
Targeting the CD47-SIRPα Innate Immune Checkpoint to Potentiate Antibody Therapy in Cancer by Neutrophils

Abstract
Simple Summary
Abstract
1. Introduction
2. Neutrophils and Their Role in Cancer
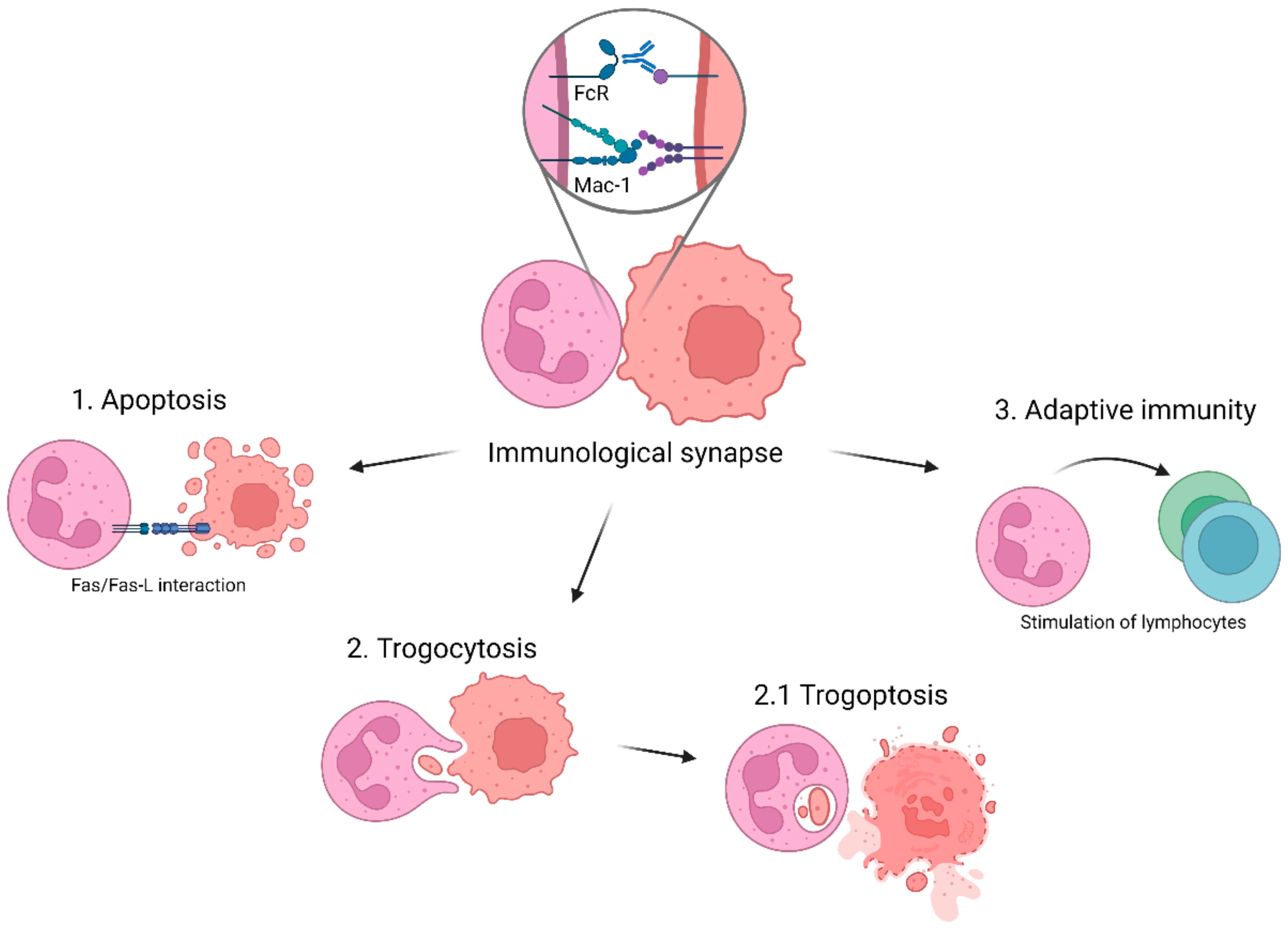
3. Cytotoxic Effector Functions of Neutrophils
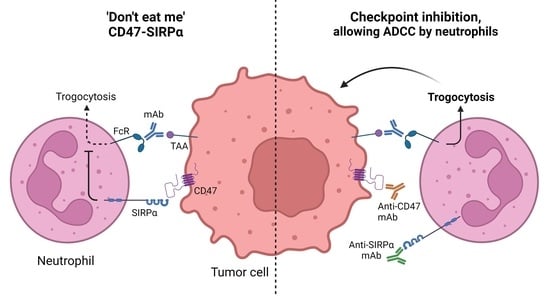
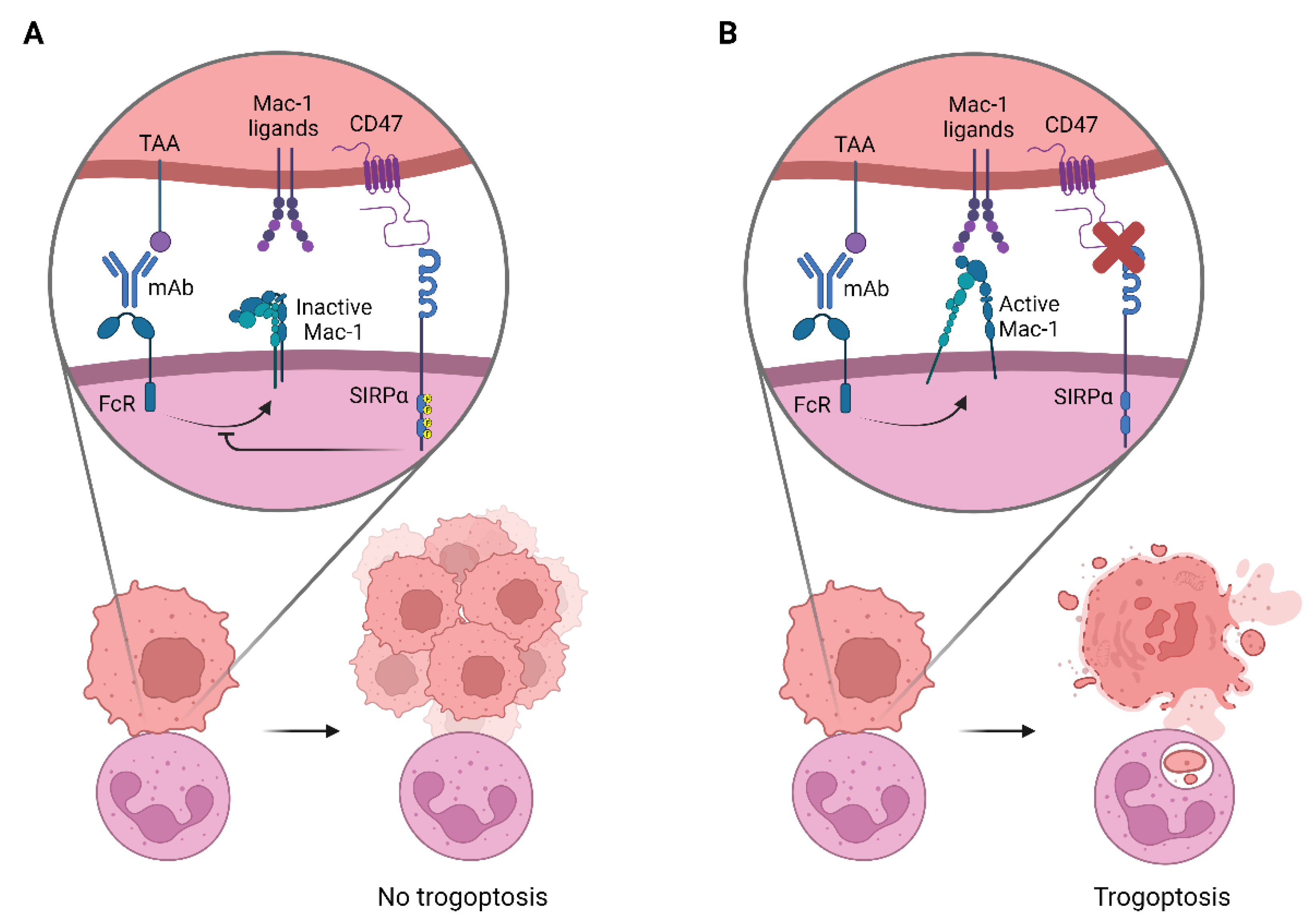
4. CD47-SIRPα as an Innate Immune Checkpoint in Neutrophil-Mediated Tumor Killing
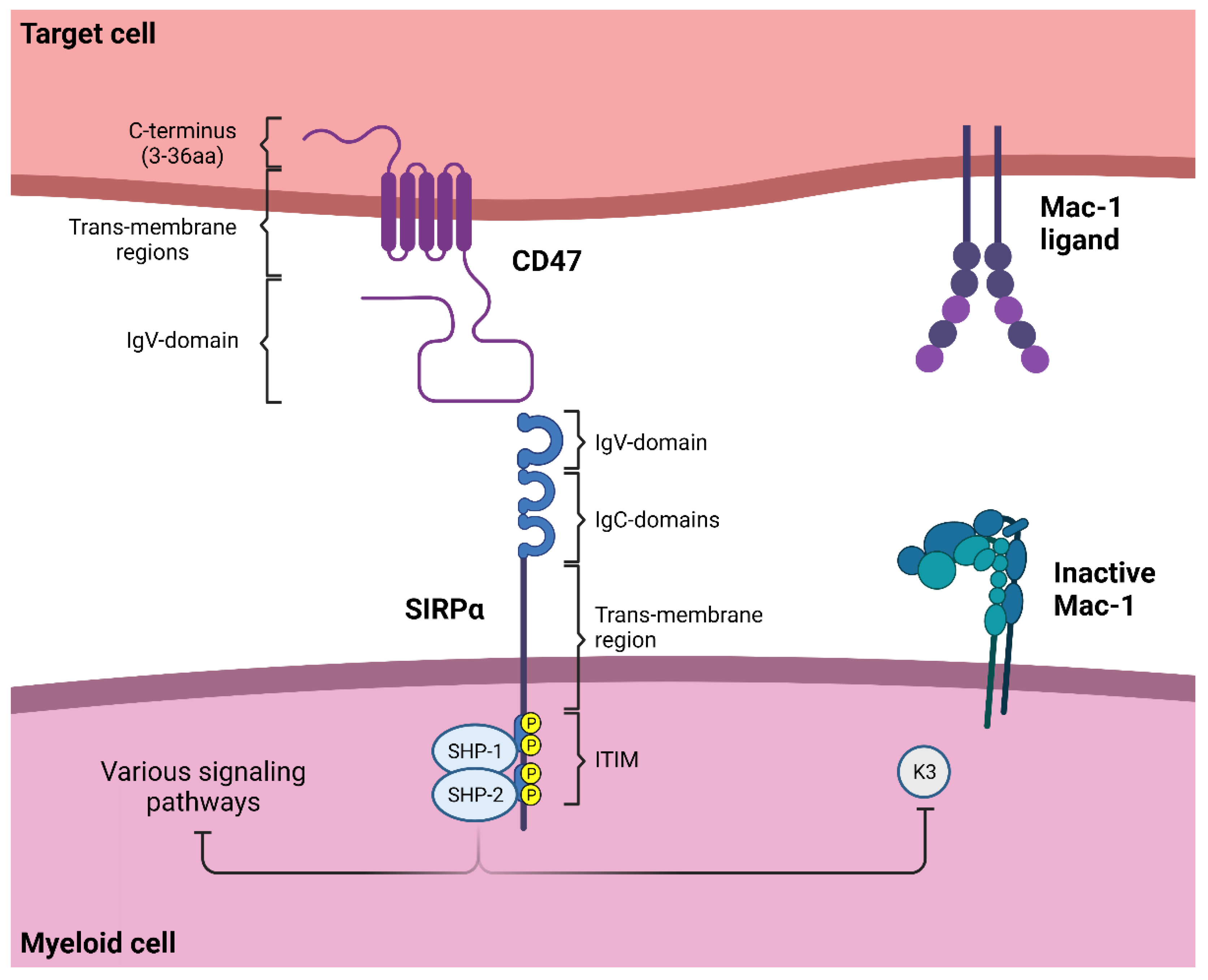
4.1. SIRPα Signaling
4.2. Neutrophil Effector Functions Influenced by CD47-SIRPα
4.3. The Innate Immune Checkpoint CD47-SIRPα in Cancer
5. Targeting CD47-SIRPα to Potentiate Antibody Therapy
5.1. CD47-Targeting Agents
5.2. Anti-SIRPα mAbs
5.3. Alternative Ways to Disrupt CD47-SIRPα Interactions
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-CD47 mAbs | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Type | Phase | Trial Identifier | Name | Indication | Treatment | Results | Ref | |
| Toxicity (≥Grade 3) | ORR (DCR) | ||||||||
| Magrolimab/GS-4721/ (Hu5F9-G4) | anti-CD47 IgG4 | 1/1b | NCT02678338 | CAMELLIA | r/r AML and hrMDS | Mono | 72% anemia 6% lymphopenia | AML = 0% (56%) | [196,236] |
| 1 | NCT03248479 | AML (65% TP53 mut) | +Azacitidine | 31% anemia 19% hyperbilirubinemia 19% neutropenia 17% thrombocytopenia | 65% (97%) | [198,199,200] | |||
| int/high risk MDS (13% TP53 mut) | +Azacitidine | 38% anemia 19% neutropenia 18% thrombocytopenia | 91% (100%) | [198,199,200] | |||||
| 1 | NCT02216409 | Solid cancer | Mono | 18% lymphopenia 14% anemia 7% Hyperbilirubinemia | 2/44 (OvC and FTC) | [195] | |||
| 1b/2 | NCT02953782 | CRC KRAS wt/mut | +Cetuximab | 12% anemia 12% hyperbilirubinemia | wtKRAS = 6% (50%) mKRAS = 0% (38%) | [237] | |||
| 1b/2 | NCT02953509 | NHL | +Rituximab | 42% anemia 15% neutropenia 36% infusion reactions | NHL = 45% (62%) Indolent = 61% (85%) DLBCL = 36% (48%) | [193,194] | |||
| NHL | +Rituximab + Gemcitabine + Oxaliplatin | [192,193,194] | |||||||
| 1 | NCT03558139 | Ovarian cancer | +Avelumab | ||||||
| 1b/2 | NCT03869190 | MORPEUS-UC | Urothelial & Bladder cancer | +Atezolizumab | |||||
| 1b | NCT03922477 | AML | +Atezolizumab | ||||||
| 1 | NCT03527147 | PRISM | NHL | +Rituximab + Acalabrutinib | |||||
| 2 | NCT04788043 | HL | +Pembrolizumab | ||||||
| 1 | NCT04599634 | VENOM | r/r indNHL/CLL | +Obinutuzumab + Venetoclax | |||||
| 1 | NCT04751383 | High-risk & rrNeuroblastoma/rOsteosarcoma | +Dinutuximab | ||||||
| +Dinutuximab + surgery | |||||||||
| 1b/2 | NCT04541017 | CTCL (Myc. Fungoides & Sezary Syndr.) | +Docetaxel + Mogamilizumab | ||||||
| Mogamilizumab mono | |||||||||
| 2 | NCT04827576 | Solid (mNSCLC, mSCLC, mUC) | Mono | ||||||
| 3 | NCT04313881 | ENHANCE | Untreated hrMDS | +Azacitidine | |||||
| Azacitidine mono | |||||||||
| 3 | NCT05079230 | ENHANCE-3 | Untreated AML | +Azacitidine + Venetoclax | |||||
| Azacitidine + Venetoclax | |||||||||
| 1b/2 | NCT04435691 | r/r and nd AML | +Azacitidine + Venetoclax | ||||||
| 2 | NCT04958785 | tnmBC | +Paclitaxel- + Nab paclitaxel | ||||||
| +Sacituzumab govitecan | |||||||||
| 2 | NCT04892446 | r/r MM | +Daratumumab | ||||||
| +Pomalidomide + Dexamethasone | |||||||||
| +Bortezomib + Dexamethasone | |||||||||
| 3 | NCT04778397 | ENHANCE-2 | untreated T53mut AML | +Azacitidine | |||||
| phys choice | |||||||||
| 2 | NCT04854499 | untreated HNSCC | +Pembrolizumab | ||||||
| +Pembrolizumab + chemo/Docetaxel | |||||||||
| 2 | NCT04778410 | newly diagnosed or r/r AML | +Azacitidine + Venetoclax | ||||||
| +MEC chemo | |||||||||
| +CC-486 | |||||||||
| 1 | NCT05169944 | Malignant brain cancer (children & adults) | Mono | ||||||
| CC90002 | anti-CD47 IgG4PE | 1 | NCT02641002 | r/r AML and hrMDS | Mono | 36% feb neutropenia 32% thrombocytopenia 29% anemia 18% neutropenia 11% hypokalemia | AML = 0/14 (0/14) MDS = 0/3 (2/3) | [201,238] | |
| 1 | NCT02367196 | Solid cancer/MM/NHL | +/− Rituximab | 38% neutropenia | NHL = 13% (25%) | [203] | |||
| Letaplimab/IBI188 | anti-CD47 IgG4 | 1 | NCT03763149 | Solid tumors and lymphomas | Mono | 1/20 anemia 1/20 hyperbilirubinemia 1/20 thrombocytopenia | [205] | ||
| 1/1b | NCT03717103 | Solid tumors and lymphomas | Mono | ||||||
| +Rituximab | |||||||||
| 1b | NCT04861948 | Solid tumors (Lung adenocar., Osteosarc., Soft tissue sarc.) | +IBI188 (Anti-PD1)/chemo + Bevacizumab/GM-CSF | ||||||
| 1 | NCT04511975 | newly diagnosed hrMDS | +Azacitidine | ||||||
| 1b | NCT04485052 | newly diagnosed or rrAML | +Azacitidine | ||||||
| 1b | NCT04485065 | newly diagnosed hrMDS | +Azacitidine | ||||||
| IBI322 | CD47/PDL1 BsAb IgG4 | 1a | NCT04338659 | Solid tumors | Mono | ||||
| 1/1b | NCT04328831 | Solid tumors | Mono | ||||||
| 1/1b | NCT04912466 | Hematologic cancer | Mono | ||||||
| 1a/1b | NCT04795128 | Hematologic cancer | Mono | ||||||
| 1a/1b | NCT05148442 | r/r AML and r/r MDS | +Azacitidine | ||||||
| AO176 | anti-CD47 IgG2 | 1/2 | NCT03834948 | Solid tumors | Mono | ||||
| +Paclitaxel/Pembrolizumab | |||||||||
| 1/2 | NCT04445701 | Multiple myeloma | Mono | ||||||
| +DEX/Bortezomib | |||||||||
| SRF231 | anti-CD47 IgG4 | 1/1b | NCT03512340 | Solid and hematologic cancer | Mono | 1/25 feb neutropenia 1/25 hemolysis 1/21 thrombocytopenia 1/21 elevated lipase/amylase | 0% | [239] | |
| TG1801/NI-1701 | CD47/CD19 BsAb IgG4 | 1 | NCT03804996 | B lymphoma | Mono | ||||
| +Ublituximab | |||||||||
| 1/1b | NCT04806035 | CD20+ NHL and CLL | Mono | ||||||
| +Ublituximab | |||||||||
| Lemzoparlimab/TJ011133/ TJC4 | anti-CD47 IgG4 | 1 | NCT03934814 | Melanoma | Mono | 5% elevated lipase | 1/20 (4/20) | [206] | |
| NHL | +Rituximab | 11% neutropenia | NHL = 71% Indolent = 75% DLBCL = 50% | [207] | |||||
| Solid and hematologic cancer | Mono | ||||||||
| +Rituximab | |||||||||
| +Pembrolizumab | |||||||||
| 1/2a | NCT04202003 | AML and ir/hrMDS | +Azacitidine | 1/5 thrombocytopenia | 1/5 | [208] | |||
| 1 | NCT04912063 | untreated AML and hrMDS | +Azacitidine + Venetoclax | ||||||
| 1 | NCT04895410 | r/r MM | Mono | ||||||
| +Pomalidomide + Dexamethasone | |||||||||
| +Carfilzomib + Dexamethasone | |||||||||
| +Daratumumab + Dexamethasone | |||||||||
| 1/2 | NCT05148533 | advanced, melanoma, GC, HNSCC | +Toripalimab | ||||||
| ZL1201 | anti-CD47 IgG4 | 1 | NCT04257617 | Solid cancer and lymphoma | Mono | ||||
| HX009 | CD47/PD1 BsAb IgG4 | 1 | NCT04097769 | Solid cancer | Mono | ||||
| 2 | NCT04886271 | Solid cancer | Mono | ||||||
| 1/2 | NCT05189093 | r/r Lymphoma (NHL, Hodgkin, PTCL) | Mono | ||||||
| AK117 | anti-CD47 IgG4 | 1 | NCT04349969 | Solid cancer and NHL | Mono | ||||
| 1/2 | NCT04900350 | MDS | +Azacitidine | ||||||
| 1 | NCT04728334 | r/r Solid cancer and NHL | Mono | ||||||
| 1b/2 | NCT04980885 | AML | +Azacitidine | ||||||
| 1b/2 | NCT05214482 | adv. Solid cancer | +AK112 (anti-PD-1/VEGF BsAb) +/− chemo | ||||||
| 1b/2 | NCT05229497 | adv. Solid cancer (Ph2: Adv HNSCC) | + AK112 (anti-PD-1/VEGF BsAb) + chemo | ||||||
| 1b/2 | NCT05235542 | adv. solid cancer (Ph2: Adv GEJ or Esoph Cancer) | + AK104 (anti-PD-1/CTLA4 BsAb) +/− chemo | ||||||
| IMC-002 | anti-CD47 IgG4 | 1 | NCT04306224 | Solid and hematological | Mono | ||||
| 1 | NCT05276310 | adv. solid cancer | |||||||
