
Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors

 ,
,  ,
,  and
and 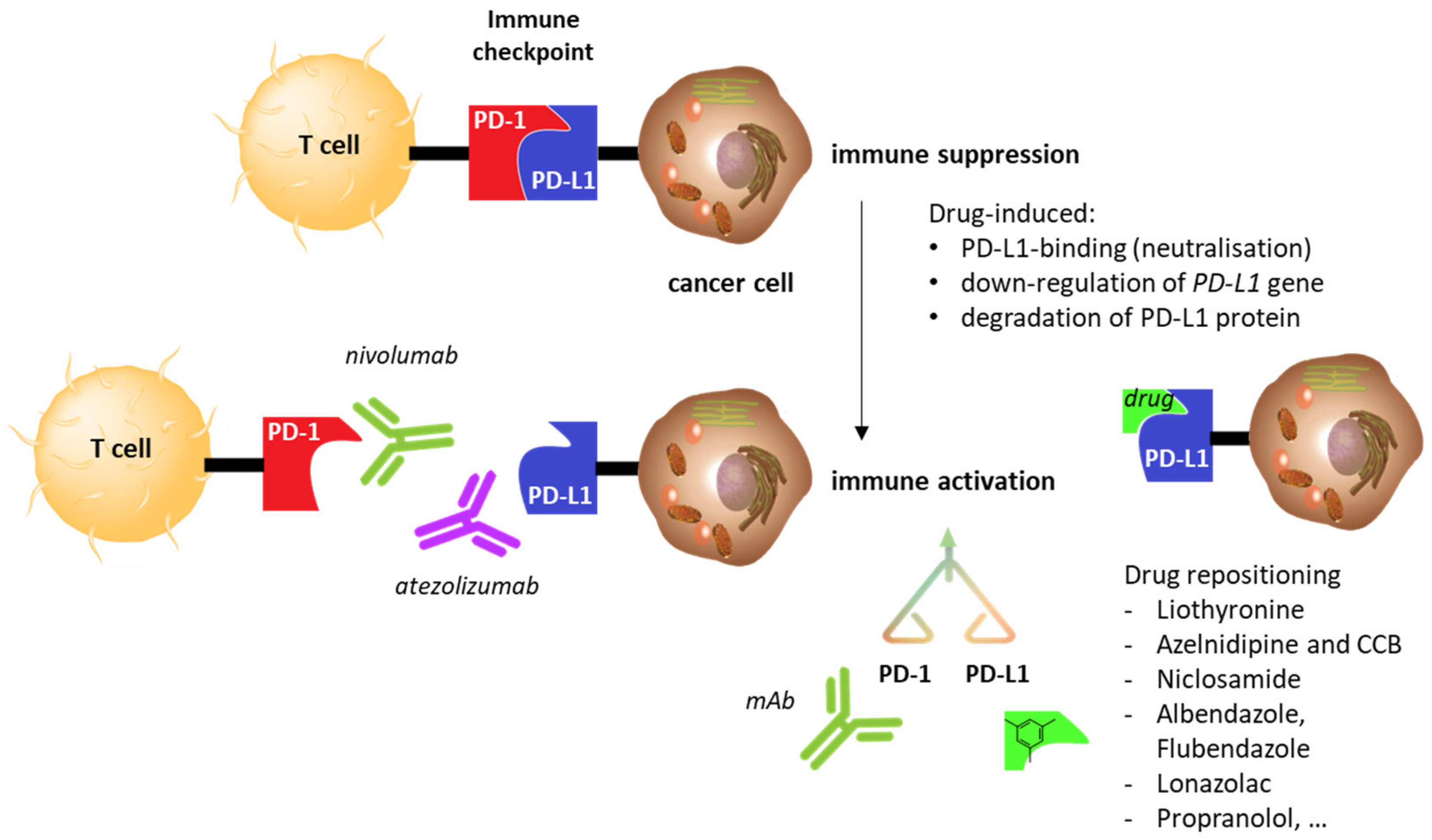{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
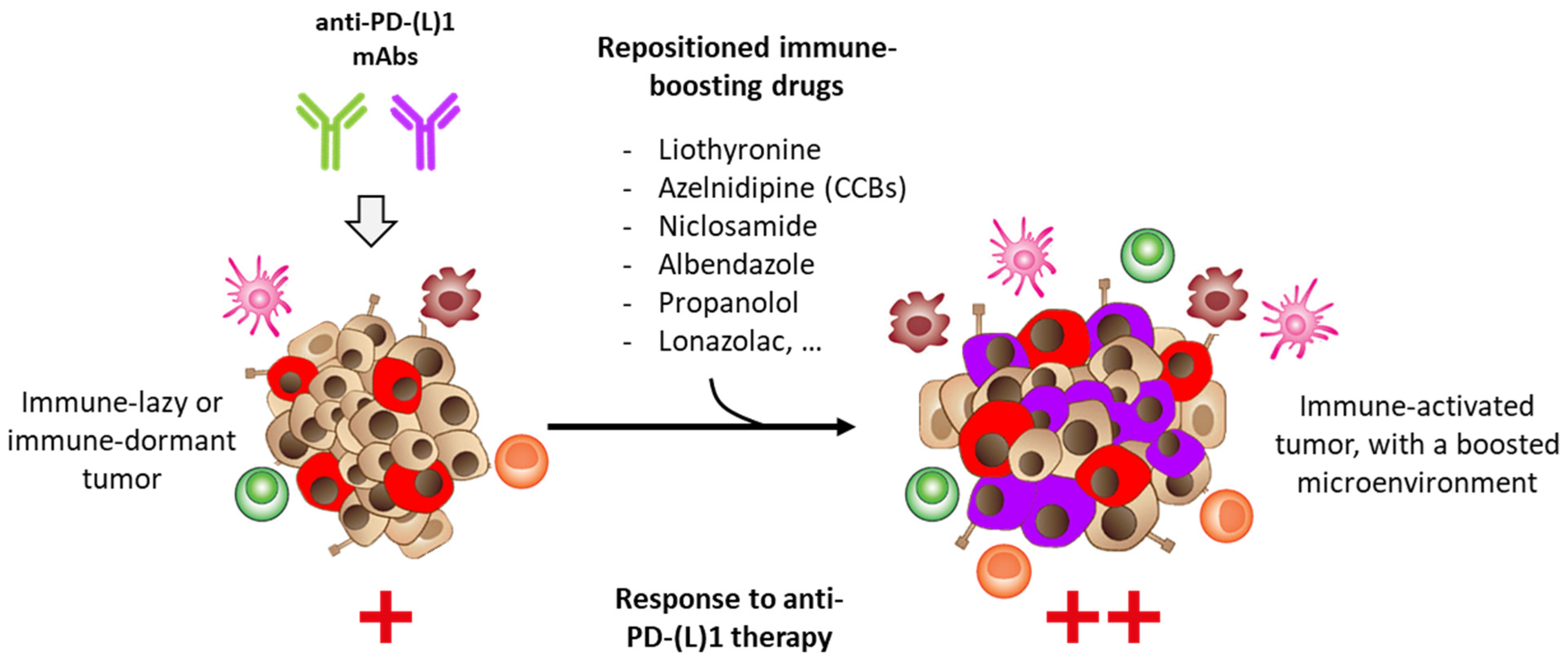
2. Drug Repositioning to Target the PD-1/PD-L1 Checkpoint
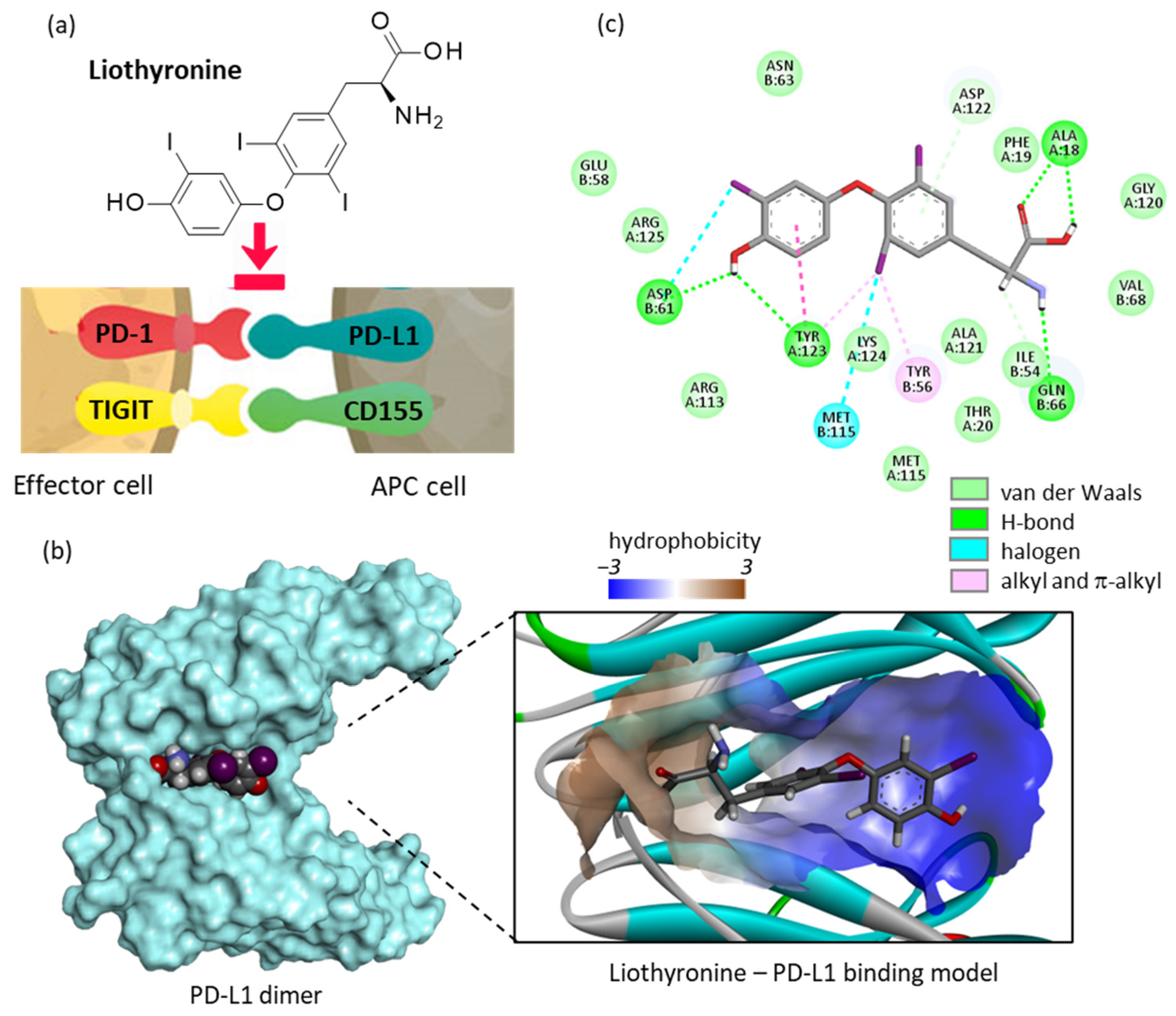
2.1. Repositioning of Liothyronine as a PD-L1 Binding Agent
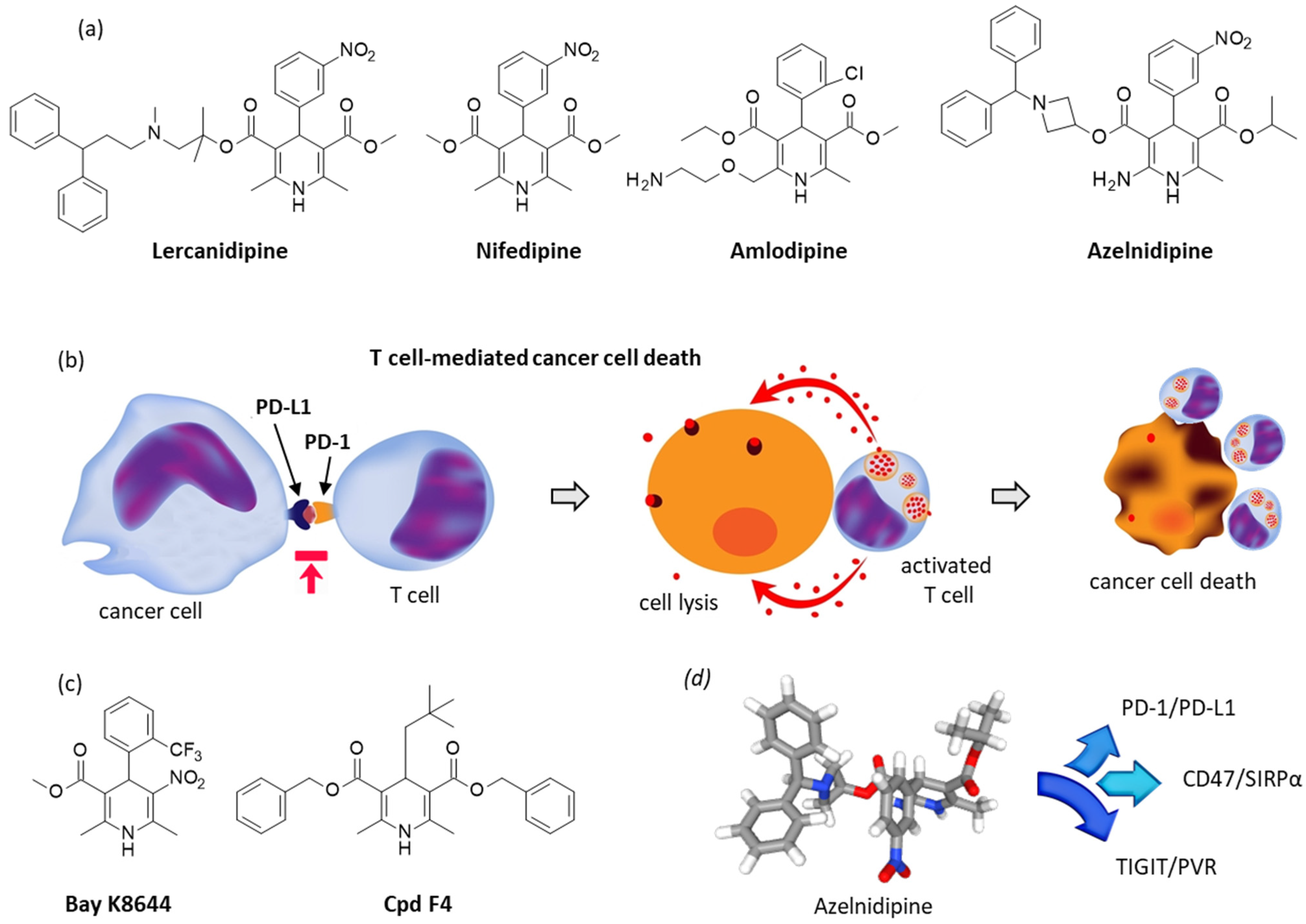
2.2. Repositioning of Dihydropyridine Calcium Channel Blockers to Treat Cancer
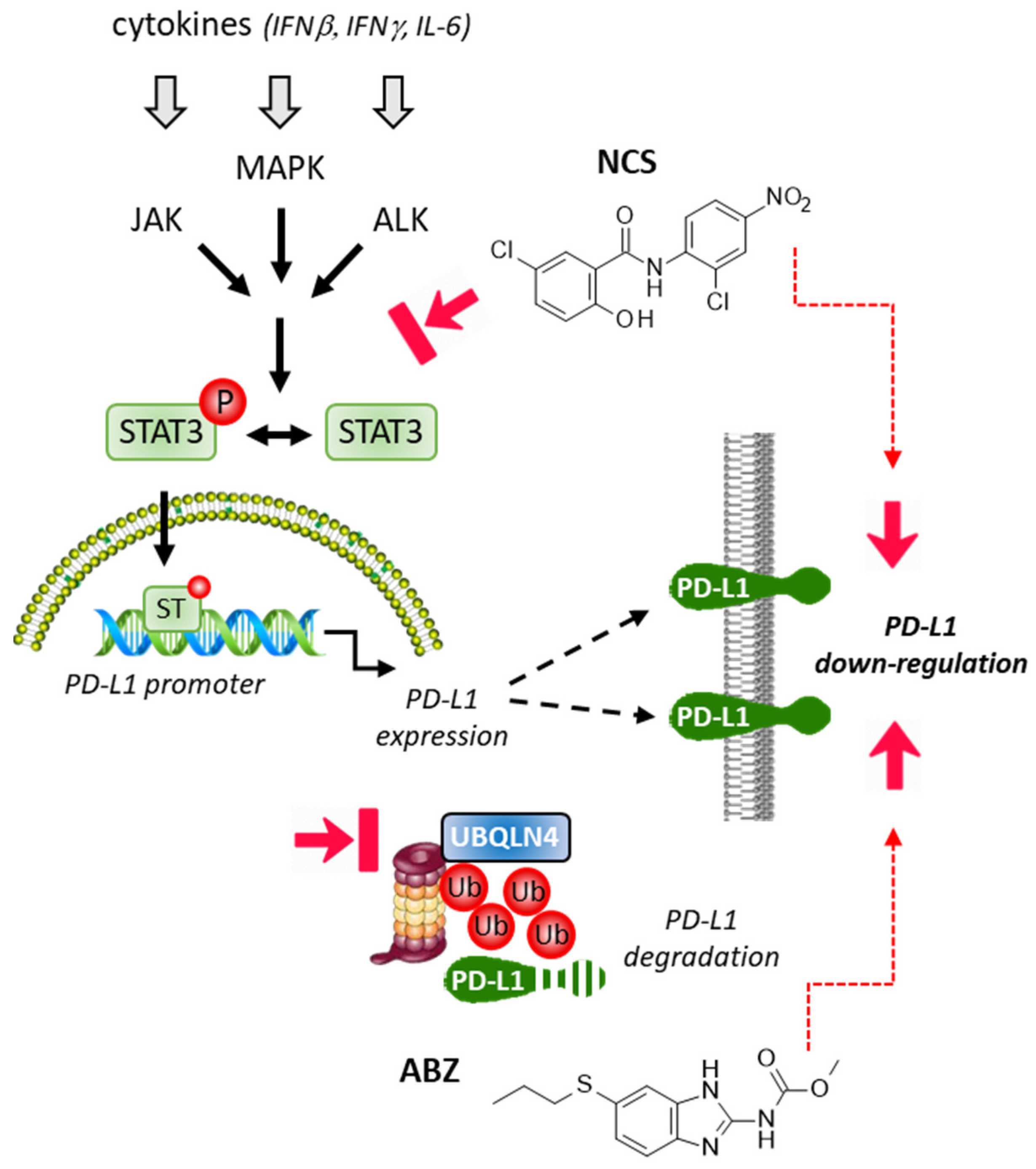
2.3. Repositioning of Niclosamide as a STAT3-Dependent Regulator of the PD-1/PD-L1 Checkpoint
2.4. Albendazole and Flubendazole to Modulate the PD-1/PD-L1 Checkpoint
2.5. Other Repositioning Drug Candidates Affecting the PD-1/PD-L1 Checkpoint
- (i)
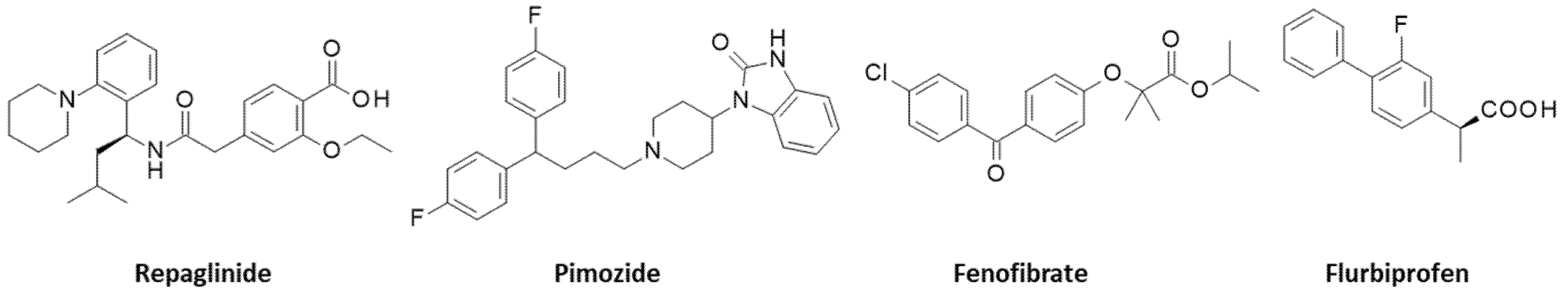
- The antidiabetic drug repaglinide is commonly used to stimulate insulin secretion from pancreatic beta cells. The drug has been shown recently to inhibit the transcription factor FOXO3 and to reduce cancer cell migration [119]. The drug has the capacity to down-regulate PD-L1 expression in glioblastoma cells [120].
- (ii)
- The dopamine antagonist pimozide, used to treat various psychiatric diseases, has been found to display interesting antileukemic and anticancer properties. The drug can reduce activation of STAT3 and STAT5 [121,122,123]. The combination of pimozide and an anti-PD-1 agent profoundly down-regulated expression of phospho-STAT5, leading to a major antitumor response in a model of melanoma. The drug enhanced PD-1 expression, rendering the tumor more sensitive to the anti-PD-1 agent [124].
- (iii)
- A repurposing of the lipid-lowering drug fenofibrate for the treatment of cancer has been proposed [125]. A recent work demonstrates that the drug can reprogram the immune microenvironment in a model of head and neck cancer. The drug was found to down-regulate PD-L1 expression in UMSCC47 cancer cells cultivated under hypoxic conditions (1% O2), probably due to a down-regulation of the upstream target hypoxia-inducible factor-1α (HIF1α) [126]. Fenofibrate has been tested in patients with multiple myeloma (NCT01965834), but not in association with an anti-PD-(L)1 mAb.
- (iv)
- In general, β-blockers are considered not to alter the antitumor response in patients with melanoma treated with anti-PD-1 therapy [127]. It has been reported that the use of β2-adrenergic receptor (βAR) antagonists (β-blockers) can improve overall survival in metastatic melanoma patients who received immunotherapy [128]. The βAR is a regulator of CD8+ T cell frequency and functional orientation within the tumor microenvironment [129]. The repurposing of β-blockers for the treatment of triple-negative breast cancer (TNBC) has been proposed previously, based on the high expression of βAR in TNBC cell lines and the capacity of β-blockers such as propranolol to reduce their proliferation, migration, and invasion [130]. β-blockers do not directly target the PD-1/PD-L1 system, but they exert useful antiangiogenic effects. Recent clinical trials have indicated that a combination of β-blockers with anti-PD-1/PD-L1 mAbs could be useful to improve progression-free survival in patients with NSCLC [131]. The β-blocker propranolol is particularly interesting, as it has been shown not only to upregulate PD-L1 expressed on tumor-associated macrophages (TAM), but also to enhance efficacy of an anti-CTLA4 therapy in soft tissue sarcoma [132]. A recent phase 1 trial in patients with metastatic melanoma has shown that the combination of propranolol and pembrolizumab (anti-PD1) is safe (no dose-limiting toxicity observed) and preliminary signs of activity were reported (increased interferon-γ, decreased interleukin-6) [133].

3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Redondo, A.; Gallego, A.; Mendiola, M. Dostarlimab for the treatment of advanced endometrial cancer. Expert Rev. Clin. Pharmacol. 2022, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sung, E.; Ko, M.; Won, J.Y.; Jo, Y.; Park, E.; Kim, H.; Choi, E.; Jung, U.J.; Jeon, J.; Kim, Y.; et al. LAG-3xPD-L1 bispecific antibody potentiates antitumor responses of T cells through dendritic cell activation. Mol. Ther. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Faroudi, M.; Allen, N.L.; Kmiecik, K.; Gliddon, D.; Seal, C.; Koers, A.; Wydro, M.M.; Batey, S.; Winnewisser, J.; et al. FS118, a bispecific antibody targeting LAG-3 and PD-L1, enhances T-cell activation resulting in potent antitumor activity. Clin. Cancer Res. 2020, 26, 3333–3344. [Google Scholar] [CrossRef]
- You, G.; Won, J.; Lee, Y.; Moon, D.; Park, Y.; Lee, S.H.; Lee, S.W. Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines 2021, 9, 724. [Google Scholar] [CrossRef]
- Al-Showbaki, L.; Nadler, M.B.; Desnoyers, A.; Almugbel, F.A.; Cescon, D.W.; Amir, E. Network Meta-analysis Comparing Efficacy, Safety and Tolerability of Anti-PD-1/PD-L1 Antibodies in Solid Cancers. J. Cancer 2021, 12, 4372–4378. [Google Scholar] [CrossRef]
