Samstein, R.M.; Li, H.; Russell, J.; Zhang, Z.; Drill, E.; Bodden, C.; Chen, Q.; Powell, S.N.; et al. An Antitumor Immune Response Is Evoked by Partial-Volume Single-Dose Radiation in 2 Murine Models. Int. J. Radiat. Oncol. Biol. Phys. 2018, 103, 697–708. [Google Scholar] [CrossRef]
- Yasmin-Karim, S.; Ziberi, B.; Wirtz, J.; Bih, N.; Moreau, M.; Guthier, R.; Ainsworth, V.; Hesser, J.; Makrigiorgos, G.; Chuong, M.; et al. Boosting the abscopal effect using immunogenic biomaterials with varying radiotherapy field sizes. Int. J. Radiat. Oncol. Biol. Phys. 2021, 112, 475–486. [Google Scholar] [CrossRef]
- Johnsrud, A.J.; Jenkins, S.V.; Jamshidi-Parsian, A.; Quick, C.M.; Galhardo, E.P.; Dings, R.P.; Vang, K.B.; Narayanasamy, G.; Makhoul, I.; Griffin, R.J. Evidence for Early Stage Anti-Tumor Immunity Elicited by Spatially Fractionated Radiotherapy-Immunotherapy Combinations. Radiat. Res. 2020, 194, 688–697. [Google Scholar] [CrossRef]
- Asur, R.; Butterworth, K.; Penagaricano, J.A.; Prise, K.; Griffin, R.J. High dose bystander effects in spatially fractionated radiation therapy. Cancer Lett. 2013, 356, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.J.; Yarchoan, M.; Hopkins, A.; Mehra, R.; Grossman, S.; Kang, H. Association between pretreatment lymphocyte count and response to PD1 inhibitors in head and neck squamous cell carcinomas. J. Immunother. Cancer 2018, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; DeMaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Korpics, M.C.; Polley, M.-Y.; Bhave, S.R.; Redler, G.; Pitroda, S.P.; Luke, J.J.; Chmura, S.J. A Validated T Cell Radiomics Score Is Associated With Clinical Outcomes Following Multisite SBRT and Pembrolizumab. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 189–195. [Google Scholar] [CrossRef]
- Kim, H.; Park, S.; Jeong, S.; Lee, Y.; Lee, H.; Kim, C.; Kim, K.; Hong, S.; Lee, J.; Kim, S.; et al. 4-1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8 T Cells in Hepatocellular Carcinoma. Hepatology 2020, 71, 955–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Hellman, S.; Weichselbaum, R. Oligometastases. J. Clin. Oncol. 1995, 13, 8–10. [Google Scholar] [CrossRef]
- Hendriks, L.E.; Dooms, C.; Berghmans, T.; Novello, S.; Levy, A.; De Ruysscher, D.; Hasan, B.; Levra, M.G.; Levra, N.G.; Besse, B.; et al. Defining oligometastatic non-small cell lung cancer: A simulated multidisciplinary expert opinion. Eur. J. Cancer 2019, 123, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Guckenberger, M.; Lievens, Y.; Bouma, A.B.; Collette, L.; Dekker, A.; Desouza, N.M.; Dingemans, A.-M.C.; Fournier, B.; Hurkmans, C.; Lecouvet, F.E.; et al. Characterisation and classification of oligometastatic disease: A European Society for Radiotherapy and Oncology and European Organisation for Research and Treatment of Cancer consensus recommendation. Lancet Oncol. 2020, 21, e18–e28. [Google Scholar] [CrossRef] [Green Version]
- Villaruz, L.C.; Kubicek, G.J.; Socinski, M.A. Management of Non-Small Cell Lung Cancer with Oligometastasis. Curr. Oncol. Rep. 2012, 14, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Ho, U.-C.; Kuo, L.-T. Oligometastatic Disease in Non-Small-Cell Lung Cancer: An Update. Cancers 2022, 14, 1350. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, A.-M.C.; Hendriks, L.E.; Berghmans, T.; Levy, A.; Hasan, B.; Faivre-Finn, C.; Giaj-Levra, M.; Giaj-Levra, N.; Girard, N.; Greillier, L.; et al. Definition of Synchronous Oligometastatic Non–Small Cell Lung Cancer—A Consensus Report. J. Thorac. Oncol. 2019, 14, 2109–2119. [Google Scholar] [CrossRef]