| SGN-CD47M | anti-CD47 ADC | 1 | NCT03957096 | Solid cancer | Mono | ||||
| PF-07257876 | CD47/PDL1 BsAb IgG4 | 1 | NCT04881045 | NSCLC, SCCHN, ovarian cancer | Mono | ||||
| TQB2928 | CD47-SIRPα NME | 1 | NCT04854681 | Solid and hematological cancer | Mono | ||||
| H4C1/ SHR-1603 | anti-CD47 | 1/2 | NCT04588324 | Solid cancer | +SHR2150 (TLR7 agonist) + chemo + Anti-PD-1 | ||||
| STI-6643 | anti-CD47 IgG4 (228P) | 1 | NCT04900519 | Solid cancer | Mono | ||||
| sB24M | anti-CD47/TNF-α | 1 | NCT04895566 | Severe Pyoderma | Mono (local application) | ||||
| IMM0306 | CD47/CD20 BsAb IgG1 | 1 | NCT04746131 | NHL | Mono | ||||
| SHR-1603 | anti-CD47 IgG4 | 1 | NCT03722186 | Solid cancer and rrLymphoma | Mono | ||||
| anti-CD47 | anti-CD47 | 1 | NCT05266274 | recurr AML after transplantation | +Azacitidine | ||||
| Gentulizumab | anti-CD47 | 1 | NCT05221385 | Adv solid cancer and NHL | Mono | ||||
| 1a | NCT05263271 | r/r AML and MDS | Mono | ||||||
| TQB2928 | anti-CD47 | 1 | NCT05192512 | Adv solid cancer and lymphoma | Mono | ||||
| BAT7104 | CD47/PDL1 BsAb | 1 | NCT05200013 | Adv solid cancer | Mono | ||||
| SG2501 | CD47/CD38 BsAb | 1 | NCT05293912 | r/r hematologic malignancies | Mono | ||||
| SIRPα-Fc | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Type | Phase | Trial Identifier | Name | Indication | Treatment | Results | Ref | |
| Toxicity (≥Grade 3) | ORR (DCR) | ||||||||
| TTI-621 | SIRPα-IgG1Fc | 1/1b | NCT02663518 | Lymphoma | Mono | 9% neutropenia 20% thrombocytopenia 9% anemia | NHL = 10% (62%) Indolent = 0% (78%) DLBCL = 29% (29–50%) | [211,240] | |
| +Rituximab | 9% neutropenia 20% thrombocytopenia 9% anemia | NHL = 23% (62%) Indolent = 50% (100%) DLBCL = 20% (57%) CTCL = 18% PTCL = 26% Hodgkin = 13% (63%) | [211,241] | ||||||
| +Nivolumab | 9% neutropenia 20% thrombocytopenia 9% anemia | NHL = 50% (50%) | [211] | ||||||
| 1 | NCT02890368 | CTCL | Mono | None reported | CTCL = 90% (34%) | [212] | |||
| Solid tumors, melanoma, breast ca, soft tissue sarcoma, CTCL | Mono | ||||||||
| +Anti-PD-1/PD-L1 (nivolumab, pembrolizumab, durvalumab, avelumab, or atezolizumab) | |||||||||
| +PEG-IFN-α2a | |||||||||
| +T-VEC | |||||||||
| +radiation | |||||||||
| 1/2 | NCT04996004 | Leiomyosarcoma | +Doxorubicin | ||||||
| TTI-622 | SIRPα-IgG4Fc | 1/1b | NCT03530683 | r/r NHL | Mono | 9% neutropenia 5% thrombocytopenia 2% anemia | NHL = 24% (53%) Indolent = 33% (33%) DLBCL = 21% (43%) | ||
| r/r NHL, r/r MM, newly diagnosed AML T53wt/mut | Mono | ||||||||
| +Azacitidine +/− Venetoclax | |||||||||
| +Carfilzomib + Dexamethasone | |||||||||
| 1/2 | NCT05261490 | OvC | +Pegylated Doxoribicin | ||||||
| Both TTI-621 and TTI-622 | 1 | NCT05139225 | r/r MM | +Daratumumab Hyaloronidase-fihj | |||||
| ALX-148/Evorpacept | affinity-enhanced SIRPα-FcDEAD | 1 | NCT03013218 | ASPEN-1 | Hematologic and SCLC | +Rituximab | 6% neutropenia 3% anemia | NHL = 48% (60%) Indolent = 61% (91%) DLBCL = 38% (48%) | [242,243] |
| Mono | |||||||||
| +Nivolumab | |||||||||
| Gastric cancer | +Trastuzumab + Ramucirumab + Paclitaxel | 44% neutropenia 33% hypertension 22% anemia | 72% (89%) | [244,245,246] | |||||
| +Trastuzumab | 6% thrombocytopenia 6% neutropenia <5% feb neutropenia WBC count | 21% (47%) | [217,244,245] | ||||||
| HNSCC | +Pembrolizumab + 5FU/Platinum | 38% neutropenia 31% anemia | 39% (89%) | [245,246] | |||||
| +Pembrolizumab | Rare (<5%) anemia, thrombocytopenia, AIHA, neutropenia, pancytopenia | 20–40% | [217,245,246] | ||||||
| NSCLC | +Pembrolizumab | Rare (<5%) anemia, thrombocytopenia, AIHA, neutropenia, pancytopenia | 5% (40%) | [217] | |||||
| 1/2 | NCT04417517 | ASPEN-2 | untreated or r/r hrMDS | +Azacitidine | 18% neutropenia 18% feb neutropenia 14% thrombocytopenia 9% anemia | 55% (80%) | [247] | ||
| 2/3 | NCT05002127 | ASPEN-6 | HER-2+ GC/GJC | +Trastuzumab + Ramucirumab + Paclitaxel | |||||
| Trastuzumab + Ramucirumab + Paclitaxel | |||||||||
| Ramucirumab + Paclitaxel | |||||||||
| 1/2 | NCT04755244 | ASPEN-5 | AML | +Venetoclax + Azacitidine | |||||
| 2 | NCT04675333 | ASPEN-4 | HNSCC | +Pembrolizumab + 5FU/Platinum | |||||
| 2 | NCT04675294 | ASPEN-3 | HNSCC | +Pembrolizumab | |||||
| Pembrolizumab mono | |||||||||
| 2 | NCT05167409 | Microsatellite stable rCRC | +Cetuximab + Pembrolizumab | ||||||
| 1/2 | NCT05025800 | NHL | +Rituximab + Lenolidamide | ||||||
| 1/2 | NCT05027139 | metastatic/inoperable HER-2+ BC/GEC cancer | +Zanidatamab (anti-HER-2 BsAb) | ||||||
| 2 | NCT05167409 | MSS CRC | +Cetuximab + Pembrolizumab | ||||||
| SL-172154 | SIRPα-Fc-CD40L | 1 | NCT04406623 | Ovarian, Fallopian tube, peritoneal cancer | Mono | ||||
| 1 | NCT04502888 | HNSCC & CSCC | Mono (intratumorally) | ||||||
| CPO107/JMT601 | SIRPα ECD/anti-CD20 BsAb IgG1 | 1/2 | NCT04853329 | NHL | Mono | ||||
| IMM01 | SIRPα-Fc | 1/2 | NCT05140811 | AML and MDS | +Azacitidine | ||||
| IMM2902 | SIRPαECD/anti-HER-2 | 1 | NCT05076591 | HER-2+ BC and GC | Mono | ||||
| DSP107 | SIRPα-41BB fusion | 1/2 | NCT04440735 | Solid/NSCLC | Mono | ||||
| +Atezolizumab | |||||||||
| Anti-SIRPα mAbs | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Type | Phase | Trial Identifier | Name | Indication | Treatment | Results | Ref | |
| Toxicity (≥Grade 3) | ORR (DCR) | ||||||||
| GS-0189 | anti-SIRPα IgG1 N297A | 1 | NCT04502706 | NHL | Mono | ||||
| +Rituximab | |||||||||
| CC-95251 | anti-SIRPα IgG1 K322A | 1 | NCT03783403 | Solid and hematologic cancer | Mono | 53% neutropenia 6% thrombocytopenia | 56% (56%) | [219] | |
| +Cetuximab/Rituximab | |||||||||
| 1 | NCT05168202 | r/r AML/MDS | Mono | ||||||
| +azacitidine | |||||||||
| BI765063/ OSE-172 | anti-SIRPαBIT IgG4 S229P-L445P | 1 | NCT03990233 | Solid cancer | Mono | None reported | 1/50 PR (HCC) | [220] | |
| +BI754091 (anti-PD-1) | 1 rash maculo-papular | 19% (25%) | [220] | ||||||
| 1 | NCT04653142 | Solid cancer | Mono | ||||||
| +BI754091 (anti-PD-1) | |||||||||
| 1 | NCT05068102 | Melanoma, HNSCC, NSCLC | Mono (Biodistribution, imaging with 89Zr-BI765063) | ||||||
| 1 | NCT05249426 | HNSCC | +BI754091 (anti-PD-1) +/− Cetuximab/chemo | ||||||
| HCC | +BI754091 (anti-PD-1) +/− BI836880 (anti-VEGF/Ang2 BsAb) | ||||||||
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Santos, E.d.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.d.B.V.; da Silva, I.J., Jr.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; et al. Anti-vascular endothelial growth factor receptor-1 antagonist antibody as a therapeutic agent for cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.M.; Veeramani, S.; Weiner, G.J. Complement in monoclonal antibody therapy of cancer. Immunol. Res. 2014, 59, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, B.S.; Ackerman, M.E. Antibody-mediated complement activation in pathology and protection. Immunol. Cell Biol. 2020, 98, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J.; Wu, J.; Walcheck, B. Engineering Anti-Tumor Monoclonal Antibodies and Fc Receptors to Enhance ADCC by Human NK Cells. Cancers 2021, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Gül, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bögels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.M.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, N.; van Egmond, M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12962. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Ravetch, J.V. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- French, R.R.; Chan, H.T.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000, 60, 5514–5521. [Google Scholar] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: Results of >2 years of follow-up. Ann. Oncol. 2019, 30, 970–976. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Weng, C.H.; Merghoub, T. Innate immune checkpoints for cancer immunotherapy: Expanding the scope of non T cell targets. Ann. Transl. Med. 2020, 8, 1031. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Albanesi, M.; Mancardi, D.A.; Jonsson, F.; Iannascoli, B.; Fiette, L.; Di Santo, J.P.; Lowell, C.A.; Bruhns, P. Neutrophils mediate antibody-induced antitumor effects in mice. Blood 2013, 122, 3160–3164. [Google Scholar] [CrossRef] [PubMed]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.M.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef]