- Huang, Q.; Zheng, Y.; Gao, Z.; Yuan, L.; Sun, Y.; Chen, H. Comparative Efficacy and Safety of PD-1/PD-L1 Inhibitors for Patients with Solid Tumors: A Systematic Review and Bayesian Network Meta-analysis. J. Cancer 2021, 12, 1133–1143. [Google Scholar] [CrossRef]
- Pandey, P.; Khan, F.; Qari, H.A.; Upadhyay, T.K.; Alkhateeb, A.F.; Oves, M. Revolutionization in Cancer Therapeutics via Targeting Major Immune Checkpoints PD-1, PD-L1 and CTLA-4. Pharmaceuticals 2022, 15, 335. [Google Scholar] [CrossRef]
- Ozer, M.; George, A.; Goksu, S.Y.; George, T.J.; Sahin, I. The Role of Immune Checkpoint Blockade in the Hepatocellular Carcinoma: A Review of Clinical Trials. Front. Oncol. 2021, 11, 801379. [Google Scholar] [CrossRef]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. Current Landscape of Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Cancers 2022, 14, 2018. [Google Scholar] [CrossRef]
- Wong, K.M.; King, G.G.; Harris, W.P. The Treatment Landscape of Advanced Hepatocellular Carcinoma. Curr. Oncol. Rep. 2022, 24, 917–927. [Google Scholar] [CrossRef]
- Taylor, M.H.; Betts, C.B.; Maloney, L.; Nadler, E.; Algazi, A.; Guarino, M.J.; Nemunaitis, J.; Jimeno, A.; Patel, P.; Munugalavadla, V.; et al. Safety and Efficacy of Pembrolizumab in Combination with Acalabrutinib in Advanced Head and Neck Squamous Cell Carcinoma: Phase 2 Proof-of-Concept Study. Clin. Cancer Res. 2022, 28, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Espinoza-Gutarra, M.R.; Bombaci, G.; Farag, S.S.; Schwartz, J.E. Immune-Based Therapeutic Interventions for Acute Myeloid Leukemia. Cancer Treat. Res. 2022, 183, 225–254. [Google Scholar] [PubMed]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 11. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.J.; Bokov, D.; Markov, A.; Jalil, A.T.; Shalaby, M.N.; Suksatan, W.; Chupradit, S.; Al-Ghamdi, H.S.; Shomali, N.; Zamani, A.; et al. Cancer combination therapies by angiogenesis inhibitors; a comprehensive review. Cell. Commun. Signal. 2022, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Germino, E.A.; Govindarajan, A.; Sedrak, M.S.; Li, D.; Amini, A. Multimodality Treatment with Radiotherapy and Immunotherapy in Older Adults: Rationale, Evolving Data, and Current Recommendations. Semin. Radiat. Oncol. 2022, 32, 142–154. [Google Scholar] [CrossRef]
- Zhai, D.; An, D.; Wan, C.; Yang, K. Radiotherapy: Brightness and darkness in the era of immunotherapy. Transl. Oncol. 2022, 19, 101366. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- Wu, R.Y.; Kong, P.F.; Xia, L.P.; Huang, Y.; Li, Z.L.; Tang, Y.Y.; Chen, Y.H.; Li, X.; Senthilkumar, R.; Zhang, H.L.; et al. Regorafenib Promotes Antitumor Immunity via Inhibiting PD-L1 and IDO1 Expression in Melanoma. Clin. Cancer Res. 2019, 25, 4530–4541. [Google Scholar] [CrossRef]
- Zhang, E.L.; Zhang, Z.Y.; Li, J.; Huang, Z.Y. Complete Response to the Sequential Treatment with Regorafenib Followed by PD-1 Inhibitor in a Sorafenib-Refractory Hepatocellular Carcinoma Patient. OncoTargets Ther. 2020, 13, 12477–12487. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Tao, Q.; Zhang, Y.; Yi, F.; Feng, L. Efficacy and Safety of Regorafenib Combined with Toripalimab in the Third-Line and beyond Treatment of Advanced Colorectal Cancer. J. Oncol. 2021, 2021, 9959946. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Maheshwari, P.; Krishnamurthy, P.T.; Patil, V.M. Drug Repurposing Strategies for Non-Cancer to Cancer Therapeutics. Anticancer Agents Med. Chem. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Dang, S. Anti-Cancer Potential of Some Commonly Used Drugs. Curr. Pharm. Des. 2021, 27, 4530–4538. [Google Scholar] [CrossRef]
- Hua, Y.; Dai, X.; Xu, Y.; Xing, G.; Liu, H.; Lu, T.; Chen, Y.; Zhang, Y. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur. J. Med. Chem. 2022, 234, 114239. [Google Scholar] [CrossRef]
- Fu, L.; Jin, W.; Zhang, J.; Zhu, L.; Lu, J.; Zhen, Y.; Zhang, L.; Ouyang, L.; Liu, B.; Yu, H. Repurposing non-oncology small-molecule drugs to improve cancer therapy: Current situation and future directions. Acta Pharm Sin. B 2022, 12, 532–557. [Google Scholar] [CrossRef]
- Li, Y.Q.; Zheng, Z.; Liu, Q.X.; Lu, X.; Zhou, D.; Zhang, J.; Zheng, H.; Dai, J.G. Repositioning of Antiparasitic Drugs for Tumor Treatment. Front. Oncol. 2021, 11, 670804. [Google Scholar] [CrossRef]
- De Lellis, L.; Veschi, S.; Tinari, N.; Mokini, Z.; Carradori, S.; Brocco, D.; Florio, R.; Grassadonia, A.; Cama, A. Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates. Cancers 2021, 13, 3946. [Google Scholar] [CrossRef]
- Ortore, G.; Poli, G.; Martinelli, A.; Tuccinardi, T.; Rizzolio, F.; Caligiuri, I. From Anti-infective Agents to Cancer Therapy: A Drug Repositioning Study Revealed a New Use for Nitrofuran Derivatives. Med. Chem. 2022, 18, 249–259. [Google Scholar] [CrossRef]
- Tharmapoopathy, M.; Thavarajah, A.; Kenny, R.P.; Pingitore, A.; Iervasi, G.; Dark, J.; Bano, A.; Razvi, S. Efficacy and safety of Triiodothyronine (T3) treatment in Cardiac Surgery or Cardiovascular Diseases—A Systematic Review and Meta-analysis of Randomized Controlled Trials. Thyroid 2022. online ahead of print. [Google Scholar] [CrossRef]