- Kroese, T.E.; van Laarhoven, H.W.; Nilsson, M.; Lordick, F.; Guckenberger, M.; Ruurda, J.P.; D’Ugo, D.; Haustermans, K.; van Cutsem, E.; van Hillegersberg, R.; et al. Definition of oligometastatic esophagogastric cancer and impact of local oligometastasis-directed treatment: A systematic review and meta-analysis. Eur. J. Cancer 2022, 166, 254–269. [Google Scholar] [CrossRef]
- Kinj, R.; Muggeo, E.; Schiappacasse, L.; Bourhis, J.; Herrera, F.G. Stereotactic Body Radiation Therapy in Patients with Oligometastatic Disease: Clinical State of the Art and Perspectives. Cancers 2022, 14, 1152. [Google Scholar] [CrossRef]
- Jasper, K.; Stiles, B.; McDonald, F.; Palma, D.A. Practical Management of Oligometastatic Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schoenhals, J.; Christie, A.; Mohamad, O.; Wang, C.; Bowman, I.; Singla, N.; Hammers, H.; Courtney, K.; Bagrodia, A.; et al. Stereotactic Ablative Radiation Therapy (SAbR) Used to Defer Systemic Therapy in Oligometastatic Renal Cell Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Palma, D.A.; Olson, R.; Harrow, S.; Gaede, S.; Louie, A.V.; Haasbeek, C.; Mulroy, L.; Lock, M.; Rodrigues, P.G.B.; Yaremko, B.P.; et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): A randomised, phase 2, open-label trial. Lancet 2019, 393, 2051–2058. [Google Scholar] [CrossRef]
- Palma, D.A.; Olson, R.; Harrow, S.; Gaede, S.; Louie, A.V.; Haasbeek, C.; Mulroy, L.; Lock, M.; Rodrigues, G.B.; Yaremko, B.P.; et al. Stereotactic Ablative Radiotherapy for the Comprehensive Treatment of Oligometastatic Cancers: Long-Term Results of the SABR-COMET Phase II Randomized Trial. J. Clin. Oncol. 2020, 38, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.R.; Blumenschein, G.R., Jr.; Lee, J.J.; Hernández, M.; Ye, R.; Camidge, D.R.; Doebele, R.C.; Skoulidis, F.; Gaspar, L.E.; Gibbons, D.L.; et al. Local consolidative therapy versus maintenance therapy or observation for patients with oligometastatic non-small-cell lung cancer without progression after first-line systemic therapy: A multicentre, randomised, controlled, phase 2 study. Lancet Oncol. 2016, 17, 1672–1682. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Xu, J.; Liu, M.; Xing, H.; Wang, Z.; Huang, L.; Mellor, A.L.; Wang, W.; Wu, S. Adoptive CD8+ T cell therapy against cancer:Challenges and opportunities. Cancer Lett. 2019, 462, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-L.; Qin, D.-Y.; Mo, Z.-M.; Li, Y.; Wei, W.; Wang, Y.-S.; Wang, W.; Wei, Y.-Q. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci. China Life Sci. 2016, 59, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Authors | Mice and Cell Line | Number of Tumor Sites | RT Regimen | Immunotherapy | Results |
|---|---|---|---|---|---|
| H Barsoumian et al. [19] | 129Sv/Ev mice 344SQ parental lung adenocarcinoma cell line | 2 | 12 Gy*3 HDRT to the primary tumor + 1 Gy*2 LDRT to the secondary tumor (3 days after HDRT) | anti-CTLA-4 anti-PD1 | Delayed growth in both primary and secondary tumors. Enhanced natural killer cell activation, increased M1 macrophages and CD4 + T-cells, and decreased TGF-β in secondary tumors. |