- Dancey, J.T.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil kinetics in man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef]
- Treffers, L.W.; Hiemstra, I.H.; Kuijpers, T.W.; Berg, T.K.; Matlung, H.L. Neutrophils in cancer. Immunol. Rev. 2016, 273, 312–328. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef]
- Mukaida, N.; Sasaki, S.-I.; Baba, T. Two-Faced Roles of Tumor-Associated Neutrophils in Cancer Development and Progression. Int. J. Mol. Sci. 2020, 21, 3457. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. The dual role of neutrophils in cancer. Semin. Immunol. 2021, 57, 101582. [Google Scholar] [CrossRef]
- Siwicki, M.; Pittet, M.J. Versatile neutrophil functions in cancer. Semin. Immunol. 2021, 57, 101538. [Google Scholar] [CrossRef]
- Taucher, E.; Taucher, V.; Fink-Neuboeck, N.; Lindenmann, J.; Smolle-Juettner, F.-M. Role of Tumor-Associated Neutrophils in the Molecular Carcinogenesis of the Lung. Cancers 2021, 13, 5972. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef]
- Guthrie, G.J.K.; Charles, K.A.; Roxburgh, C.S.D.; Horgan, P.G.; McMillan, D.C.; Clarke, S.J. The systemic inflammation-based neutrophil–lymphocyte ratio: Experience in patients with cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 218–230. [Google Scholar] [CrossRef]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.T.; Goicoechea, I.; Alameda, D.; San Jose-Eneriz, E.; Merino, J.; Rodriguez-Otero, P.; et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Golay, J.; Valgardsdottir, R.; Musaraj, G.; Giupponi, D.; Spinelli, O.; Introna, M. Human neutrophils express low levels of FcgammaRIIIA, which plays a role in PMN activation. Blood 2019, 133, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, S.Q.; Kuijpers, T.W. Immunomodulation by IVIg and the Role of Fc-Gamma Receptors: Classic Mechanisms of Action after all? Front. Immunol. 2014, 5, 674. [Google Scholar] [CrossRef]
- Tsang, A.S.M.W.; Nagelkerke, S.Q.; Bultink, I.E.; Geissler, J.; Tanck, M.W.; Tacke, C.E.; Ellis, J.A.; Zenz, W.; Bijl, M.; Berden, J.H.; et al. Fc-gamma receptor polymorphisms differentially influence susceptibility to systemic lupus erythematosus and lupus nephritis. Rheumatology 2016, 55, 939–948. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mocsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef]
- Valerius, T.; Repp, R.; de Wit, T.P.; Berthold, S.; Platzer, E.; Kalden, J.R.; Gramatzki, M.; van de Winkel, J.G. Involvement of the high-affinity receptor for IgG (Fc gamma RI.; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood 1993, 82, 931–939. [Google Scholar] [CrossRef]
- Treffers, L.W.; Van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; Van Der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.J.J.H.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcγRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils. Front. Immunol. 2019, 10, 3124. [Google Scholar] [CrossRef]
- Treffers, L.W.; Zhao, X.W.; van der Heijden, J.; Nagelkerke, S.Q.; van Rees, D.J.; Gonzalez, P.; Geissler, J.; Verkuijlen, P.; van Houdt, M.; de Boer, M.; et al. Genetic variation of human neutrophil Fcγ receptors and SIRPα in antibody-dependent cellular cytotoxicity towards cancer cells. Eur. J. Immunol. 2018, 48, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Valerius, T.; Wurflein, D.; Stockmeyer, B.; Repp, R.; Kalden, J.R.; Gramatzki, M. Activated neutrophils as effector cells for bispecific antibodies. Cancer Immunol. Immunother. 1997, 45, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Van Rees, D.J.; Brinkhaus, M.; Klein, B.; Verkuijlen, P.; Tool, A.T.J.; Schornagel, K.; Treffers, L.W.; van Houdt, M.; Kater, A.P.; Vidarsson, G.; et al. Sodium stibogluconate and CD47-SIRPalpha blockade overcome resistance of anti-CD20-opsonized B cells to neutrophil killing. Blood Adv. 2022, 6, 2156–2166. [Google Scholar] [CrossRef]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018, 23, 3946–3959.e6. [Google Scholar] [CrossRef]
- Otten, M.A.; Rudolph, E.; Dechant, M.; Tuk, C.W.; Reijmers, R.M.; Beelen, R.H.; van de Winkel, J.G.; van Egmond, M. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J. Immunol. 2005, 174, 5472–5480. [Google Scholar] [CrossRef]
- Dechant, M.; Beyer, T.; Schneider-Merck, T.; Weisner, W.; Peipp, M.; van de Winkel, J.G.; Valerius, T. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J. Immunol. 2007, 179, 2936–2943. [Google Scholar] [CrossRef]
- Bakema, J.E.; van Egmond, M. The human immunoglobulin A Fc receptor FcalphaRI: A multifaceted regulator of mucosal immunity. Mucosal Immunol. 2011, 4, 612–624. [Google Scholar] [CrossRef]
- Bakema, J.E.; van Egmond, M. Immunoglobulin A: A next generation of therapeutic antibodies? mAbs 2011, 3, 352–361. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rosner, T.; Valerius, T.; Leusen, J.H.W.; Ten Broeke, T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef]
- Otten, M.A.; Bakema, J.E.; Tuk, C.W.; Glennie, M.J.; Tutt, A.L.; Beelen, R.H.; van de Winkel, J.G.; van Egmond, M. Enhanced FcalphaRI-mediated neutrophil migration towards tumour colonies in the presence of endothelial cells. Eur. J. Immunol. 2012, 42, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Van der Steen, L.; Tuk, C.W.; Bakema, J.E.; Kooij, G.; Reijerkerk, A.; Vidarsson, G.; Bouma, G.; Kraal, G.; de Vries, H.E.; Beelen, R.H.; et al. Immunoglobulin A: Fc(alpha)RI interactions induce neutrophil migration through release of leukotriene B4. Gastroenterology 2009, 137, 2018–2029.e3. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Heitzmann, A.; Viel, S.; Nicolas, A.; Sastre-Garau, X.; Oppezzo, P.; Pritsch, O.; Osinaga, E.; Amigorena, S. Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 2011, 71, 5134–5143. [Google Scholar] [CrossRef]
- Van Spriel, A.B.; Leusen, J.H.; van Egmond, M.; Dijkman, H.B.; Assmann, K.J.; Mayadas, T.N.; van de Winkel, J.G. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 2001, 97, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, M.; van Vuuren, A.J.; Morton, H.C.; van Spriel, A.B.; Shen, L.; Hofhuis, F.M.; Saito, T.; Mayadas, T.N.; Verbeek, J.S.; van de Winkel, J.G. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood 1999, 93, 4387–4394. [Google Scholar] [CrossRef]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef]