- Rubingh, J.; van der Spek, A.; Fliers, E.; Boelen, A. The Role of Thyroid Hormone in the Innate and Adaptive Immune Response during Infection. Compr. Physiol. 2020, 10, 1277–1287. [Google Scholar] [PubMed]
- Deligiorgi, M.V.; Sagredou, S.; Vakkas, L.; Trafalis, D.T. The Continuum of Thyroid Disorders Related to Immune Checkpoint Inhibitors: Still Many Pending Queries. Cancers 2021, 13, 5277. [Google Scholar] [CrossRef] [PubMed]
- Goyal, I.; Pandey, M.R.; Sharma, R.; Chaudhuri, A.; Dandona, P. The side effects of immune checkpoint inhibitor therapy on the endocrine system. Indian J. Med. Res. 2021, 154, 559–570. [Google Scholar] [PubMed]
- Iwama, S.; Kobayashi, T.; Yasuda, Y.; Arima, H. Immune checkpoint inhibitor-related thyroid dysfunction. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 101660, online ahead of print. [Google Scholar] [CrossRef]
- Alamino, V.A.; Montesinos, M.D.M.; Soler, M.F.; Giusiano, L.; Gigena, N.; Fozzatti, L.; Maller, S.M.; Méndez-Huergo, S.P.; Rabinovich, G.A.; Pellizas, C.G. Dendritic Cells Exposed to Triiodothyronine Deliver Pro-Inflammatory Signals and Amplify IL-17-Driven Immune Responses. Cell. Physiol. Biochem. 2019, 52, 354–367. [Google Scholar]
- Vasilopoulou, E.; Loubière, L.S.; Lash, G.E.; Ohizua, O.; McCabe, C.J.; Franklyn, J.A.; Kilby, M.D.; Chan, S.Y. Triiodothyronine regulates angiogenic growth factor and cytokine secretion by isolated human decidual cells in a cell-type specific and gestational age-dependent manner. Hum. Reprod. 2014, 29, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- De Vito, P.; Incerpi, S.; Pedersen, J.Z.; Luly, P.; Davis, F.B.; Davis, P.J. Thyroid hormones as modulators of immune activities at the cellular level. Thyroid 2011, 21, 879–890. [Google Scholar] [CrossRef]
- Montesinos, M.M.; Alamino, V.A.; Mascanfroni, I.D.; Susperreguy, S.; Gigena, N.; Masini-Repiso, A.M.; Rabinovich, G.A.; Pellizas, C.G. Dexamethasone counteracts the immunostimulatory effects of triiodothyronine (T3) on dendritic cells. Steroids 2012, 77, 67–76. [Google Scholar] [CrossRef]
- Alamino, V.A.; Mascanfroni, I.D.; Montesinos, M.M.; Gigena, N.; Donadio, A.C.; Blidner, A.G.; Milotich, S.I.; Cheng, S.Y.; Masini-Repiso, A.M.; Rabinovich, G.A.; et al. Antitumor Responses Stimulated by Dendritic Cells Are Improved by Triiodothyronine Binding to the Thyroid Hormone Receptor β. Cancer Res. 2015, 75, 1265–1274. [Google Scholar] [CrossRef]
- Alamino, V.A.; Montesinos, M.M.; Rabinovich, G.A.; Pellizas, C.G. The thyroid hormone triiodothyronine reinvigorates dendritic cells and potentiates anti-tumor immunity. Oncoimmunology 2015, 5, e1064579. [Google Scholar] [CrossRef][Green Version]
- Pourzardosht, N.; Hashemi, Z.S.; Mard-Soltani, M.; Jahangiri, A.; Rahbar, M.R.; Zakeri, A.; Mirzajani, E.; Khalili, S. Liothyronine could block the programmed death-ligand 1 (PDL1) activity: An e-Pharmacophore modeling and virtual screening study. J. Recept. Signal Transduct. Res. 2022, 42, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Mejías, C.; Guirola, O. Pharmacophore model of immunocheckpoint protein PD-L1 by cosolvent molecular dynamics simulations. J. Mol. Graph. Model. 2019, 91, 105–111. [Google Scholar] [CrossRef] [PubMed]
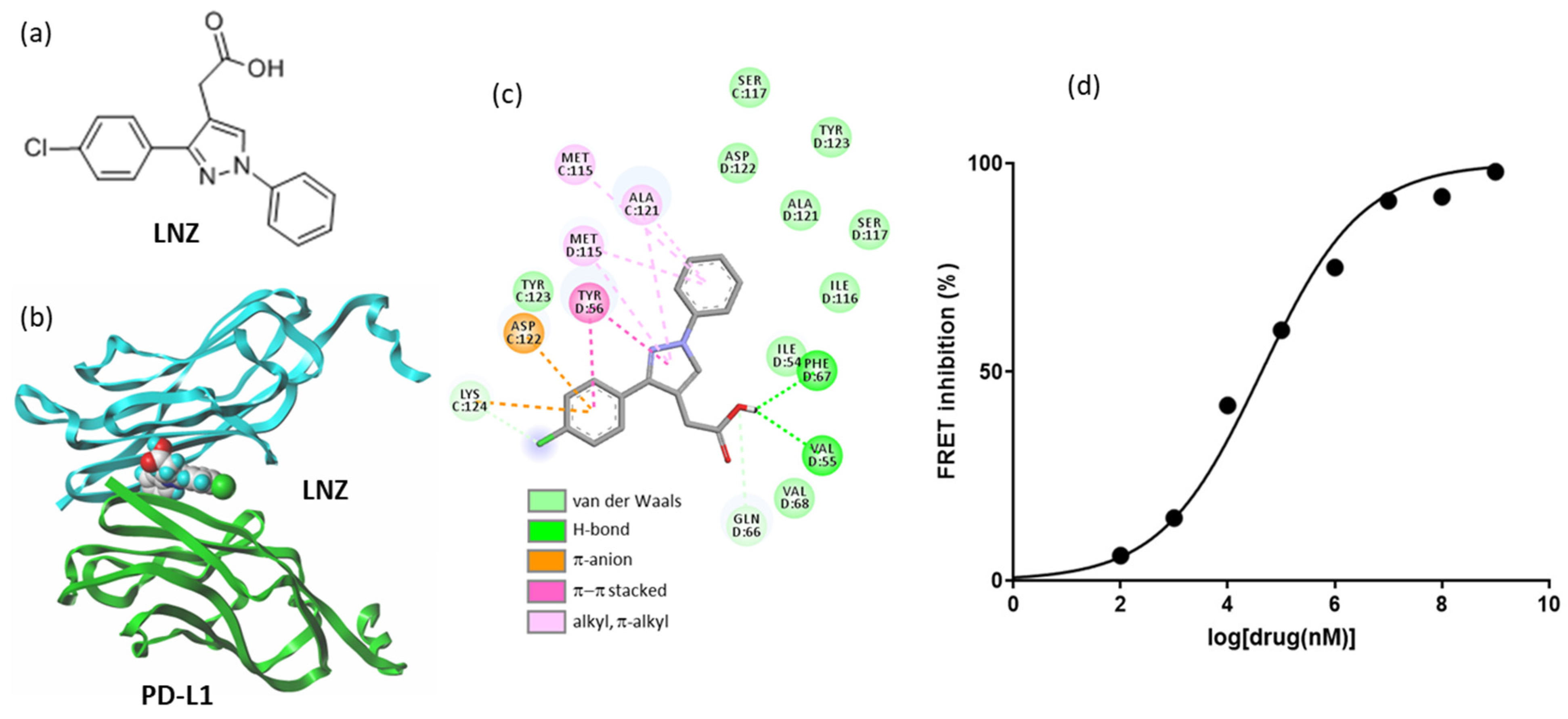
- Bailly, C.; Vergoten, G. Flurbiprofen as a biphenyl scaffold for the design of small molecules binding to PD-L1 protein dimer. Biochem. Pharmacol. 2020, 178, 114042. [Google Scholar] [CrossRef] [PubMed]
- Muir, C.A.; Tsang, V.H.M.; Menzies, A.M.; Clifton-Bligh, R.J. Immune Related Adverse Events of the Thyroid—A Narrative Review. Front. Endocrinol. 2022, 13, 886930. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yao, J.; Yuan, G.; Gao, Y.; Zhang, J.; Guo, X. Immune Checkpoint Inhibitor-related New-onset Thyroid Dysfunction: A Retrospective Analysis Using the US FDA Adverse Event Reporting System. Oncologist 2022, 27, 126–132. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Liu, L.; You, X.; Han, S.; Sun, Y.; Zhang, J.; Zhang, Y. CD155/TIGIT, a novel immune checkpoint in human cancers (Review). Oncol. Rep. 2021, 45, 835–845. [Google Scholar] [CrossRef]
- Ge, Z.; Peppelenbosch, M.P.; Sprengers, D.; Kwekkeboom, J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front. Immunol. 2021, 12, 699895. [Google Scholar] [CrossRef]
- Zhou, X.; Du, J.; Wang, H.; Chen, C.; Jiao, L.; Cheng, X.; Zhou, X.; Chen, S.; Gou, S.; Zhao, W.; et al. Repositioning liothyronine for cancer immunotherapy by blocking the interaction of immune checkpoint TIGIT/PVR. Cell. Commun. Signal. 2020, 18, 142. [Google Scholar] [CrossRef]
- Robles, N.R.; Fici, F.; Grassi, G. Dihydropyridine calcium channel blockers and renal disease. Hypertens. Res. 2017, 40, 21–28. [Google Scholar] [CrossRef]
- Peters, J.; Booth, A.; Peters, R. Potential for specific dihydropyridine calcium channel blockers to have a positive impact on cognitive function in humans: A systematic review. Ther. Adv. Chronic Dis. 2015, 6, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Little, H.J. L-Type Calcium Channel Blockers: A Potential Novel Therapeutic Approach to Drug Dependence. Pharmacol. Rev. 2021, 73, 127–154. [Google Scholar] [CrossRef]
