
Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Donor Characteristics
2.2. Patient Therapies and Blood Sampling
2.3. Peripheral Blood Mononuclear Cells (PBMCs) Isolation and Cell Culture
2.4. Flow Cytometry
2.5. IFN-γ Secretion Assay
2.6. Cytokine Multiplex Array
2.7. Human Cytomegalovirus (HCMV) Testing
2.8. Statistical Analysis
3. Results
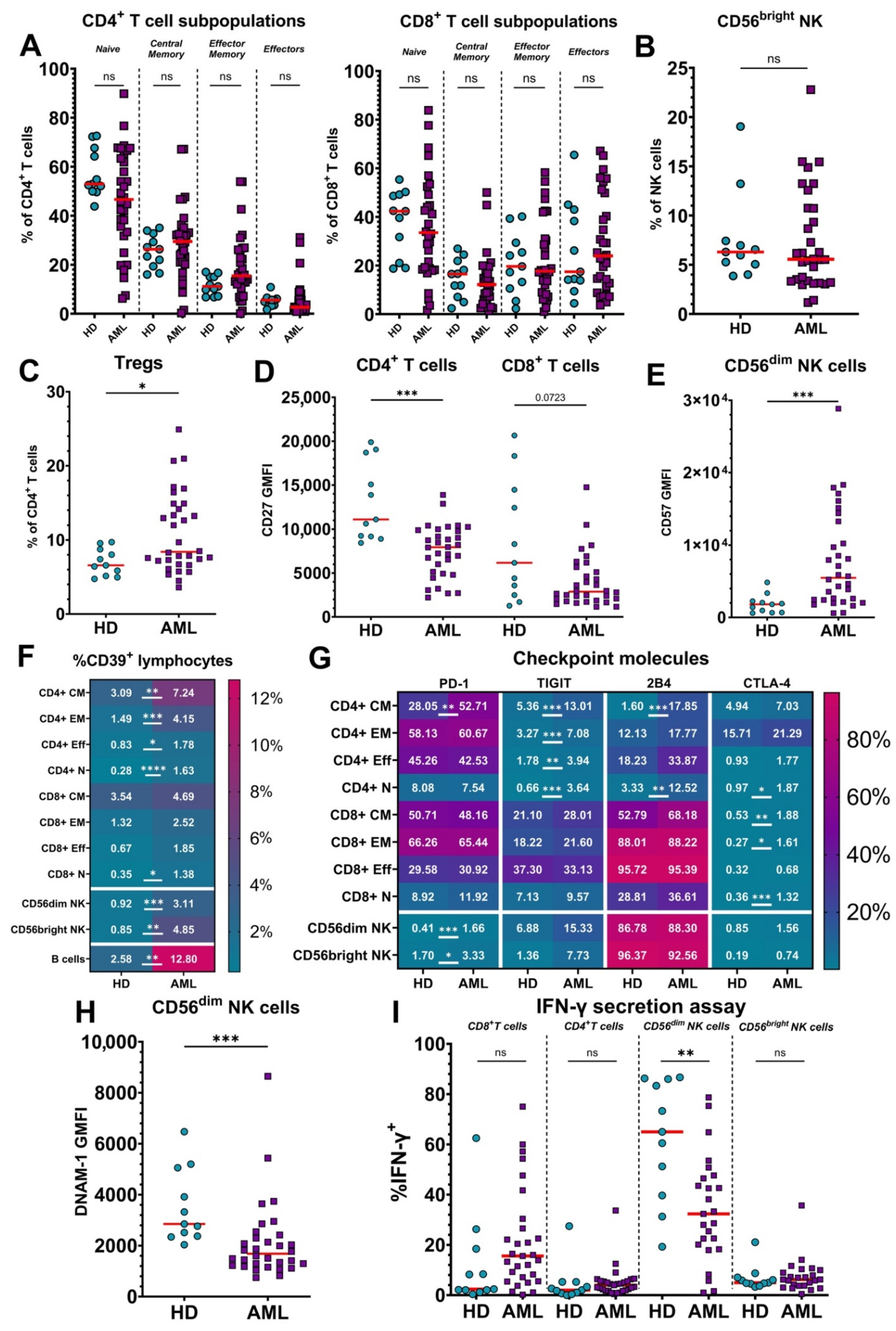
3.1. Lymphocytes from Untreated AML Patients Show an Exhausted Phenotype Compared to Healthy Donors
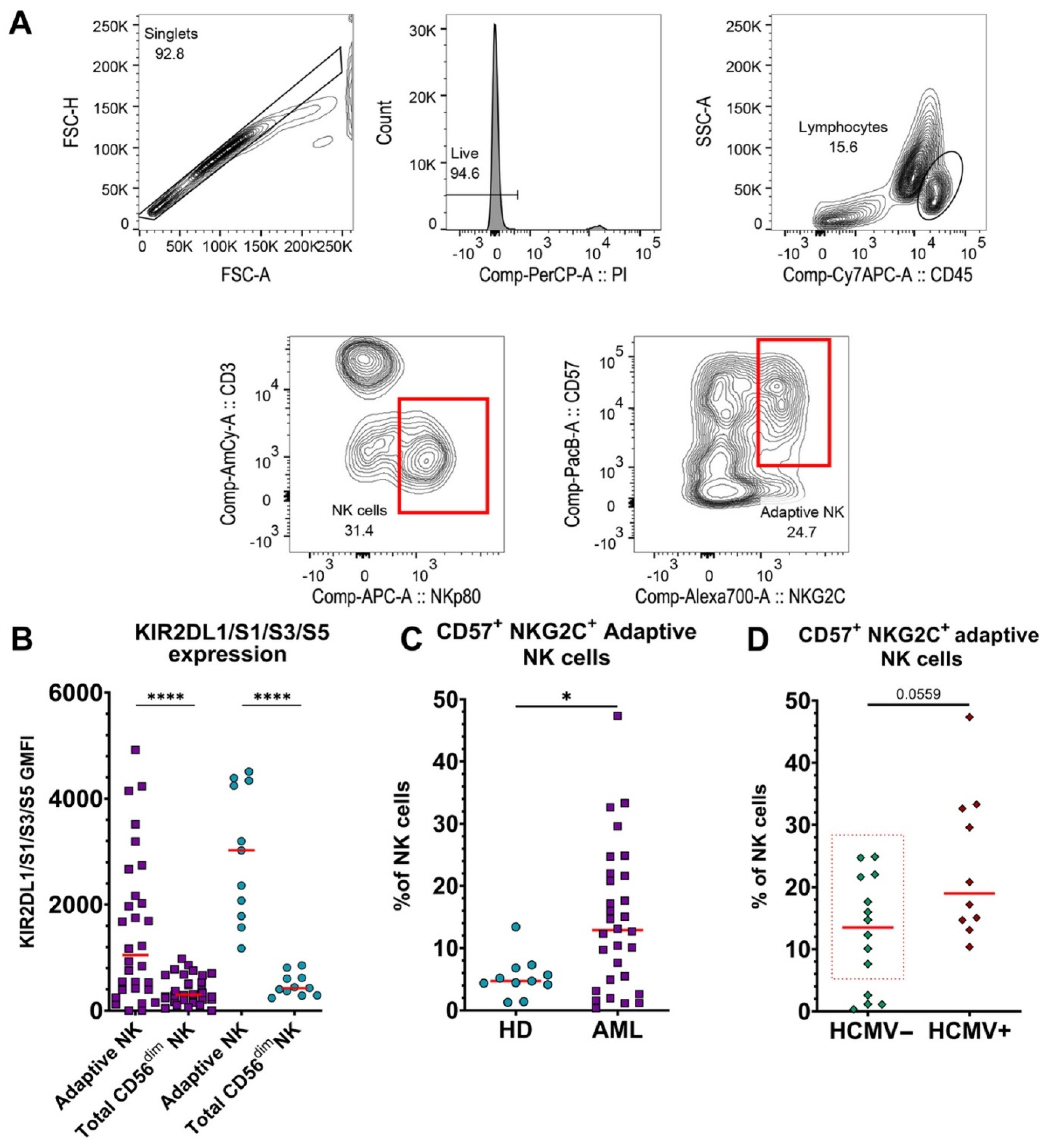
3.2. Adaptive NK Cells Are Significantly Increased in Blood of AML Patients, Independent of HCMV Serotype
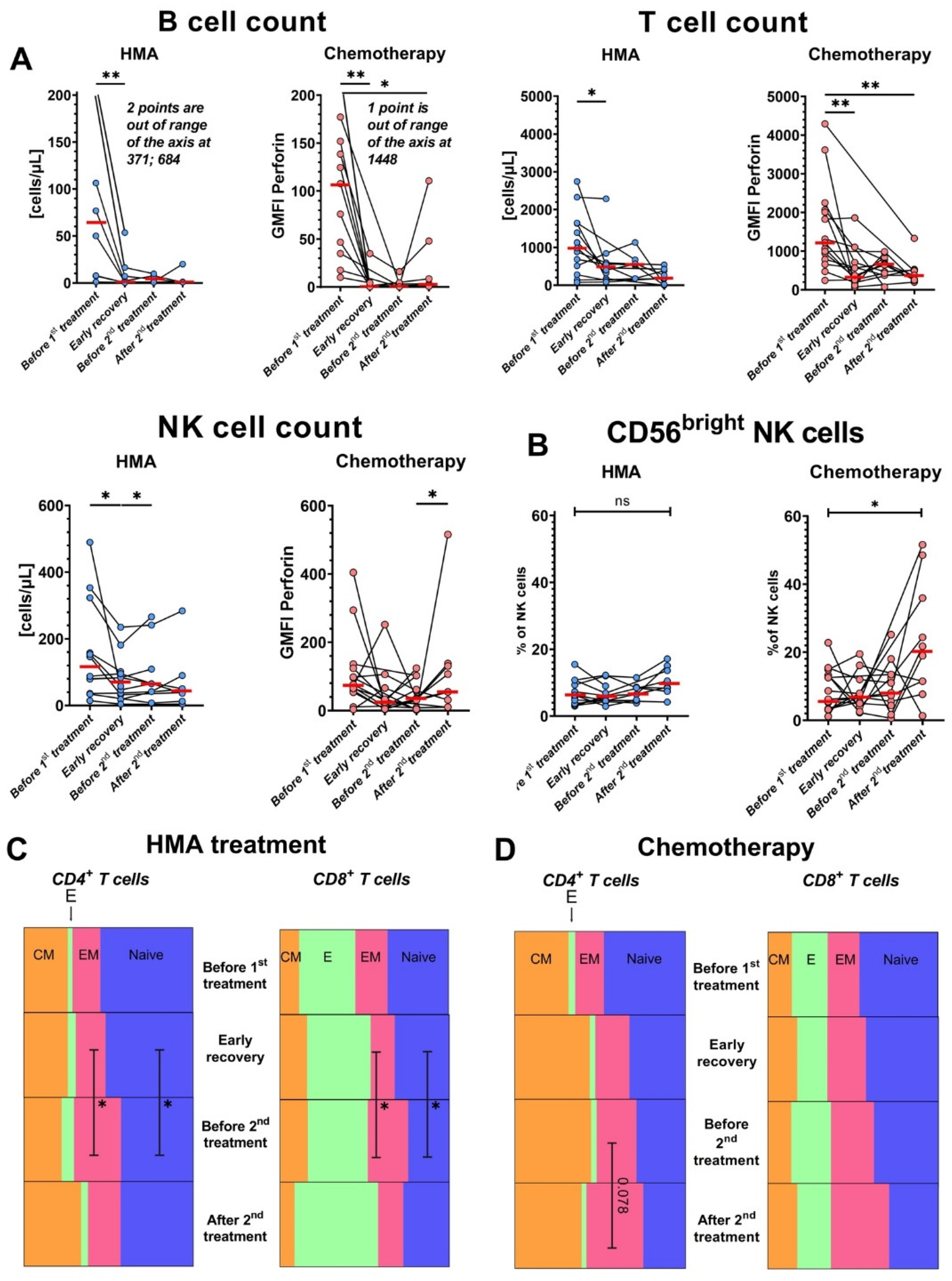
3.3. Both IC and HMA/Venetoclax Aggressively Deplete Lymphocytes, and HMA/Venetoclax Therapy Increases the Frequencies of T cells with an Effector Memory Phenotype
3.4. HMA/Venetoclax Therapy Significantly Inhibits IFN-γ Production by CD8+ T Cells
3.5. Levels of Perforin Increase in NK Cells after HMA/Venetoclax Treatment
3.6. PD-1 and 2B4 Surface Exposure Is Lowered on CD4+ T Cells from Patients Treated with HMA/Venetoclax
3.7. Treg Proliferation and CTLA-4 Expression Are Enhanced after HMA/Venetoclax Therapy
3.8. NK Cells Become Activated and Alter Receptor Expression after Treatments
3.9. Venetoclax-Resistant Patients Exhibit a Distinct T Cell Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ngai, L.L.; Kelder, A.; Janssen, J.J.W.M.; Ossenkoppele, G.J.; Cloos, J. MRD tailored therapy in AML: What we have learned so far. Front. Oncol. 2021, 10, 603636. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.D.; Langston, A.A.; Heffner, L.T., Jr. Acute myeloid leukemia in young adults: Does everyone need a transplant? J. Oncol. Pract. 2019, 15, 315–320. [Google Scholar] [CrossRef]