- Borregaard, N.; Cowland, J.B. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 1997, 89, 3503–3521. [Google Scholar] [CrossRef]
- Sengelov, H.; Kjeldsen, L.; Diamond, M.S.; Springer, T.A.; Borregaard, N. Subcellular localization and dynamics of Mac-1 (alpha m beta 2) in human neutrophils. J. Clin. Investig. 1993, 92, 1467–1476. [Google Scholar] [CrossRef]
- Clark, R.A.; Olsson, I.; Klebanoff, S.J. Cytotoxicity for tumor cells of cationic proteins from human neutrophil granules. J. Cell Biol. 1976, 70, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A. Mechanism of mammalian cell lysis mediated by peptide defensins. Evidence for an initial alteration of the plasma membrane. J. Clin. Investig. 1991, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Seignez, C.; Racoeur, C.; Isambert, N.; Mabrouk, N.; Scagliarini, A.; Reveneau, S.; Arnould, L.; Bettaieb, A.; Jeannin, J.F.; et al. Tumor-derived granzyme B-expressing neutrophils acquire antitumor potential after lipid A treatment. Oncotarget 2018, 9, 28364–28378. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Chakraborty, K.; Tang, X.A.; Zhou, G.; Schoenfelt, K.Q.; Becker, K.M.; Hoffman, A.; Chang, Y.F.; Blank, A.; Reardon, C.A.; et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 2021, 184, 3163–3177.e21. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Treffers, L.W.; Ten Broeke, T.; Rosner, T.; Jansen, J.H.M.; van Houdt, M.; Kahle, S.; Schornagel, K.; Verkuijlen, P.; Prins, J.M.; Franke, K.; et al. IgA-Mediated Killing of Tumor Cells by Neutrophils Is Enhanced by CD47-SIRPalpha Checkpoint Inhibition. Cancer Immunol. Res. 2020, 8, 120–130. [Google Scholar] [CrossRef]
- Hofmann, A.; Blau, H.M. Death of solid tumor cells induced by Fas ligand expressing primary myoblasts. Somat. Cell Mol. Genet. 1997, 23, 249–257. [Google Scholar] [CrossRef]
- Liles, W.C.; Kiener, P.A.; Ledbetter, J.A.; Aruffo, A.; Klebanoff, S.J. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: Implications for the regulation of apoptosis in neutrophils. J. Exp. Med. 1996, 184, 429–440. [Google Scholar] [CrossRef]
- Sun, B.; Qin, W.; Song, M.; Liu, L.; Yu, Y.; Qi, X.; Sun, H. Neutrophil Suppresses Tumor Cell Proliferation via Fas/Fas Ligand Pathway Mediated Cell Cycle Arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Berger-Achituv, S.; Brinkmann, V.; Abed, U.A.; Kuhn, L.I.; Ben-Ezra, J.; Elhasid, R.; Zychlinsky, A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 2013, 4, 48. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Roberts, R.E.; Hallett, M.B. Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. Int. J. Mol. Sci. 2019, 20, 1383. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Bologna, L.; Gotti, E.; Da Roit, F.; Intermesoli, T.; Rambaldi, A.; Introna, M.; Golay, J. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J. Immunol. 2013, 190, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Valgardsdottir, R.; Cattaneo, I.; Klein, C.; Introna, M.; Figliuzzi, M.; Golay, J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood 2017, 129, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Bouti, P.; Zhao, X.W.; Verkuijlen, P.J.J.H.; Tool, A.T.J.; van Houdt, M.; Köker, N.; Köker, M.Y.; Keskin, O.; Akbayram, S.; van Bruggen, R.; et al. Kindlin3-dependent CD11b/CD18-integrin activation is required for potentiation of neutrophil cytotoxicity by CD47-SIRPa checkpoint disruption. Cancer Immunol. Res. 2021, 9, 147–155. [Google Scholar] [CrossRef]
- Van Rees, D.J.; Bouti, P.; Klein, B.; Verkuijlen, P.J.H.; van Houdt, M.; Schornagel, K.; Tool, A.T.J.; Venet, D.; Sotiriou, C.; El-Abed, S.; et al. Cancer cells resist antibody-mediated destruction by neutrophils through activation of the exocyst complex. J. Immunother. Cancer 2022, 10, e004820. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Chtanova, T.; Schaeffer, M.; Han, S.J.; van Dooren, G.G.; Nollmann, M.; Herzmark, P.; Chan, S.W.; Satija, H.; Camfield, K.; Aaron, H.; et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity 2008, 29, 487–496. [Google Scholar] [CrossRef]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef]
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32. [Google Scholar] [CrossRef]
- Meinderts, S.M.; Baker, G.; van Wijk, S.; Beuger, B.M.; Geissler, J.; Jansen, M.H.; Saris, A.; Ten Brinke, A.; Kuijpers, T.W.; van den Berg, T.K.; et al. Neutrophils acquire antigen-presenting cell features after phagocytosis of IgG-opsonized erythrocytes. Blood Adv. 2019, 3, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.M.; Cochrane, F.; Barclay, A.N.; van den Berg, T.K. Signal Regulatory Proteins in the Immune System. J. Immunol. 2005, 175, 7781–7787. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; van der Laan, L.J.; Vernon-Wilson, E.; Renardel de Lavalette, C.; Dopp, E.A.; Dijkstra, C.D.; Simmons, D.L.; van den Berg, T.K. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J. Immunol. 1998, 161, 1853–1859. [Google Scholar] [PubMed]
- Van den Berg, T.K.; Yoder, J.A.; Litman, G.W. On the origins of adaptive immunity: Innate immune receptors join the tale. Trends Immunol. 2004, 25, 11–16. [Google Scholar] [CrossRef]
- Hatherley, D.; Harlos, K.; Dunlop, D.C.; Stuart, D.I.; Barclay, A.N. The structure of the macrophage signal regulatory protein alpha (SIRPalpha) inhibitory receptor reveals a binding face reminiscent of that used by T cell receptors. J. Biol. Chem. 2007, 282, 14567–14575. [Google Scholar] [CrossRef]
- Reinhold, M.I.; Lindberg, F.P.; Plas, D.; Reynolds, S.; Peters, M.G.; Brown, E.J. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47). J. Cell Sci. 1995, 108, 3419–3425. [Google Scholar] [CrossRef]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Brown, E.; Hooper, L.; Ho, T.; Gresham, H. Integrin-associated protein: A 50-kD plasma membrane antigen physically and functionally associated with integrins. J. Cell Biol. 1990, 111, 2785–2794. [Google Scholar] [CrossRef]
- Campbell, I.G.; Freemont, P.S.; Foulkes, W.; Trowsdale, J. An ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains. Cancer Res. 1992, 52, 5416–5420. [Google Scholar]
- Oldenborg, P.A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef]
- Jiang, P.; Lagenaur, C.F.; Narayanan, V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J. Biol. Chem. 1999, 274, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, M.; Cant, C.; Chen, Z.; Rappold, I.; Brugger, W.; Kanz, L.; Brown, E.J.; Ullrich, A.; Buhring, H.J. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 1999, 94, 3633–3643. [Google Scholar] [CrossRef] [PubMed]
- Vernon-Wilson, E.F.; Kee, W.J.; Willis, A.C.; Barclay, A.N.; Simmons, D.L.; Brown, M.H. CD47 is a ligand for rat macrophage membrane signal regulatory protein SIRP (OX41) and human SIRPalpha 1. Eur. J. Immunol. 2000, 30, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, D.; Graham, S.C.; Turner, J.; Harlos, K.; Stuart, D.I.; Barclay, A.N. Paired Receptor Specificity Explained by Structures of Signal Regulatory Proteins Alone and Complexed with CD47. Mol. Cell 2008, 31, 266–277. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Jansen, J.H.M.; Raaben, M.; Toebes, M.; Franke, K.; Brandsma, A.M.; Matlung, H.L.; Fauster, A.; Gomez-Eerland, R.; Bakker, N.A.M.; et al. Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRPα axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. [Google Scholar] [CrossRef]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Polymorphisms in the human inhibitory signal-regulatory protein alpha do not affect binding to its ligand CD47. J. Biol. Chem. 2014, 289, 10024–10028. [Google Scholar] [CrossRef]