- Principe, D.R.; Aissa, A.F.; Kumar, S.; Pham, T.N.D.; Underwood, P.W.; Nair, R.; Ke, R.; Rana, B.; Trevino, J.G.; Munshi, H.G.; et al. Calcium channel blockers potentiate gemcitabine chemotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2200143119. [Google Scholar] [CrossRef] [PubMed]
- Alandağ, C.; Karaman, E.; Yüce, E. Amlodipine improves the outcomes of regorafenib in metastatic colorectal cancer. Anticancer Drugs 2022, 33, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Saito, T.; Tojo, K. Efonidipine, a Ca2+-channel blocker, enhances the production of dehydroepiandrosterone sulfate in NCI-H295R human adrenocortical carcinoma cells. Tohoku J. Exp. Med. 2011, 224, 263–271. [Google Scholar] [CrossRef][Green Version]
- Shaughnessy, M.; Lamuraglia, G.; Klebanov, N.; Ji, Z.; Rajadurai, A.; Kumar, R.; Flaherty, K.; Tsao, H. Selective uveal melanoma inhibition with calcium channel blockade. Int. J. Oncol. 2019, 55, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Taghizadehghalehjoughi, A.; Sezen, S.; Hacimuftuoglu, A.; Güllüce, M. Vincristine combination with Ca+2 channel blocker increases antitumor effects. Mol. Biol. Rep. 2019, 46, 2523–2528. [Google Scholar] [CrossRef]
- Panneerpandian, P.; Rao, D.B.; Ganesan, K. Calcium channel blockers lercanidipine and amlodipine inhibit YY1/ERK/TGF-β mediated transcription and sensitize the gastric cancer cells to doxorubicin. Toxicol. In Vitro 2021, 74, 105152. [Google Scholar] [CrossRef]
- Shiozaki, A.; Katsurahara, K.; Kudou, M.; Shimizu, H.; Kosuga, T.; Ito, H.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; et al. Amlodipine and Verapamil, Voltage-Gated Ca2+ Channel Inhibitors, Suppressed the Growth of Gastric Cancer Stem Cells. Ann. Surg. Oncol. 2021, 28, 5400–5411. [Google Scholar] [CrossRef]
- Mohapatra, P.K.; Srivastava, R.; Varshney, K.K.; Babu, S.H. Formulation and Evaluation of Isradipine Nanosuspension and Exploring its Role as a Potential Anticancer Drug by Computational Approach. Anticancer Agents Med. Chem. 2022, 22, 1984–2001. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.W.; Kim, D.K.; Choi, D.K.; Lee, S.; Yu, J.H.; Kwon, O.B.; Lee, J.; Lee, D.S.; Kim, J.H.; et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 2327. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, J.W.; Lee, D.S.; Min, S.H. Combined Poziotinib with Manidipine Treatment Suppresses Ovarian Cancer Stem-Cell Proliferation and Stemness. Int. J. Mol. Sci. 2020, 21, 7379. [Google Scholar] [CrossRef]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, R.; Guo, H.; Zhang, W.; Xu, X.; Chen, X.; Ding, L. Dihydropyridine Calcium Channel Blockers Suppress the Transcription of PD-L1 by Inhibiting the Activation of STAT1. Front. Pharmacol. 2021, 11, 539261. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yao, H.; Wang, H.; Fang, J.Y.; Xu, J. Repurposing screen identifies Amlodipine as an inducer of PD-L1 degradation and antitumor immunity. Oncogene 2021, 40, 1128–1146. [Google Scholar] [CrossRef]
- Wu, L.; Lin, W.; Liao, Q.; Wang, H.; Lin, C.; Tang, L.; Lian, W.; Chen, Z.; Li, K.; Xu, L.; et al. Calcium Channel Blocker Nifedipine Suppresses Colorectal Cancer Progression and Immune Escape by Preventing NFAT2 Nuclear Translocation. Cell. Rep. 2020, 33, 108327. [Google Scholar] [CrossRef]
- Segovia, M.; Russo, S.; Jeldres, M.; Mahmoud, Y.D.; Perez, V.; Duhalde, M.; Charnet, P.; Rousset, M.; Victoria, S.; Veigas, F.; et al. Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell 2019, 35, 767–781. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Murata, T.; Ramos, J.W. The calcium channel agonist Bay K 8644 promotes the growth of human liver cancer HepG2 cells in vitro: Suppression with overexpressed regucalcin. Mol. Cell Biochem. 2020, 472, 173–185. [Google Scholar] [CrossRef]
- Wu, L.; Wang, R.; Karpinski, E.; Pang, P.K. Bay K-8644 in different solvents acts as a transient calcium channel antagonist and a long-lasting calcium channel agonist. J. Pharmacol. Exp. Ther. 1992, 260, 966–973. [Google Scholar]
- Zhou, X.; Jiao, L.; Qian, Y.; Dong, Q.; Sun, Y.; Zheng, W.V.; Zhao, W.; Zhai, W.; Qiu, L.; Wu, Y.; et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules 2021, 11, 706. [Google Scholar] [CrossRef]
- Liu, H.; Xiang, Y.; Zong, Q.B.; Dai, Z.T.; Wu, H.; Zhang, H.M.; Huang, Y.; Shen, C.; Wang, J.; Lu, Z.X.; et al. TDO2 modulates liver cancer cell migration and invasion via the Wnt5a pathway. Int. J. Oncol. 2022, 60, 72. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Luo, M.; Lu, Y.; Pan, X.; Chen, X.; Ding, L.; Che, J.; He, Q.; Dong, X. Design, synthesis and biological evaluation of new dihydropyridine derivatives as PD-L1 degraders for enhancing antitumor immunity. Bioorg. Chem. 2022, 125, 105820. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Niyomdecha, N.; Suptawiwat, O.; Boonarkart, C.; Thitithanyanont, A.; Auewarakul, P. Repurposing of antiparasitic niclosamide to inhibit respiratory syncytial virus (RSV) replication. Virus Res. 2021, 295, 198277. [Google Scholar] [CrossRef]