- Schiffer, C.A. Optimal dose and schedule of consolidation in AML: Is there a standard? Best Pract. Res. Clin. Haematol. 2014, 27, 259–264. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Stanchina, M.; Soong, D.; Zheng-Lin, B.; Watts, J.M.; Taylor, J. Advances in acute myeloid leukemia: Recently approved therapies and drugs in development. Cancers 2020, 12, 3225. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2 (Suppl. S1), S4–S11. [Google Scholar] [CrossRef]
- Talati, C.; Dhulipala, V.C.; Extermann, M.T.; Al Ali, N.; Kim, J.; Komrokji, R.; Sweet, K.; Kuykendall, A.; Sehovic, M.; Reljic, T.; et al. Comparisons of commonly used front-line regimens on survival outcomes in patients aged 70 years and older with acute myeloid leukemia. Haematologica 2020, 105, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Vachhani, P.; Al Yacoub, R.; Miller, A.; Zhang, F.; Cronin, T.L.; Ontiveros, E.P.; Thompson, J.E.; Griffiths, E.A.; Wang, E.S. Intensive chemotherapy vs. hypomethylating agents in older adults with newly diagnosed high-risk acute myeloid leukemia: A single center experience. Leuk. Res. 2018, 75, 29–35. [Google Scholar] [CrossRef]
- Santini, V.; Ossenkoppele, G.J. Hypomethylating agents in the treatment of acute myeloid leukemia: A guide to optimal use. Crit. Rev. Oncol. Hematol. 2019, 140, 1–7. [Google Scholar] [CrossRef]
- Samra, B.; Konopleva, M.; Isidori, A.; Daver, N.; Dinardo, C. Venetoclax-based combinations in acute myeloid leukemia: Current evidence and future directions. Front. Oncol. 2020, 10, 562558. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Swatler, J.; Turos-Korgul, L.; Kozlowska, E.; Piwocka, K. Immunosuppressive cell subsets and factors in myeloid leukemias. Cancers 2021, 13, 1203. [Google Scholar] [CrossRef] [PubMed]