- Zhao, X.W.; Van Beek, E.M.; Schornagel, K.; Van Der Maaden, H.; Van Houdt, M.; Otten, M.A.; Finetti, P.; Van Egmond, M.; Matozaki, T.; Kraal, G.; et al. CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. USA 2011, 108, 18342–18347. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Kern, N.; Vale, R.D. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 2020, 53, 290–302.e6. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Gresham, H.D.; Lindberg, F.P. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J. Exp. Med. 2001, 193, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Suk, K.; Lee, W.H. SHPS-1 and a synthetic peptide representing its ITIM inhibit the MyD88, but not TRIF, pathway of TLR signaling through activation of SHP and PI3K in THP-1 cells. Inflamm. Res. 2013, 62, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Thibaudeau, E.; Latour, S. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J. Biol. Chem. 1998, 273, 22719–22728. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Costell, M.; Fassler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Franke, K.; Pillai, S.Y.; Hoogenboezem, M.; Gijbels, M.J.J.; Matlung, H.L.; Geissler, J.; Olsman, H.; Pottgens, C.; van Gorp, P.J.; Ozsvar-Kozma, M.; et al. SIRPalpha on Mouse B1 Cells Restricts Lymphoid Tissue Migration and Natural Antibody Production. Front. Immunol. 2020, 11, 570963. [Google Scholar] [CrossRef]
- Cooper, D.; Lindberg, F.P.; Gamble, J.R.; Brown, E.J.; Vadas, M.A. Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc. Natl. Acad. Sci. USA 1995, 92, 3978–3982. [Google Scholar] [CrossRef]
- Parkos, C.A.; Colgan, S.P.; Liang, T.W.; Nusrat, A.; Bacarra, A.E.; Carnes, D.K.; Madara, J.L. CD47 mediates post-adhesive events required for neutrophil migration across polarized intestinal epithelia. J. Cell Biol. 1996, 132, 437–450. [Google Scholar] [CrossRef]
- Lindberg, F.P.; Bullard, D.C.; Caver, T.E.; Gresham, H.D.; Beaudet, A.L.; Brown, E.J. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science 1996, 274, 795–798. [Google Scholar] [CrossRef]
- Meinderts, S.M.; Oldenborg, P.A.; Beuger, B.M.; Klei, T.R.L.; Johansson, J.; Kuijpers, T.W.; Matozaki, T.; Huisman, E.J.; de Haas, M.; van den Berg, T.K.; et al. Human and murine splenic neutrophils are potent phagocytes of IgG-opsonized red blood cells. Blood Adv. 2017, 1, 875–886. [Google Scholar] [CrossRef]
- Liu, Y.; Merlin, D.; Burst, S.L.; Pochet, M.; Madara, J.L.; Parkos, C.A. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J. Biol. Chem. 2001, 276, 40156–40166. [Google Scholar] [CrossRef] [PubMed]
- Zen, K.; Liu, Y. Role of different protein tyrosine kinases in fMLP-induced neutrophil transmigration. Immunobiology 2008, 213, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Buhring, H.J.; Zen, K.; Burst, S.L.; Schnell, F.J.; Williams, I.R.; Parkos, C.A. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J. Biol. Chem. 2002, 277, 10028–10036. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; O’Connor, M.B.; Mandell, K.J.; Zen, K.; Ullrich, A.; Buhring, H.J.; Parkos, C.A. Peptide-mediated inhibition of neutrophil transmigration by blocking CD47 interactions with signal regulatory protein alpha. J. Immunol. 2004, 172, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Zarate, J.; Matlung, H.L.; Matozaki, T.; Kuijpers, T.W.; Maridonneau-Parini, I.; van den Berg, T.K. Regulation of Phagocyte Migration by Signal Regulatory Protein-Alpha Signaling. PLoS ONE 2015, 10, e0127178. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Ishikawa-Sekigami, T.; Kaneko, Y.; Okazawa, H.; Tomizawa, T.; Okajo, J.; Saito, Y.; Okuzawa, C.; Sugawara-Yokoo, M.; Nishiyama, U.; Ohnishi, H.; et al. SHPS-1 promotes the survival of circulating erythrocytes through inhibition of phagocytosis by splenic macrophages. Blood 2006, 107, 341–348. [Google Scholar] [CrossRef]
- Olsson, M.; Bruhns, P.; Frazier, W.A.; Ravetch, J.V.; Oldenborg, P.A. Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood 2005, 105, 3577–3582. [Google Scholar] [CrossRef]
- Yamao, T.; Noguchi, T.; Takeuchi, O.; Nishiyama, U.; Morita, H.; Hagiwara, T.; Akahori, H.; Kato, T.; Inagaki, K.; Okazawa, H.; et al. Negative regulation of platelet clearance and of the macrophage phagocytic response by the transmembrane glycoprotein SHPS-1. J. Biol. Chem. 2002, 277, 39833–39839. [Google Scholar] [CrossRef]
- Blazar, B.R.; Lindberg, F.P.; Ingulli, E.; Panoskaltsis-Mortari, A.; Oldenborg, P.A.; Iizuka, K.; Yokoyama, W.M.; Taylor, P.A. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J. Exp. Med. 2001, 194, 541–549. [Google Scholar] [CrossRef]
- Wang, H.; Madariaga, M.L.; Wang, S.; Van Rooijen, N.; Oldenborg, P.A.; Yang, Y.G. Lack of CD47 on nonhematopoietic cells induces split macrophage tolerance to CD47null cells. Proc. Natl. Acad. Sci. USA 2007, 104, 13744–13749. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 8, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, T.K.; van der Schoot, C.E. Innate immune ‘self’ recognition: A role for CD47-SIRPalpha interactions in hematopoietic stem cell transplantation. Trends Immunol. 2008, 29, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.S.; Brown, M.H.; Barclay, A.N.; Hatherley, D. Signal-regulatory protein alpha from the NOD mouse binds human CD47 with an exceptionally high affinity—Implications for engraftment of human cells. Immunology 2014, 143, 61–67. [Google Scholar] [CrossRef]
- Yamauchi, T.; Takenaka, K.; Urata, S.; Shima, T.; Kikushige, Y.; Tokuyama, T.; Iwamoto, C.; Nishihara, M.; Iwasaki, H.; Miyamoto, T.; et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood 2013, 121, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Fu, F.; Zhang, Y.; Gao, Z.; Zhao, Y.; Wen, Z.; Han, H.; Li, Y.; Hu, H.; Chen, H. Combination of CD47 and CD68 expression predicts survival in eastern-Asian patients with non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 739–747. [Google Scholar] [CrossRef]
- Barclay, A.N.; Van Den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Edris, B.; Weiskopf, K.; Weissman, I.L.; van de Rijn, M. Flipping the script on macrophages in leiomyosarcoma. Oncoimmunology 2012, 1, 1202–1204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, J.F.; Pan, X.H.; Zhang, S.J.; Zhao, C.; Qiu, B.S.; Gu, H.F.; Hong, J.F.; Cao, L.; Chen, Y.; Xia, B.; et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget 2015, 6, 23662–23670. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Dai, M.; Xu, Q.; Zhu, X.; Zhou, Y.; Jiang, S.; Wang, Y.; Ai, Z.; Ma, L.; Zhang, Y.; et al. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-kappaB-CD47 axis. Oncogene 2018, 37, 2394–2409. [Google Scholar] [CrossRef]
- Tan, M.; Zhu, L.; Zhuang, H.; Hao, Y.; Gao, S.; Liu, S.; Liu, Q.; Liu, D.; Liu, J.; Lin, B. Lewis Y antigen modified CD47 is an independent risk factor for poor prognosis and promotes early ovarian cancer metastasis. Am. J. Cancer Res. 2015, 5, 2777–2787. [Google Scholar]