- Singh, S.; Weiss, A.; Goodman, J.; Fisk, M.; Kulkarni, S.; Lu, I.; Gray, J.; Smith, R.; Sommer, M.; Cheriyan, J. Niclosamide-A promising treatment for COVID-19. Br. J. Pharmacol. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Cairns, D.M.; Dulko, D.; Griffiths, J.K.; Golan, Y.; Cohen, T.; Trinquart, L.; Price, L.L.; Beaulac, K.R.; Selker, H.P. Efficacy of Niclosamide vs Placebo in SARS-CoV-2 Respiratory Viral Clearance, Viral Shedding, and Duration of Symptoms Among Patients With Mild to Moderate COVID-19: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open. 2022, 5, 2144942. [Google Scholar] [CrossRef]
- Abdulamir, A.S.; Gorial, F.I.; Saadi, S.J.; Maulood, M.F.; Hashim, H.A.; Alnuaimi, A.S.; Abdulrrazaq, M.K. A randomised controlled trial of effectiveness and safety of Niclosamide as add on therapy to the standard of care measures in COVID-19 management. Ann. Med. Surg. 2021, 69, 102779. [Google Scholar] [CrossRef]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef]
- Newton, P.T. New insights into niclosamide action: Autophagy activation in colorectal cancer. Biochem J. 2019, 476, 779–781. [Google Scholar] [CrossRef]
- Wang, L.H.; Xu, M.; Fu, L.Q.; Chen, X.Y.; Yang, F. The Antihelminthic Niclosamide Inhibits Cancer Stemness, Extracellular Matrix Remodeling, and Metastasis through Dysregulation of the Nuclear β-catenin/c-Myc axis in OSCC. Sci. Rep. 2018, 8, 12776. [Google Scholar] [CrossRef]
- Kaushal, J.B.; Bhatia, R.; Kanchan, R.K.; Raut, P.; Mallapragada, S.; Ly, Q.P.; Batra, S.K.; Rachagani, S. Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers 2021, 13, 3105, Correction in Cancers 2021, 13, 5591. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhu, H.; Xiao, Y.; Guo, H.; Lin, M.; Yuan, Z.; Yang, X.; Huang, Y.; Zhang, Q.; Bai, Y. The anthelmintic drug niclosamide induces GSK-β-mediated β-catenin degradation to potentiate gemcitabine activity, reduce immune evasion ability and suppress pancreatic cancer progression. Cell Death Dis. 2022, 13, 112, Erratum in Cell Death Dis. 2022, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.C.; Shim, J.K.; Park, J.; Lee, J.H.; Choi, R.J.; Kim, N.H.; Kim, H.S.; Moon, J.H.; Kim, E.H.; Chang, J.H.; et al. Combined effects of niclosamide and temozolomide against human glioblastoma tumorspheres. J. Cancer Res. Clin. Oncol. 2020, 146, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- Valdez, L.; Cheng, B.; Gonzalez, D.; Rodriguez, R.; Campano, P.; Tsin, A.; Fang, X. Combined treatment with niclosamide and camptothecin enhances anticancer effect in U87 MG human glioblastoma cells. Oncotarget 2022, 13, 642–658. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, C.; Fu, Q.; Lv, H. Combined chemotherapy for triple negative breast cancer treatment by paclitaxel and niclosamide nanocrystals loaded thermosensitive hydrogel. Eur. J. Pharm. Sci. 2021, 167, 105992. [Google Scholar] [CrossRef]
- Lohiya, G.; Katti, D.S. Mesoporous Silica Nanoparticle-Based Combination of Niclosamide and Doxorubicin: Effect of Treatment Regimens on Breast Cancer Subtypes. ACS Appl. Bio Mater. 2021, 4, 7811–7824. [Google Scholar] [CrossRef]
- Parikh, M.; Liu, C.; Wu, C.Y.; Evans, C.P.; Dall’Era, M.; Robles, D.; Lara, P.N.; Agarwal, N.; Gao, A.C.; Pan, C.X. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 6377. [Google Scholar] [CrossRef]
- Yeh, L.T.; Lin, C.W.; Lu, K.H.; Hsieh, Y.H.; Yeh, C.B.; Yang, S.F.; Yang, J.S. Niclosamide Suppresses Migration and Invasion of Human Osteosarcoma Cells by Repressing TGFBI Expression via the ERK Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 484. [Google Scholar] [CrossRef]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of Niclosamide as a Novel Anticancer Agent for Adrenocortical Carcinoma. Clin. Cancer Res. 2016, 22, 3458–3466. [Google Scholar] [CrossRef]
- Wu, M.M.; Zhang, Z.; Tong, C.W.S.; Yan, V.W.; Cho, W.C.S.; To, K.K.W. Repurposing of niclosamide as a STAT3 inhibitor to enhance the anticancer effect of chemotherapeutic drugs in treating colorectal cancer. Life Sci. 2020, 262, 118522. [Google Scholar] [CrossRef]
- Zhang, X.H.; Hsiang, J.; Rosen, S.T. Flavopiridol (Alvocidib), a Cyclin-dependent Kinases (CDKs) Inhibitor, Found Synergy Effects with Niclosamide in Cutaneous T-cell Lymphoma. J. Clin. Haematol. 2021, 2, 48–61. [Google Scholar] [PubMed]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Hu, R.; Wang, Q.; Xu, K.; Yang, H.; Cui, Z.; Zhang, Y.; Teng, M.; Xia, L.; Chen, J.; et al. Interleukin-22 promotes PD-L1 expression via STAT3 in colon cancer cells. Oncol. Lett. 2021, 22, 716. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Lee, J.M.; Jang, K.J. Mechanistic Insights of Anti-Immune Evasion by Nobiletin through Regulating miR-197/STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer (NSCLC) Cells. Int. J. Mol. Sci. 2021, 22, 9843. [Google Scholar] [CrossRef] [PubMed]