- Aldarouish, M.; Su, X.; Qiao, J.; Gao, C.; Chen, Y.; Dai, A.; Zhang, T.; Shu, Y.; Wang, C. Immunomodulatory effects of chemotherapy on blood lymphocytes and survival of patients with advanced non-small cell lung cancer. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419839592. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Waidhauser, J.; Schuh, A.; Trepel, M.; Schmälter, A.-K.; Rank, A. Chemotherapy markedly reduces B cells but not T cells and NK cells in patients with cancer. Cancer Immunol. Immunother. 2020, 69, 147–157. [Google Scholar] [CrossRef]
- Kanakry, C.G.; Hess, A.D.; Gocke, C.D.; Thoburn, C.; Kos, F.; Meyer, C.; Briel, J.; Luznik, L.; Smith, B.D.; Levitsky, H.; et al. Early lymphocyte recovery after intensive timed sequential chemotherapy for acute myelogenous leukemia: Peripheral oligoclonal expansion of regulatory T cells. Blood 2011, 117, 608–617. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, A.W., IV; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. OncoImmunology 2017, 6, e1330235. [Google Scholar] [CrossRef]
- Balança, C.-C.; Salvioni, A.; Scarlata, C.-M.; Michelas, M.; Martinez-Gomez, C.; Gomez-Roca, C.; Sarradin, V.; Tosolini, M.; Valle, C.; Pont, F.; et al. PD-1 blockade restores helper activity of tumor-infiltrating, exhausted PD-1hiCD39+ CD4 T cells. JCI Insight 2021, 6, e142513. [Google Scholar] [CrossRef] [PubMed]
- Brauneck, F.; Seubert, E.; Wellbrock, J.; Wiesch, J.S.Z.; Duan, Y.; Magnus, T.; Bokemeyer, C.; Koch-Nolte, F.; Menzel, S.; Fiedler, W. Combined blockade of TIGIT and CD39 or A2AR enhances NK-92 cell-mediated cytotoxicity in AML. Int. J. Mol. Sci. 2021, 22, 12919. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiró, F.; Muller, L.; Funk, S.; Jackson, E.; Battastini, A.; Whiteside, T. Phenotypic and functional characteristics of CD39highhuman regulatory B cells (Breg). OncoImmunology 2016, 5, e1082703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkstrom, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Bjorklund, A.T.; Flodstrom-Tullberg, M.; Michaelsson, J.; Rottenberg, M.E.; et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueff, J.; Medinger, M.; Heim, D.; Passweg, J.; Stern, M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol. Blood Marrow Transplant. 