- Chao, M.P.; Tang, C.; Pachynski, R.K.; Chin, R.; Majeti, R.; Weissman, I.L. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 2011, 118, 4890–4901. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Martinez-Sanz, P.; Hoogendijk, A.J.; Verkuijlen, P.; Schornagel, K.; van Bruggen, R.; van den Berg, T.K.; Tytgat, G.A.M.; Franke, K.; Kuijpers, T.W.; Matlung, H.L. CD47-SIRPalpha Checkpoint Inhibition Enhances Neutrophil-Mediated Killing of Dinutuximab-Opsonized Neuroblastoma Cells. Cancers 2021, 13, 4261. [Google Scholar] [CrossRef]
- Zhao, X.W.; Kuijpers, T.W.; van den Berg, T.K. Is targeting of CD47-SIRPalpha enough for treating hematopoietic malignancy? Blood 2012, 119, 4333–4334. [Google Scholar] [CrossRef]
- Zhao, X.W.; Matlung, H.L.; Kuijpers, T.W.; van den Berg, T.K. On the mechanism of CD47 targeting in cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E2843. [Google Scholar] [CrossRef]
- Kuo, T.C.; Chen, A.; Harrabi, O.; Sockolosky, J.T.; Zhang, A.; Sangalang, E.; Doyle, L.V.; Kauder, S.E.; Fontaine, D.; Bollini, S.; et al. Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J. Hematol. Oncol. 2020, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Rosner, T.; Kahle, S.; Montenegro, F.; Matlung, H.L.; Jansen, J.H.M.; Evers, M.; Beurskens, F.; Leusen, J.H.W.; van den Berg, T.K.; Valerius, T. Immune Effector Functions of Human IgG2 Antibodies against EGFR. Mol. Cancer Ther. 2019, 18, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanz, P.; van Rees, D.J.; van Zogchel, L.M.J.; Klein, B.; Bouti, P.; Olsman, H.; Schornagel, K.; Kok, I.; Sunak, A.; Leeuwenburg, K.; et al. G-CSF as a suitable alternative to GM-CSF to boost dinutuximab-mediated neutrophil cytotoxicity in neuroblastoma treatment. J. Immunother. Cancer 2021, 9, e002259. [Google Scholar] [CrossRef]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef]
- Du, K.; Li, Y.; Liu, J.; Chen, W.; Wei, Z.; Luo, Y.; Liu, H.; Qi, Y.; Wang, F.; Sui, J. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol. Ther. 2021, 29, 1572–1584. [Google Scholar] [CrossRef]
- Hendriks, M.; Ploeg, E.M.; Koopmans, I.; Britsch, I.; Ke, X.; Samplonius, D.F.; Helfrich, W. Bispecific antibody approach for EGFR-directed blockade of the CD47-SIRPalpha “don’t eat me” immune checkpoint promotes neutrophil-mediated trogoptosis and enhances antigen cross-presentation. Oncoimmunology 2020, 9, 1824323. [Google Scholar] [CrossRef]
- Hernandez-Ilizaliturri, F.J.; Jupudy, V.; Ostberg, J.; Oflazoglu, E.; Huberman, A.; Repasky, E.; Czuczman, M.S. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin. Cancer Res. 2003, 9, 5866–5873. [Google Scholar]
- Siders, W.M.; Shields, J.; Garron, C.; Hu, Y.; Boutin, P.; Shankara, S.; Weber, W.; Roberts, B.; Kaplan, J.M. Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk. Lymphoma 2010, 51, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhu, E.F.; Gai, S.A.; Opel, C.F.; Kwan, B.H.; Surana, R.; Mihm, M.C.; Kauke, M.J.; Moynihan, K.D.; Angelini, A.; Williams, R.T.; et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 2015, 27, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Yanagita, T.; Murata, Y.; Tanaka, D.; Motegi, S.I.; Arai, E.; Daniwijaya, E.W.; Hazama, D.; Washio, K.; Saito, Y.; Kotani, T.; et al. Anti-SIRPalpha antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2017, 2, e89140. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, R.C.; Krishnan, S.; Wu, R.C.; Boda, A.R.; Liu, A.; Winkler, M.; Hsu, W.H.; Lin, S.H.; Hung, M.C.; Chan, L.C.; et al. ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci. Immunol. 2022, 7, eabl9330. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef]
- Gauttier, V.; Pengam, S.; Durand, J.; Biteau, K.; Mary, C.; Morello, A.; Neel, M.; Porto, G.; Teppaz, G.; Thepenier, V.; et al. Selective SIRP alpha blockade reverses tumor T cell exclusion and overcomes cancer immunotherapy resistance. J. Clin. Investig. 2020, 130, 6109–6123. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Y.; Deng, Y.; Wei, F.; Zhao, Q.; Liu, Y.; Liu, Z.; Yu, B.; Huang, Z. Delivery of CD47 blocker SIRPalpha-Fc by CAR-T cells enhances antitumor efficacy. J. Immunother. Cancer 2022, 10, e003737. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C.; et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, T.K.; Valerius, T. Myeloid immune-checkpoint inhibition enters the clinical stage. Nat. Rev. Clin. Oncol. 2019, 16, 275–276. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M. The First-In-Class Anti-CD47 Antibody HU5F9-G4 with Rituximab induces Durable Responses in Relapsed/Refractory DLBCL and Indolent Lymphoma: Interim Phase 1b/2 Results; EHA Meeting Report; EHA: Hague, The Netherlands, 2019. [Google Scholar]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients with Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Vyas, P.; Knapper, S.; Kelly, R.; Salim, R.; Lubowiecki, M.; Royston, D.; Johnson, H.; Roberts, C.; Chen, J.; Agoram, B.; et al. Initial Phase 1 Results of the First-In-Class Anti-CD47 Antibody HU5F9-G4 in Relapsed/Refractory Acute Myeloid Leukemia Patients; EHA Meeting Report; EHA: Hague, The Netherlands, 2018. [Google Scholar]
- Sallman, D.A.; Donnellan, W.B.; Asch, A.S.; Lee, D.J.; Al Malki, M.; Marcucci, G.; Pollyea, D.A.; Kambhampati, S.; Komrokji, R.S.; Van Elk, J.; et al. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: Initial phase 1b results. J. Clin. Oncol. 2019, 37, 7009. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al Malki, M.; Asch, A.S.; Lee, D.J.; Kambhampati, S.; Donnellan, W.B.; Bradley, T.J.; Vyas, P.; Jeyakumar, D.; Marcucci, G.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: Phase Ib results. J. Clin. Oncol. 2020, 38, 7507. [Google Scholar] [CrossRef]
- Sallman, D.; Asch, A.; Kambhampati, S.; Al Malki, M.; Zeidner, J.; Donnellan, W.; Lee, D.; Vyas, P.; Jeyakumar, D.; Mannis, G.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab in Combination with Azacitidine Is Well Tolerated and Effective in AML Patients: Phase 1b Results. Clin. Lymph. Myeloma Leuk. 2021, 21, S290. [Google Scholar] [CrossRef]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.; Seet, C.S.; Tallman, M.S.; Wei, X.; Raymon, H.; Sriraman, P.; Kopytek, S.; Bewersdorf, J.P.; et al. Phase 1 study of anti-CD47 monoclonal antibody CC-90002 in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. Ann. Hematol. 2022, 101, 557–569. [Google Scholar] [CrossRef]
- Narla, R.K.; Modi, H.; Wong, L.; Abassian, M.; Bauer, D.; Desai, P.; Gaffney, B.; Jackson, P.; Leisten, J.; Liu, J.; et al. The humanized anti-CD47 monclonal antibody, CC-90002, has antitumor activity in vitro and in vivo. Cancer Res. 2017, 77, 4694. [Google Scholar] [CrossRef]
- Abrisqueta, P.; Sancho, J.M.; Cordoba, R.; Persky, D.O.; Andreadis, C.; Huntington, S.F.; Carpio, C.; Giles, D.M.; Wei, X.; Li, Y.F.; et al. Anti-CD47 Antibody, CC-90002, in Combination with Rituximab in Subjects with Relapsed and/or Refractory Non-Hodgkin Lymphoma (R/R NHL). Blood 2019, 134, 4089. [Google Scholar] [CrossRef]
- Ni, H.; Cao, L.; Wu, Z.; Wang, L.; Zhou, S.; Guo, X.; Gao, Y.; Jing, H.; Wu, M.; Liu, Y.; et al. Combined strategies for effective cancer immunotherapy with a novel anti-CD47 monoclonal antibody. Cancer Immunol. Immunother. 2022, 71, 353–363. [Google Scholar] [CrossRef]