- Wondergem, N.E.; Nijenhuis, D.N.L.M.; Poell, J.B.; Leemans, C.R.; Brakenhoff, R.H.; van de Ven, R. At the Crossroads of Molecular Biology and Immunology: Molecular Pathways for Immunological Targeting of Head and Neck Squamous Cell Carcinoma. Front. Oral Health 2021, 2, 647980. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, Y.; Jiang, S.; Dang, Y.; Yang, Y.; Zuo, W.; Zhu, Q.; Liu, P.; Gao, Y.; Lu, S. Traditional Chinese medicine CFF-1 exerts a potent anti-tumor immunity to hinder tumor growth and metastasis in prostate cancer through EGFR/JAK1/STAT3 pathway to inhibit PD-1/PD-L1 checkpoint signaling. Phytomedicine 2022, 99, 153939. [Google Scholar] [CrossRef]
- Tu, J.; Xu, H.; Ma, L.; Li, C.; Qin, W.; Chen, X.; Yi, M.; Sun, L.; Liu, B.; Yuan, X. Nintedanib enhances the efficacy of PD-L1 blockade by upregulating MHC-I and PD-L1 expression in tumor cells. Theranostics 2022, 12, 747–766. [Google Scholar] [CrossRef]
- Venkatraman, S.; Meller, J.; Hongeng, S.; Tohtong, R.; Chutipongtanate, S. Transcriptional Regulation of Cancer Immune Checkpoints: Emerging Strategies for Immunotherapy. Vaccines 2020, 8, 735. [Google Scholar] [CrossRef]
- Lodagekar, A.; Borkar, R.M.; Thatikonda, S.; Chavan, R.B.; Naidu, V.G.M.; Shastri, N.R.; Srinivas, R.; Chella, N. Formulation and evaluation of cyclodextrin complexes for improved anticancer activity of repurposed drug: Niclosamide. Carbohydr. Polym. 2019, 212, 252–259. [Google Scholar] [CrossRef]
- Lohiya, G.; Katti, D.S. A Synergistic Combination of Niclosamide and Doxorubicin as an Efficacious Therapy for All Clinical Subtypes of Breast Cancer. Cancers 2021, 13, 3299. [Google Scholar] [CrossRef]
- Al Amin, A.S.M.; Wadhwa, R. Helminthiasis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Thakare, R.; Dasgupta, A.; Chopra, S. Triclabendazole for the treatment of fascioliasis. Drugs Today 2019, 55, 743–752. [Google Scholar] [CrossRef] [PubMed]
- de Moraes, J.; Geary, T.G. FDA-Approved Antiparasitic Drugs in the 21st Century: A Success for Helminthiasis? Trends Parasitol. 2020, 36, 573–575. [Google Scholar] [CrossRef] [PubMed]
- Dokla, E.M.E.; Abdel-Aziz, A.K.; Milik, S.N.; McPhillie, M.J.; Minucci, S.; Abouzid, K.A.M. Discovery of a benzimidazole-based dual FLT3/TrKA inhibitor targeting acute myeloid leukemia. Bioorg. Med. Chem. 2022, 56, 116596. [Google Scholar] [CrossRef]
- Son, D.S.; Lee, E.S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Sultana, T.; Jan, U.; Lee, J.I. Double Repositioning: Veterinary Antiparasitic to Human Anticancer. Int. J. Mol. Sci. 2022, 23, 4315. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M. Emerging insights on functions of the anthelmintic flubendazole as a repurposed anticancer agent. Cancer Lett. 2021, 522, 57–62. [Google Scholar] [CrossRef]
- Chen, C.; Ding, Y.; Liu, H.; Sun, M.; Wang, H.; Wu, D. Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers. Int. J. Mol. Sci. 2022, 23, 519. [Google Scholar] [CrossRef]
- Nath, J.; Paul, R.; Ghosh, S.K.; Paul, J.; Singha, B.; Debnath, N. Drug repurposing and relabeling for cancer therapy: Emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 2020, 258, 118189. [Google Scholar] [CrossRef]
- Li, Y.; Acharya, G.; Elahy, M.; Xin, H.; Khachigian, L.M. The anthelmintic flubendazole blocks human melanoma growth and metastasis and suppresses programmed cell death protein-1 and myeloid-derived suppressor cell accumulation. Cancer Lett. 2019, 459, 268–276. [Google Scholar] [CrossRef]
- Lin, S.; Yang, L.; Yao, Y.; Xu, L.; Xiang, Y.; Zhao, H.; Wang, L.; Zuo, Z.; Huang, X.; Zhao, C. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J. Exp. Clin. Cancer Res. 2019, 38, 293. [Google Scholar] [CrossRef]
- Zhu, L.; Kuang, X.; Zhang, G.; Liang, L.; Liu, D.; Hu, B.; Xie, Z.; Li, H.; Liu, H.; Ye, M.; et al. Albendazole induces immunotherapy response by facilitating ubiquitin-mediated PD-L1 degradation. J. Immunother. Cancer 2022, 10, 3819. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jebbawi, F.; Bellanger, A.P.; Beldi, G.; Millon, L.; Gottstein, B. Immunotherapy of alveolar echinococcosis via PD-1/PD-L1 immune checkpoint blockade in mice. Parasite Immunol. 2018, 40, 12596. [Google Scholar] [CrossRef]
- Jebbawi, F.; Bellanger, A.P.; Lunström-Stadelmann, B.; Rufener, R.; Dosch, M.; Goepfert, C.; Gottstein, B.; Millon, L.; Grandgirard, D.; Leib, S.L.; et al. Innate and adaptive immune responses following PD-L1 blockade in treating chronic murine alveolar echinococcosis. Parasite Immunol. 2021, 43, 12834. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ding, J.; Cui, X.; Wu, M.; Huang, C.; Zhang, R.; Wang, J.; Li, X.; Cen, S.; Zhou, J. Screening of kinase inhibitors downregulating PD-L1 expression via on/in cell quantitative immunoblots. Eur. J. Pharm. Sci. 2020, 142, 105088. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Matsushima, T.; Kurimoto, R.; Chiba, T.; Inutani, Y.; Asahara, H. Identification of chemical compounds regulating PD-L1 by introducing HiBiT-tagged cells. FEBS Lett. 2021, 595, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yuan, D.; Ma, J. Advances of biphenyl small-molecule inhibitors targeting PD-1/PD-L1 interaction in cancer immunotherapy. Future Med. Chem. 2022, 14, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Xiao, Y.; Xue, M.; Cao, H.; Chen, J. Recent Advances in the Development of PD-L1 Modulators: Degraders, Downregulators, and Covalent Inhibitors. J. Med. Chem. 2020, 63, 15389–15398. [Google Scholar] [CrossRef] [PubMed]