2014, 20, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Pazina, T.; Shemesh, A.; Brusilovsky, M.; Porgador, A.; Campbell, K.S. Regulation of the functions of natural cytotoxicity receptors by interactions with diverse ligands and alterations in splice variant expression. Front. Immunol. 2017, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Béziat, V.; Hilton, H.; Norman, P.; Traherne, J.A. Deciphering the killer-cell immunoglobulin-like receptor system at super-resolution for natural killer and T-cell biology. Immunology 2016, 150, 248–264. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, K.E.; Goswami, M.; Hourigan, C.S.; Oetjen, K.A. Immunological effects of hypomethylating agents. Expert Rev. Hematol. 2017, 10, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.-C.S.; Rancan, C.; et al. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef]
- Xu, L.; Liu, L.; Yao, D.; Zeng, X.; Zhang, Y.; Lai, J.; Zhong, J.; Zha, X.; Zheng, R.; Lu, Y.; et al. PD-1 and TIGIT are highly co-expressed on CD8+ T cells in AML patient bone marrow. Front. Oncol. 2021, 11, 686156. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Tian, Z. NK cell exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.E.; Diamond, D.; et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2015, 30, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Merino, A.; Zhang, B.; Dougherty, P.; Luo, X.; Wang, J.; Blazar, B.R.; Miller, J.S.; Cichocki, F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig. 2019, 129, 3770–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ali, H.K.; Jaekel, N.; Niederwieser, D. The role of hypomethylating agents in the treatment of elderly patients with AML. J. Geriatr. Oncol. 2014, 5, 89–105. [Google Scholar] [CrossRef] [Green Version]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef]