- Lakhani, N.; Orloff, M.; Fu, S.Q.; Liu, Y.; Wang, Y.; Zhou, H.; Lin, K.D.; Liu, F.; Yan, S.L.; Patnaik, A. First-in-Human Phase I Trial of Ibi188, an Anti-Cd47 Targeting Monoclonal Antibody, in Patients with Advanced Solid Tumors and Lymphomas. J. Immunother. Cancer 2020, 8, A180. [Google Scholar] [CrossRef]
- Berlin, J.; Harb, W.; Adjei, A.; Xing, Y.; Swiecicki, P.; Seetharam, M.; Nandagopal, L.; Gopal, A.; Xu, C.; Meng, Y.; et al. A First-in-Human Study of Lemzoparlimab, a Differentiated Anti-Cd47 Antibody, in Subjects with Relapsed/Refractory Malignancy: Initial Monotherapy Results. J. Immunother. Cancer 2020, 8, A233–A234. [Google Scholar] [CrossRef]
- Mehta, A.; Harb, W.; Xu, C.; Meng, Y.; Lee, L.; Yuan, V.; Wang, Z.; Song, P.; Shen, J.H.; Gopal, A.K. Lemzoparlimab, a Differentiated Anti-CD47 Antibody in Combination with Rituximab in Relapsed and Refractory Non-Hodgkin’s Lymphoma: Initial Clinical Results. Blood 2021, 138, 3542. [Google Scholar] [CrossRef]
- Qi, J.Y.; Li, J.; Jiang, B.; Jiang, B.; Liu, H.Z.; Cao, X.X.; Zhang, M.X.; Meng, Y.; Ma, X.Y.; Jia, Y.M.; et al. A Phase I/IIa Study of Lemzoparlimab, a Monoclonal Antibody Targeting CD47, in Patients with Relapsed and/or Refractory Acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome (MDS): Initial Phase I Results. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef]
- Lin, G.H.Y.; Chai, V.; Lee, V.; Dodge, K.; Truong, T.; Wong, M.; Johnson, L.D.; Linderoth, E.; Pang, X.; Winston, J.; et al. TTI-621 (SIRPalphaFc), a CD47-blocking cancer immunotherapeutic, triggers phagocytosis of lymphoma cells by multiple polarized macrophage subsets. PLoS ONE 2017, 12, e0187262. [Google Scholar] [CrossRef]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.M.; Advani, A.S.; et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2021, 27, 2190–2199. [Google Scholar] [CrossRef]
- Querfeld, C.; Thompson, J.A.; Taylor, M.H.; DeSimone, J.A.; Zain, J.M.; Shustov, A.R.; Johns, C.; McCann, S.; Lin, G.H.Y.; Petrova, P.S.; et al. Intralesional TTI-621, a novel biologic targeting the innate immune checkpoint CD47, in patients with relapsed or refractory mycosis fungoides or Sezary syndrome: A multicentre, phase 1 study. Lancet Haematol. 2021, 8, e808–e817. [Google Scholar] [CrossRef]
- Patel, K.; Ramchandren, R.; Maris, M.; Lesokhin, A.M.; von Keudell, G.R.; Cheson, B.D.; Zonder, J.; Seymour, E.K.; Catalano, T.; Lin, G.H.Y.; et al. Investigational CD47-Blocker TTI-622 Shows Single-Agent Activity in Patients with Advanced Relapsed or Refractory Lymphoma: Update from the Ongoing First-in-Human Dose Escalation Study. Blood 2020, 136, 46–47. [Google Scholar] [CrossRef]
- Patel, K.; Zonder, J.A.; Sano, D.; Maris, M.; Lesokhin, A.; von Keudell, G.R.; Lai, C.; Ramchandren, R.; Catalano, T.; Lin, G.H.Y.; et al. CD47-Blocker TTI-622 Shows Single-Agent Activity in Patients with Advanced Relapsed or Refractory Lymphoma: Update from the Ongoing First-in-Human Dose Escalation Study. Blood 2021, 138, 3560. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Kauder, S.E.; Kuo, T.C.; Harrabi, O.; Chen, A.; Sangalang, E.; Doyle, L.; Rocha, S.S.; Bollini, S.; Han, B.; Sim, J.; et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE 2018, 13, e0201832. [Google Scholar] [CrossRef]
- Lakhani, N.J.; Chow, L.Q.M.; Gainor, J.F.; LoRusso, P.; Lee, K.W.; Chung, H.C.; Lee, J.; Bang, Y.J.; Hodi, F.S.; Kim, W.S.; et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): A first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021, 22, 1740–1751. [Google Scholar] [CrossRef]
- Chan, H.; Trout, C.; Mikolon, D.; Adams, P.; Guzman, R.; Fenalti, G.; Mavrommatis, K.; Abbasian, M.; Dearth, L.; Fox, B.; et al. Discovery and Preclinical Characterization of CC-95251, an Anti-SIRPα Antibody That Enhances Macrophage-Mediated Phagocytosis of Non-Hodgkin Lymphoma (NHL) Cells When Combined with Rituximab. Blood 2021, 138, 2271. [Google Scholar] [CrossRef]
- Strati, P.; Hawkes, E.; Ghosh, N.; Tuscano, J.M.; Chu, Q.; Anderson, M.A.; Patel, A.; Burgess, M.R.; Hege, K.; Chhagan, S.; et al. Interim Results from the First Clinical Study of CC-95251, an Anti-Signal Regulatory Protein-Alpha (SIRPα) Antibody, in Combination with Rituximab in Patients with Relapsed and/or Refractory Non-Hodgkin Lymphoma (R/R NHL). Blood 2021, 183, 2493. [Google Scholar] [CrossRef]
- Champiat, S.; Cassier, P.A.; Kotecki, N.; Korakis, I.; Vinceneux, A.; Jungels, C.; Blatchford, J.; Elgadi, M.M.; Clarke, N.; Fromond, C.; et al. Safety, Pharmacokinetics, Efficacy, and Preliminary Biomarker Data of First-In-Class BI 765063, a Selective SIRPα Inhibitor: Results of Monotherapy Dose Escalation in Phase 1 Study in Patients with Advanced Solid Tumors; ASCO Meeting Report; ASCO: Alexandria, VA, USA, 2021. [Google Scholar]
- Kotecki, N.; Champiat, S.; Delord, J.; Vinceneux, A.; Jungels, C.; Marabelle, A.; Korakis, I.; Wojciekowski, S.; Block, E.; Clarke, N.; et al. Phase I Dose Escalation Study in Patients (pts) with Advanced Solid Tumours Receiving First-In-Class BI 765063, a Selective Signal-Regulatory Protein α (SIRPα) Inhibitor, in Combination with Ezabenlimab (BI 754091), a Programmed Cell Death Protein 1 (PD-1) Inhibitor; EMSO Meeting Report; EMSO: London, UK, 2021. [Google Scholar]
- Irie, A.; Yamauchi, A.; Kontani, K.; Kihara, M.; Liu, D.; Shirato, Y.; Seki, M.; Nishi, N.; Nakamura, T.; Yokomise, H.; et al. Galectin-9 as a prognostic factor with antimetastatic potential in breast cancer. Clin. Cancer Res. 2005, 11, 2962–2968. [Google Scholar] [CrossRef]
- Ustyanovska Avtenyuk, N.; Choukrani, G.; Ammatuna, E.; Niki, T.; Cendrowicz, E.; Lourens, H.J.; Huls, G.; Wiersma, V.R.; Bremer, E. Galectin-9 Triggers Neutrophil-Mediated Anticancer Immunity. Biomedicines 2021, 10, 66. [Google Scholar] [CrossRef]
- Oronsky, B.; Guo, X.; Wang, X.; Cabrales, P.; Sher, D.; Cannizzo, L.; Wardle, B.; Abrouk, N.; Lybeck, M.; Caroen, S.; et al. Discovery of RRx-001, a Myc and CD47 Downregulating Small Molecule with Tumor Targeted Cytotoxicity and Healthy Tissue Cytoprotective Properties in Clinical Development. J. Med. Chem. 2021, 64, 7261–7271. [Google Scholar] [CrossRef]
- Oronsky, B.; Cabrales, P.; Caroen, S.; Guo, X.; Scribner, C.; Oronsky, A.; Reid, T.R. RRx-001, a downregulator of the CD47- SIRPalpha checkpoint pathway, does not cause anemia or thrombocytopenia. Expert Opin. Drug Metab. Toxicol. 2021, 17, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-alpha on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Oronsky, B.; Scicinski, J.; Scribner, C.L.; Knox, S.J.; Ning, S.; Peehl, D.M.; Korn, R.; Stirn, M.; Carter, C.A.; et al. Safety and activity of RRx-001 in patients with advanced cancer: A first-in-human, open-label, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1133–1142. [Google Scholar] [CrossRef]