- Salcher, S.; Spoden, G.; Huber, J.M.; Golderer, G.; Lindner, H.; Ausserlechner, M.J.; Kiechl-Kohlendorfer, U.; Geiger, K.; Obexer, P. Repaglinide Silences the FOXO3/Lumican Axis and Represses the Associated Metastatic Potential of Neuronal Cancer Cells. Cells 2019, 9, 1. [Google Scholar] [CrossRef]
- Xiao, Z.X.; Chen, R.Q.; Hu, D.X.; Xie, X.Q.; Yu, S.B.; Chen, X.Q. Identification of repaglinide as a therapeutic drug for glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 488, 33–39. [Google Scholar] [CrossRef]
- Ranjan, A.; Kaushik, I.; Srivastava, S.K. Pimozide Suppresses the Growth of Brain Tumors by Targeting STAT3-Mediated Autophagy. Cells 2020, 9, 2141. [Google Scholar] [CrossRef]
- Subramaniam, D.; Angulo, P.; Ponnurangam, S.; Dandawate, P.; Ramamoorthy, P.; Srinivasan, P.; Iwakuma, T.; Weir, S.J.; Chastain, K.; Anant, S. Suppressing STAT5 signaling affects osteosarcoma growth and stemness. Cell Death Dis. 2020, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Pontiggia, L.; Jasmin, J.F.; Mercier, I. Phosphorylated STAT3 (Tyr705) as a biomarker of response to pimozide treatment in triple-negative breast cancer. Cancer Biol. Ther. 2020, 21, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wei, T.; Guo, J.; Wang, Y.; Shi, X.; Guo, S.; Jia, X.; Jia, H.; Feng, Z. PD-1-siRNA delivered by attenuated Salmonella enhances the antimelanoma effect of pimozide. Cell Death Dis. 2019, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Wang, G.; Zhou, H.; Zheng, Z.; Fu, Y.; Cai, L. Anticancer Properties of Fenofibrate: A Repurposing Use. J. Cancer 2018, 9, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, W.Q.; Xie, X.; Gui, S.; Yu, H.; Davenport, J.; Cartwright, T.; Storl-Desmond, M.; Ryu, E.; Chan, E.R.; Cao, S.; et al. Repositioning Fenofibrate to Reactivate p53 and Reprogram the Tumor-Immune Microenvironment in HPV+ Head and Neck Squamous Cell Carcinoma. Cancers 2022, 14, 282. [Google Scholar] [CrossRef]
- Wang, D.Y.; McQuade, J.L.; Rai, R.R.; Park, J.J.; Zhao, S.; Ye, F.; Beckermann, K.E.; Rubinstein, S.M.; Johnpulle, R.; Long, G.V.; et al. The Impact of Nonsteroidal Anti-Inflammatory Drugs, Beta Blockers, and Metformin on the Efficacy of Anti-PD-1 Therapy in Advanced Melanoma. Oncologist 2020, 25, 602–605. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2017, 7, e1405205. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.; Messmer, M.N.; et al. β-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8+ T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef]
- Spini, A.; Roberto, G.; Gini, R.; Bartolini, C.; Bazzani, L.; Donnini, S.; Crispino, S.; Ziche, M. Evidence of β-blockers drug repurposing for the treatment of triple negative breast cancer: A systematic review. Neoplasma 2019, 66, 963–970. [Google Scholar] [CrossRef]
- Oh, M.S.; Guzner, A.; Wainwright, D.A.; Mohindra, N.A.; Chae, Y.K.; Behdad, A.; Villaflor, V.M. The Impact of Beta Blockers on Survival Outcomes in Patients With Non-small-cell Lung Cancer Treated with Immune Checkpoint Inhibitors. Clin. Lung Cancer 2021, 22, 57–62. [Google Scholar] [CrossRef]
- Fjæstad, K.Y.; Rømer, A.M.A.; Goitea, V.; Johansen, A.Z.; Thorseth, M.L.; Carretta, M.; Engelholm, L.H.; Grøntved, L.; Junker, N.; Madsen, D.H. Blockade of beta-adrenergic receptors reduces cancer growth and enhances the response to anti-CTLA4 therapy by modulating the tumor microenvironment. Oncogene 2022, 41, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Pandey, M.R.; Attwood, K.; Ji, W.; Witkiewicz, A.K.; Knudsen, E.S.; Allen, C.; Tario, J.D.; Wallace, P.K.; Cedeno, C.D.; et al. Phase I Clinical Trial of Combination Propranolol and Pembrolizumab in Locally Advanced and Metastatic Melanoma: Safety, Tolerability, and Preliminary Evidence of Antitumor Activity. Clin. Cancer Res. 2021, 27, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Garbán, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in combination with immune checkpoint inhibitors in breast cancer immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Adib, E.; Weise, N.; Curran, C.; Stewart, T.; Freeman, D.; Nassar, A.H.; Abou Alaiwi, S.; Bakouny, Z.; McGregor, B.A.; et al. Impact of renin-angiotensin system inhibitors on outcomes in patients with metastatic renal cell carcinoma treated with immune-checkpoint inhibitors. Clin. Genitourin Cancer 2022, S1558–S7673, online ahead of print. [Google Scholar] [CrossRef]
- Son, S.; Shin, J.M.; Shin, S.; Kim, C.H.; Lee, J.A.; Ko, H.; Lee, E.S.; Jung, J.M.; Kim, J.; Park, J.H. Repurposing macitentan with nanoparticle modulates tumor microenvironment to potentiate immune checkpoint blockade. Biomaterials 2021, 276, 121058. [Google Scholar] [CrossRef]
