
Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Normal Fibroblasts and CAFs
1.2. Hypoxia Signaling Pathways
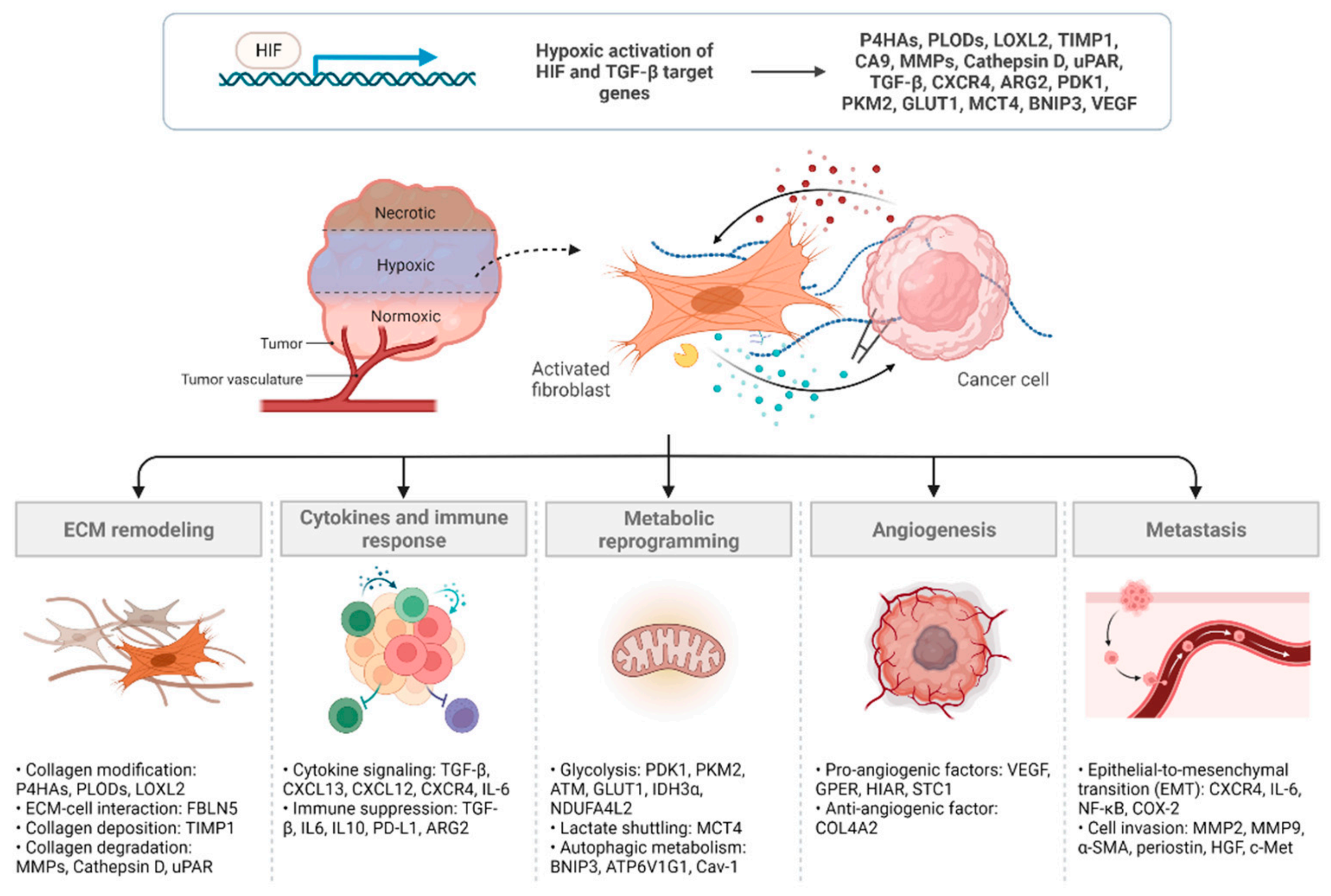
2. Mechanisms Underlying CAF Regulation and Function in Hypoxia
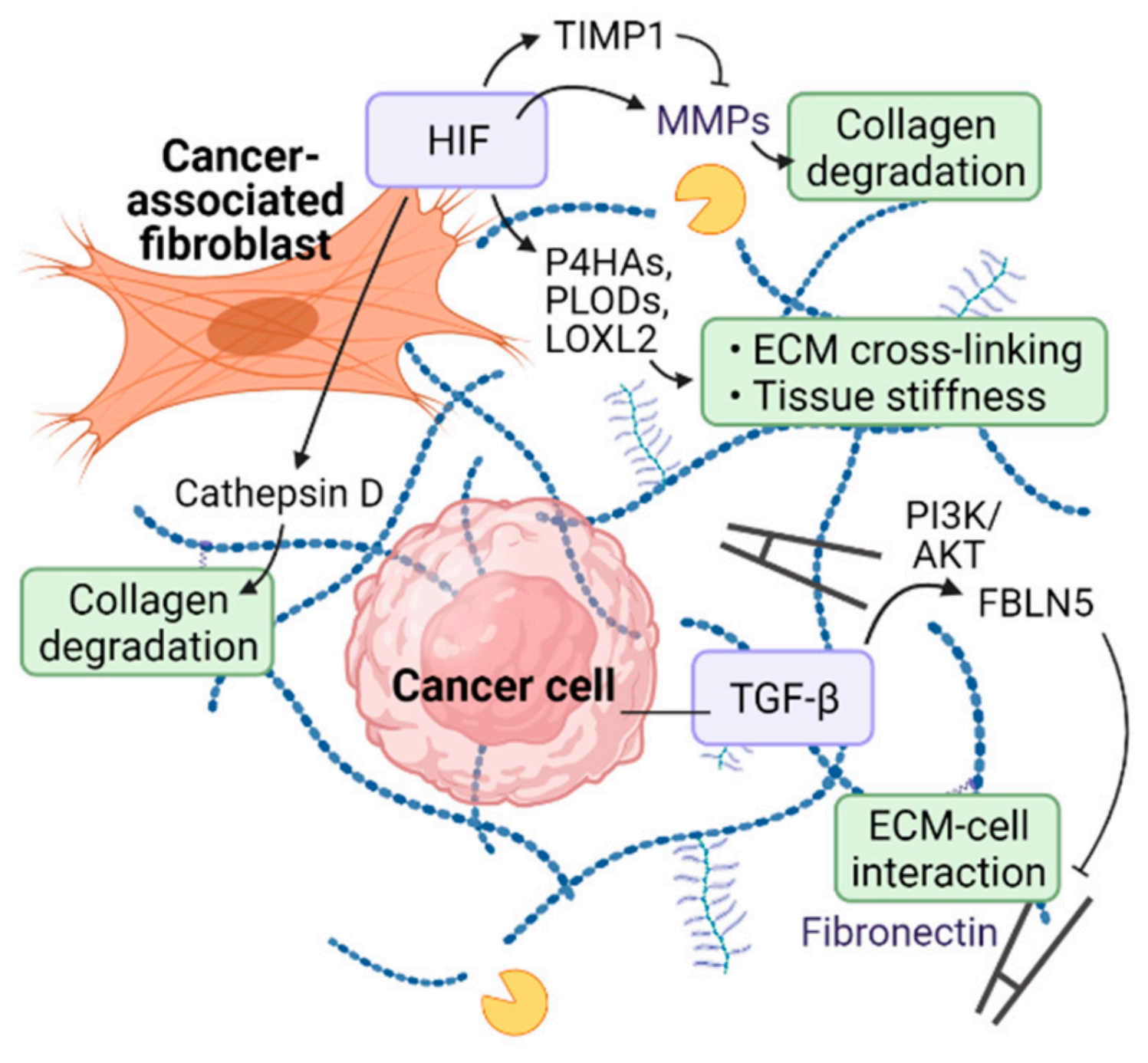
2.1. ECM Remodeling
2.2. Cytokines and Immune Response
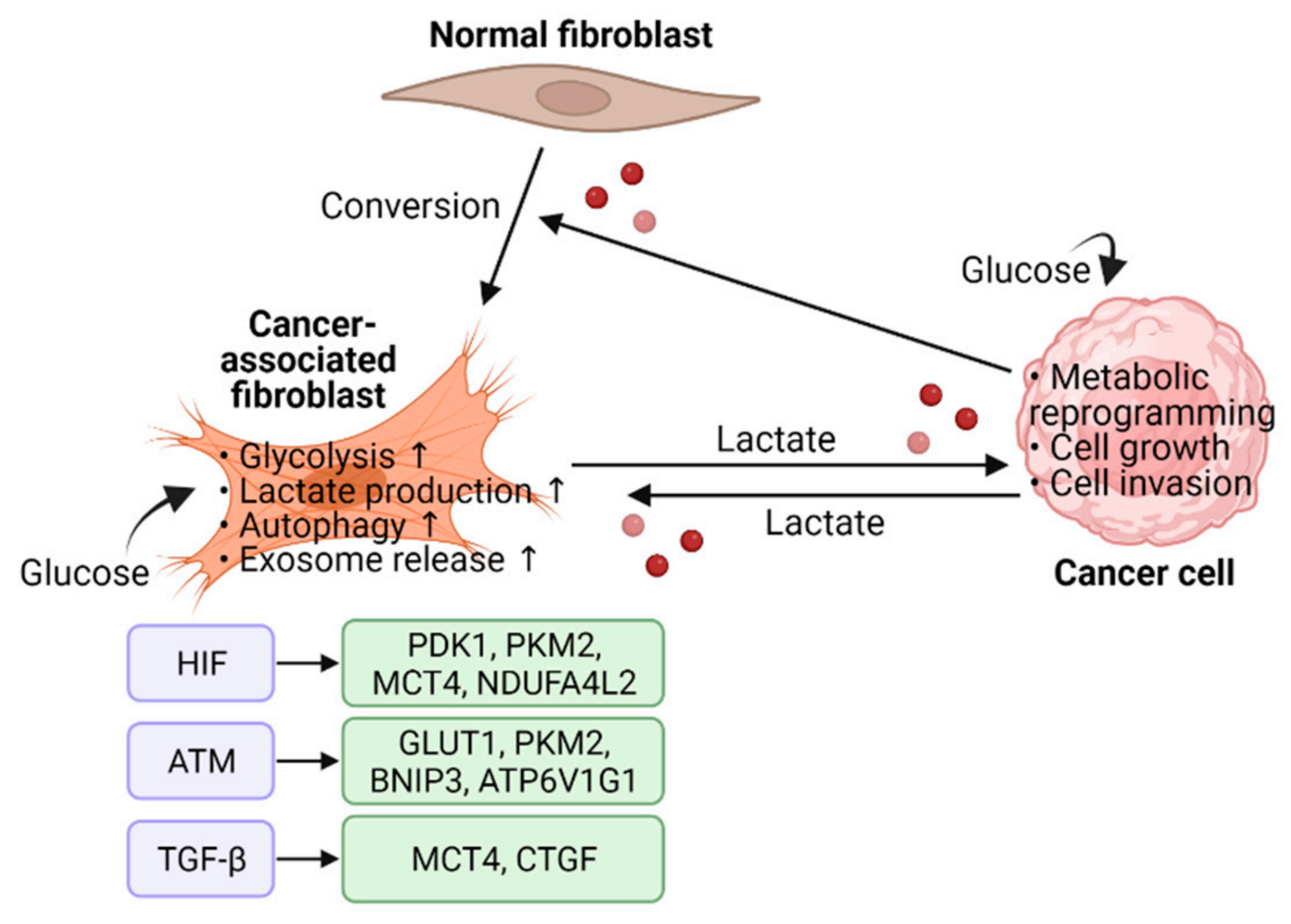
2.3. Metabolic Reprogramming
2.4. Angiogenesis
2.5. Metastasis
3. Targeting CAFs for Cancer Therapy
3.1. Targeting CAF Activation and Function
3.2. CAF Depletion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lynch, M.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Ping, Q.R.; Yan, R.P.; Cheng, X.; Wang, W.J.; Zhong, Y.M.; Hou, Z.L.; Shi, Y.Q.; Wang, C.H.; Li, R.H. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 1074. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast–Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 609653. [Google Scholar] [CrossRef]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Van Linthout, S.; Miteva, K.; Tschöpe, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, L.; Liu, X.; Kammertoens, T.; Blankenstein, T.; Qin, Z. Fibroblast-Specific Protein 1/S100A4–Positive Cells Prevent Carcinoma through Collagen Production and Encapsulation of Carcinogens. Cancer Res. 2013, 73, 2770–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.; Hsu, K.