| H Barsoumian et al. [20] | 129Sv/Ev mice 344SQ parental lung adenocarcinoma cell line | 2 | 12 Gy*3 HDRT to the primary tumor + 1 Gy*2 LDRT to the secondary tumor (3 days after HDRT) | anti-TIGIT anti-PD1 | Delayed growth in both primary and secondary tumors, reduced the exhaustion of T-cells, generated effector immune memory, and prolonged survival. |
| Y Hu et al. [25] | 129Sv/Ev mice 344SQ parental lung adenocarcinoma cell line | 2 | 12 Gy*3 HDRT to the primary tumor + 1 Gy*2 LDRT to the secondary tumor (3 days after HDRT) | anti-PD1 anti-CTLA4 NBTXR3 nanoparticle | Slowed the growth of both primary and secondary tumors, suppressed the appearance of lung metastases, increased survival rates, induced robust long-term immune memory, and increased the CD8/Treg ratio in the secondary tumors |
| T Savage et al. [26] | C57BL/6 mice Lewis Lung Carcinoma, 3LL | 1 | 22 Gy*1 + 0.5 Gy*4(12 days after HDRT) to the tumor site | - | Delayed tumor growth, increased survival, reduced Tregs and M2 macrophages in the tumor microenvironment (TME), and increased systemic T-cell responses. |
| BalB/C mice breast carcinoma cell line, 4T1 | 1 | 22 Gy*1 to the tumor site + 0.5 Gy*4(12 days after HDRT) to the whole lung (metastatic prone organ) | - | Delayed local tumor progression, suppressed pulmonary metastases, remodeled the metastatic niche with decreased Tregs and increased effector T-cell infiltration in lungs, and increased survival. | |
| J Liu et al. [16] | BALB/C mice mammary carcinoma 4T1 and colon carcinoma CT26 cell lines | 2 | 0.1 Gy total body irradiation (3 days before HDRT) + 8 Gy*3 to the primary tumor | - | Delayed growth in both primary and secondary tumors, increased secondary tumor infiltration of CD8+ T-Cells, decreased myeloid-derived suppressor cells (MDSCs) and M2 macrophages in the secondary tumor, and inhibited metastasis |
| R Patel et al. [27] | C57Bl/6 and BALB/c mice B78 melanoma tumors cell line | 2 | targeted radionuclide therapy (TRT) using 50uCi90Y-NM600 (2.5 Gy) + 12 Gy external beam radiotherapy targeting the primary tumor | anti-CTLA4 | Improved tumor response at the secondary tumor not targeted by EBRT and improved overall survival, augmented response to ICIs, and induced robust long-term immune memory. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, H.; Zhou, Z. A ‘Hybrid’ Radiotherapy Regimen Designed for Immunomodulation: Combining High-Dose Radiotherapy with Low-Dose Radiotherapy. Cancers 2022, 14, 3505. https://doi.org/10.3390/cancers14143505
Ji H, Zhou Z. A ‘Hybrid’ Radiotherapy Regimen Designed for Immunomodulation: Combining High-Dose Radiotherapy with Low-Dose Radiotherapy. Cancers. 2022; 14(14):3505. https://doi.org/10.3390/cancers14143505
Chicago/Turabian StyleJi, Hongshan, and Zhiguo Zhou. 2022. "A ‘Hybrid’ Radiotherapy Regimen Designed for Immunomodulation: Combining High-Dose Radiotherapy with Low-Dose Radiotherapy" Cancers 14, no. 14: 3505. https://doi.org/10.3390/cancers14143505
APA StyleJi, H., & Zhou, Z. (2022). A ‘Hybrid’ Radiotherapy Regimen Designed for Immunomodulation: Combining High-Dose Radiotherapy with Low-Dose Radiotherapy. Cancers, 14(14), 3505. https://doi.org/10.3390/cancers14143505