- Yau, H.L.; Bell, E.; Ettayebi, I.; de Almeida, F.C.; Boukhaled, G.M.; Shen, S.Y.; Allard, D.; Morancho, B.; Marhon, S.A.; Ishak, C.A.; et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8+ T cells. Mol. Cell 2021, 81, 1469–1483.e8. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.-R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Zhang, M.; Xiao, X.Q.; Jiang, Y.F.; Liang, Y.S.; Peng, M.Y.; Xu, Y.; Gong, G.Z. DNA demethylation in PD-1 gene promoter induced by 5-azacytidine activates PD-1 expression on Molt-4 cells. Cell. Immunol. 2011, 271, 450–454. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, J.; Nie, K.; Yan, J.; Liu, Y.; Bacchi, C.E.; Queiroga, E.M.; Gualco, G.; Sample, J.T.; Orazi, A.; et al. Hypermethylation of the tumor suppressor gene PRDM1/Blimp-1 supports a pathogenetic role in EBV-positive Burkitt lymphoma. Blood Cancer J. 2014, 4, e261. [Google Scholar] [CrossRef]
- Zhang, Z.; Liang, L.; Li, D.; Nong, L.; Liu, J.; Qu, L.; Zheng, Y.; Zhang, B.; Li, T. Hypermethylation of PRDM1/Blimp-1 promoter in extranodal NK/T-cell lymphoma, nasal type: An evidence of predominant role in its downregulation. Hematol. Oncol. 2017, 35, 645–654. [Google Scholar] [CrossRef]
- Fu, S.-H.; Yeh, L.-T.; Chu, C.-C.; Yen, B.L.-J.; Sytwu, H.-K. New insights into Blimp-1 in T lymphocytes: A divergent regulator of cell destiny and effector function. J. Biomed. Sci. 2017, 24, 49. [Google Scholar] [CrossRef]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Martins, G.A.; Liao, J.; Magnusdottir, E.; Grunig, G.; Perez, R.K.; Calame, K.L. Blimp-1 attenuates Th1 differentiation by repression of ifng, tbx21, and bcl6 gene expression. J. Immunol. 2008, 181, 2338–2347. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Yao, S.; Pham, D.; Jiang, L.; Wright, J.; Sawant, D.; Dent, A.L.; Braciale, T.J.; Kaplan, M.H.; Sun, J. Cytokine-dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J. Virol. 