- Tomita, Y.; Oronsky, B.; Abrouk, N.; Cabrales, P.; Reid, T.R.; Lee, M.J.; Yuno, A.; Baker, J.; Lee, S.; Trepel, J.B. In small cell lung cancer patients treated with RRx-001, a downregulator of CD47, decreased expression of PD-L1 on circulating tumor cells significantly correlates with clinical benefit. Transl. Lung Cancer Res. 2021, 10, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Das, D.S.; Ray, A.; Das, A.; Song, Y.; Tian, Z.; Oronsky, B.; Richardson, P.; Scicinski, J.; Chauhan, D.; Anderson, K.C. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Larson, C.; Caroen, S.; Quinn, M.; Burbano, E.; Varner, G.; Thilagar, B.; Brown, B.; Coyle, A.; et al. REPLATINUM Phase III randomized study: RRx-001 + platinum doublet versus platinum doublet in third-line small cell lung cancer. Future Oncol. 2019, 15, 3427–3433. [Google Scholar] [CrossRef]
- Wu, Z.; Weng, L.; Zhang, T.; Tian, H.; Fang, L.; Teng, H.; Zhang, W.; Gao, J.; Hao, Y.; Li, Y.; et al. Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRPalpha-CD47 axis. Cell Res. 2019, 29, 502–505. [Google Scholar] [CrossRef]
- Baumann, N.; Rosner, T.; Jansen, J.H.M.; Chan, C.; Marie Eichholz, K.; Klausz, K.; Winterberg, D.; Muller, K.; Humpe, A.; Burger, R.; et al. Enhancement of epidermal growth factor receptor antibody tumor immunotherapy by glutaminyl cyclase inhibition to interfere with CD47/signal regulatory protein alpha interactions. Cancer Sci. 2021, 112, 3029–3040. [Google Scholar] [CrossRef]
- Burgess, T.L.; Amason, J.D.; Rubin, J.S.; Duveau, D.Y.; Lamy, L.; Roberts, D.D.; Farrell, C.L.; Inglese, J.; Thomas, C.J.; Miller, T.W. A homogeneous SIRPalpha-CD47 cell-based, ligand-binding assay: Utility for small molecule drug development in immuno-oncology. PLoS ONE 2020, 15, e0226661. [Google Scholar] [CrossRef]
- Evers, M.; Rosner, T.; Dunkel, A.; Jansen, J.H.M.; Baumann, N.; Ten Broeke, T.; Nederend, M.; Eichholz, K.; Klausz, K.; Reiding, K.; et al. The selection of variable regions affects effector mechanisms of IgA antibodies against CD20. Blood Adv. 2021, 5, 3807–3820. [Google Scholar] [CrossRef]
- Li, Z.; Gu, X.; Rao, D.; Lu, M.; Wen, J.; Chen, X.; Wang, H.; Cui, X.; Tang, W.; Xu, S.; et al. Luteolin promotes macrophage-mediated phagocytosis by inhibiting CD47 pyroglutamation. Transl. Oncol. 2021, 14, 101129. [Google Scholar] [CrossRef] [PubMed]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.T.; Murphy, M.F. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.A.; Lakhani, N.J.; Eng, C.; Hecht, J.R.; Bendell, J.C.; Philip, P.A.; O’Dwyer, P.J.; Johnson, B.; Kardosh, A.; Ippolito, T.M.; et al. A phase Ib/II study of the anti-CD47 antibody magrolimab with cetuximab in solid tumor and colorectal cancer patients. J. Clin. Oncol. 2020, 38, 114. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Wang, R.; Wang, X.; Shallis, R.M.; Podoltsev, N.A.; Bewersdorf, J.P.; Huntington, S.F.; Neparidze, N.; Giri, S.; Gore, S.D.; et al. Clinical Outcomes of Older Patients (pts) with Acute Myeloid Leukemia (AML) Receiving Hypomethylating Agents (HMAs): A Large Population-Based Study in the United States. Blood 2019, 134, 646. [Google Scholar] [CrossRef]
- Patnaik, A.; Spreafico, A.; Paterson, A.M.; Peluso, M.O.; Chung, J.; Bowers, B.; Niforos, D.; O’Neill, A.M.; Beeram, M.; Iafolla, M.; et al. Results of a first-in-human phase I study of SRF231, a fully human, high-affinity anti-CD47 antibody. J. Clin. Oncol. 2020, 38, 3064. [Google Scholar] [CrossRef]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Foran, J.M.; Maris, M.; Lue, J.K.; Sawas, A.; Okada, C.; Feldman, T.A.; Sokol, L.; Mei, M.; Flinn, I.W.; et al. Updates from Ongoing, First-in-Human Phase 1 Dose Escalation and Expansion Study of TTI-621, a Novel Biologic Targeting CD47, in Patients with Relapsed or Refractory Hematologic Malignancies. Blood 2021, 138, 2448. [Google Scholar] [CrossRef]
- Kim, T.M.; Lakhani, N.; Gainor, J.F.; Kamdar, M.; Fanning, P.; Squifflet, P.; Jin, F.; Forgie, A.J.; Wan, H.I.; Pons, J.; et al. ALX148, a CD47 Blocker, in Combination with Rituximab in Patients with Relapsed/Refractory (R/R) Non-Hodgkin Lymphoma; ASPEN-1. Blood 2020, 136, 13–14. [Google Scholar]
- Kim, T.M.; Lakhani, N.; Gainor, J.F.; Kamdar, M.; Fanning, P.; Squifflet, P.; Jin, F.; Forgie, A.J.; Wan, H.I.; Pons, J.; et al. ALX148, a CD47 Blocker, in Combination with Rituximab in Patients with Non-Hodgkin Lymphoma. In Proceedings of the 62nd ASH Annual Meeting and Exposition, Washington, DC, USA, 5–8 December 2020. [Google Scholar]
- Chung, H.C.; Lee, K.W.; Kim, W.S.; Gainor, J.F.; Lakhani, N.; Chow, L.Q.; Messersmith, W.A.; Fanning, P.; Squifflet, P.; Jin, F.; et al. ESMO meeting report: ASPEN-01: A phase 1 study of ALX148, a CD47 blocker, in combination with trastuzumab, ramucirumab and paclitaxel in patients with second-line HER2-positive advanced gastric or gastroesophageal junction cancer. In Proceedings of the Esmo World Congress on Gastrointestinal Cancer, Virtual Meeting, 30 June–3 July 2021. [Google Scholar]
- Lee, K.W.; Chung, H.C.; Kim, W.S.; Chow, L.Q.; Lakhani, N.; Messersmith, W.A.; Bang, Y.J.; LoRusso, P.; Fanning, P.; Squifflet, P.; et al. ALX148, a CD47 blocker, in combination with standard chemotherapy and antibody regimens in patients with gastric/gastroesophageal junction (GC) cancer and head and neck squamous cell carcinoma (HNSCC). J. Immunother. Cancer 2020, 8, A429. [Google Scholar]
- Lee, K.W.; Chung, H.C.; Kim, T.M.; Lakhani, N.; Messersmith, W.A.; Santana-Davila, R.; Kim, W.S.; LoRusso, P.; Bang, Y.J.; Chow, L.Q.; et al. Evorpacept (ALX148), a CD47 myeloid checkpoint inhibitor, in patients with head and neck squamous cell carcinoma (HNSCC) and with gastric/gastroesophageal cancer (GC); ASPEN-01. J. Immunother. Cancer 2021, 9, 498. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Erba, H.P.; Sanikommu, S.R.; Altman, J.K.; Sayar, H.; Scott, B.L.; Fong, A.P.; Guan, S.; Jin, F.; Forgie, A.J.; et al. Evorpacept (ALX148), a CD47-Blocking Myeloid Checkpoint Inhibitor, in Combination with Azacitidine: A Phase 1/2 Study in Patients with Myelodysplastic Syndrome (ASPEN-02). Blood 2021, 138, 2601. [Google Scholar] [CrossRef]



| Name | FcγRI (CD64) | FcγRIIa (CD32a) | FcγRIIb (CD32b) | FcγRIIc (CD32c) | FcγRIIIa (CD16a) | FcγRIIIb (CD16b) | FcαRI (CD89) |
|---|---|---|---|---|---|---|---|
| Structure |  | ||||||
| Affinity | High | Low | Low | Low | Low | Low | High |
| Expression on neutrophils | Induced 1 | + | Genotype-dependent 2 | Genotype-dependent 3 | −/+ 4 | + | + |
| Class | Activation | Activation | Inhibition | Activation | Decoy | Activation | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behrens, L.M.; van den Berg, T.K.; van Egmond, M. Targeting the CD47-SIRPα Innate Immune Checkpoint to Potentiate Antibody Therapy in Cancer by Neutrophils. Cancers 2022, 14, 3366. https://doi.org/10.3390/cancers14143366
Behrens LM, van den Berg TK, van Egmond M. Targeting the CD47-SIRPα Innate Immune Checkpoint to Potentiate Antibody Therapy in Cancer by Neutrophils. Cancers. 2022; 14(14):3366. https://doi.org/10.3390/cancers14143366
Chicago/Turabian StyleBehrens, Leonie M., Timo K. van den Berg, and Marjolein van Egmond. 2022. "Targeting the CD47-SIRPα Innate Immune Checkpoint to Potentiate Antibody Therapy in Cancer by Neutrophils" Cancers 14, no. 14: 3366. https://doi.org/10.3390/cancers14143366
APA StyleBehrens, L. M., van den Berg, T. K., & van Egmond, M. (2022). Targeting the CD47-SIRPα Innate Immune Checkpoint to Potentiate Antibody Therapy in Cancer by Neutrophils. Cancers, 14(14), 3366. https://doi.org/10.3390/cancers14143366