- Xia, Y.; Jia, C.; Xue, Q.; Jiang, J.; Xie, Y.; Wang, R.; Ran, Z.; Xu, F.; Zhang, Y.; Ye, T. Antipsychotic Drug Trifluoperazine Suppresses Colorectal Cancer by Inducing G0/G1 Arrest and Apoptosis. Front. Pharmacol. 2019, 10, 1029. [Google Scholar] [CrossRef]
- Hu, J.C.; Chai, X.Q.; Wang, D.; Shu, S.H.; Magnussen, C.G.; Xie, L.X.; Hu, S.S. Intraoperative Flurbiprofen Treatment Alters Immune Checkpoint Expression in Patients Undergoing Elective Thoracoscopic Resection of Lung Cancer. Med. Princ. Pract. 2020, 29, 150–159. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Fadaly, W.A.A.; Elshaier, Y.A.M.M.; Ali, W.A.M.; Kamel, G.M. Non-acidic 1,3,4-trisubstituted-pyrazole derivatives as lonazolac analogs with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2018, 77, 568–578. [Google Scholar] [CrossRef]
- Harras, M.F.; Sabour, R.; Alkamali, O.M. Discovery of new non-acidic lonazolac analogues with COX-2 selectivity as potent anti-inflammatory agents. Medchemcomm 2019, 10, 1775–1788. [Google Scholar] [CrossRef]
- Mamytbeková, A.; Hájícek, J.; Grimová, J.; Rezábek, K. Reductive effect of lonazolac on lung metastasis formation in mice. Neoplasma 1990, 37, 349–355. [Google Scholar]
- Le Biannic, R.; Magnez, R.; Klupsch, F.; Leleu-Chavain, N.; Thiroux, B.; Tardy, M.; El Bouazzati, H.; Dezitter, X.; Renault, N.; Vergoten, G.; et al. Pyrazolones as inhibitors of immune checkpoint blocking the PD-1/PD-L1 interaction. Eur. J. Med. Chem. 2022, 236, 114343. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tian, H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target 2019, 27, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Zavareh, R.B.; Spangenberg, S.H.; Woods, A.; Martínez-Peña, F.; Lairson, L.L. HSP90 Inhibition Enhances Cancer Immunotherapy by Modulating the Surface Expression of Multiple Immune Checkpoint Proteins. Cell Chem. Biol. 2021, 28, 158–168. [Google Scholar] [CrossRef]
- Delmas, D.; Hermetet, F.; Aires, V. PD-1/PD-L1 Checkpoints and Resveratrol: A Controversial New Way for a Therapeutic Strategy. Cancers 2021, 13, 4509. [Google Scholar] [CrossRef]
- Dong, S.; Guo, X.; Han, F.; He, Z.; Wang, Y. Emerging role of natural products in cancer immunotherapy. Acta Pharm. Sin. B 2022, 12, 1163–1185. [Google Scholar] [CrossRef]
- Lim, S.; Park, J.; Shim, M.K.; Um, W.; Yoon, H.Y.; Ryu, J.H.; Lim, D.K.; Kim, K. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics 2019, 9, 7906–7923. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Kozik, P.; Gros, M.; Itzhak, D.N.; Joannas, L.; Heurtebise-Chrétien, S.; Krawczyk, P.A.; Rodríguez-Silvestre, P.; Alloatti, A.; Magalhaes, J.G.; Del Nery, E.; et al. Small Molecule Enhancers of Endosome-to-Cytosol Import Augment Anti-tumor Immunity. Cell Rep. 2020, 32, 107905. [Google Scholar] [CrossRef]
- Zhou, J.; Karshalev, E.; Mundaca-Uribe, R.; Esteban-Fernández de Ávila, B.; Krishnan, N.; Xiao, C.; Ventura, C.J.; Gong, H.; Zhang, Q.; Gao, W.; et al. Physical Disruption of Solid Tumors by Immunostimulatory Microrobots Enhances Antitumor Immunity. Adv. Mater. 2021, 33, 2103505. [Google Scholar] [CrossRef]
- Kannan, S.; O’Connor, G.M.; Bakker, E.Y. Molecular Mechanisms of PD-1 and PD-L1 Activity on a Pan-Cancer Basis: A Bioinformatic Exploratory Study. Int. J. Mol. Sci. 2021, 22, 5478. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Scapozza, L.; Ruiz i Altaba, A. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H. Repurposing approved drugs for cancer therapy. Br. Med. Bull. 2021, 137, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Mottini, C.; Napolitano, F.; Li, Z.; Gao, X.; Cardone, L. Computer-aided drug repurposing for cancer therapy: Approaches and opportunities to challenge anticancer targets. Semin. Cancer Biol. 2021, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2021, 68, 258–278. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Demus, T.; Moubarak, M.M.; Daher, D.; Alvarez Moreno, J.C.; Polit, F.; Lopez, O.; Merhe, A.; Abou-Kheir, W.; Nieder, A.M.; et al. Overcoming Drug Resistance in Advanced Prostate Cancer by Drug Repurposing. Med. Sci. 2022, 10, 15. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thuru, X.; Magnez, R.; El-Bouazzati, H.; Vergoten, G.; Quesnel, B.; Bailly, C. Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors. Cancers 2022, 14, 3368. https://doi.org/10.3390/cancers14143368
Thuru X, Magnez R, El-Bouazzati H, Vergoten G, Quesnel B, Bailly C. Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors. Cancers. 2022; 14(14):3368. https://doi.org/10.3390/cancers14143368
Chicago/Turabian StyleThuru, Xavier, Romain Magnez, Hassiba El-Bouazzati, Gérard Vergoten, Bruno Quesnel, and Christian Bailly. 2022. "Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors" Cancers 14, no. 14: 3368. https://doi.org/10.3390/cancers14143368
APA StyleThuru, X., Magnez, R., El-Bouazzati, H., Vergoten, G., Quesnel, B., & Bailly, C. (2022). Drug Repurposing to Enhance Antitumor Response to PD-1/PD-L1 Immune Checkpoint Inhibitors. Cancers, 14(14), 3368. https://doi.org/10.3390/cancers14143368