2013, 87, 11884–11893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Youngblood, B.; Austin, J.W.; Mohammed, A.U.R.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Palmisiano, N.D.; Wang, M.; Jia, B.; Bayerl, M.; et al. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 124. [Google Scholar] [CrossRef]
- Lin, M.-H.; Yeh, L.-T.; Chen, S.-J.; Chiou, H.-Y.C.; Chu, C.-C.; Yen, L.B.; Lin, K.-I.; Chang, D.-M.; Sytwu, H.-K. T cell-specific BLIMP-1 deficiency exacerbates experimental autoimmune encephalomyelitis in nonobese diabetic mice by increasing Th1 and Th17 cells. Clin. Immunol. 2014, 151, 101–113. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhigarev, D.; Varshavsky, A.; MacFarlane, A.W., IV; Jayaguru, P.; Barreyro, L.; Khoreva, M.; Dulaimi, E.; Nejati, R.; Drenberg, C.; Campbell, K.S. Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology. Cancers 2022, 14, 3352. https://doi.org/10.3390/cancers14143352
Zhigarev D, Varshavsky A, MacFarlane AW IV, Jayaguru P, Barreyro L, Khoreva M, Dulaimi E, Nejati R, Drenberg C, Campbell KS. Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology. Cancers. 2022; 14(14):3352. https://doi.org/10.3390/cancers14143352
Chicago/Turabian StyleZhigarev, Dmitry, Asya Varshavsky, Alexander W. MacFarlane, IV, Prathiba Jayaguru, Laura Barreyro, Marina Khoreva, Essel Dulaimi, Reza Nejati, Christina Drenberg, and Kerry S. Campbell. 2022. "Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology" Cancers 14, no. 14: 3352. https://doi.org/10.3390/cancers14143352
APA StyleZhigarev, D., Varshavsky, A., MacFarlane, A. W., IV, Jayaguru, P., Barreyro, L., Khoreva, M., Dulaimi, E., Nejati, R., Drenberg, C., & Campbell, K. S. (2022). Lymphocyte Exhaustion in AML Patients and Impacts of HMA/Venetoclax or Intensive Chemotherapy on Their Biology. Cancers, 14(14), 3352. https://doi.org/10.3390/cancers14143352