-S.; Yu, G.-J.; et al. Tumor stroma–targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.-B.; Tobin, N.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. JNCI J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Mangolini, M.; Gotte, F.; Ammon, T.; Oelsner, M.; Hitpass, L.K.; Durig, J.; Hughes, K.; Lutzny, G.; Strobl, U.; Ringshausen, I. NOTCH-signaling in the tumor microenvironment is required for canonical WNT signaling in CLL cells. Leuk. Lymphoma 2017, 58, 125–126. [Google Scholar]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Coleman, M.; Brekken, R. Perspectives on Hypoxia Signaling in Tumor Stroma. Cancers 2021, 13, 3070. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003, 22, 15–24. [Google Scholar] [CrossRef]
- Qi, Y.; Xu, R. Roles of PLODs in Collagen Synthesis and Cancer Progression. Front. Cell Dev. Biol. 2018, 6, 66. [Google Scholar] [CrossRef]
- Rappu, P.; Salo, A.M.; Myllyharju, J.; Heino, J. Role of prolyl hydroxylation in the molecular interactions of collagens. Essays Biochem. 2019, 63, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary, B.; Savage, A.; Summers, B.; Silver, R. Collagen Prolyl 4-Hydroxylase as a Therapeutic Target in Idiopathic Pulmonary Fibrosis. Am. J. Resp. Crit. Care 2019, 199, A5390. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible Factor 1 (HIF-1) Promotes Extracellular Matrix Remodeling under Hypoxic Conditions by Inducing P4HA1, P4HA2, and PLOD2 Expression in Fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brereton, C.J.; Yao, L.; Davies, E.R.; Zhou, Y.; Vukmirovic, M.; Bell, J.A.; Wang, S.; Ridley, R.A.; Dean, L.S.; Andriotis, O.G.; et al. Pseudohypoxic HIF pathway activation dysregulates collagen structure-function in human lung fibrosis. eLife 2022, 11, e69348. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Hagopian, M.; Wang, M.; Brekken, R.A. Hypoxia and Transforming Growth Factor beta Cooperate to Induce Fibulin-5 Expression in Pancreatic Cancer. J. Biol. Chem. 2016, 291, 22244–22252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Mimura, I.; Shoji, K.; Tanaka, T.; Nangaku, M. Hypoxia and fibrosis in chronic kidney disease: Crossing at pericytes. Kidney Int. Suppl. 2014, 4, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Tamamori, M.; Ito, H.; Hiroe, M.; Marumo, F.; Hata, R. Stimulation of collagen synthesis in rat cardiac fibroblasts by exposure to hypoxic culture conditions and suppression of the effect by natriuretic peptides. Cell Biol. Int. 1997, 21, 175–180. [Google Scholar] [CrossRef]
- Kang, Y.; Roh, M.R.; Rajadurai, S.; Rajadurai, A.; Kumar, R.; Njauw, C.-N.; Zheng, Z.; Tsao, H. Hypoxia and HIF-1α Regulate Collagen Production in Keloids. J. Investig. Dermatol. 2020, 140, 2157–2165. [Google Scholar] [CrossRef]
- Colpaert, C.G.; Vermeulen, P.B.; Fox, S.; Harris, A.; Dirix, L.Y.; Van Marck, E.A. The Presence of a Fibrotic Focus in Invasive Breast Carcinoma Correlates with the Expression of Carbonic Anhydrase IX and is a Marker of Hypoxia and Poor Prognosis. Breast Cancer Res. Treat. 2003, 81, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.P.; Zelník, V.; Opavský, R.; Zat’Ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J.; et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994, 9, 2877–2888. [Google Scholar]
- Muñoz-Nájar, U.M.; Neurath, K.M.; Vumbaca, F.; Claffey, K.P. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene 2005, 25, 2379–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.C.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1alpha in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrella, B.L.; Lohi, J.; E Brinckerhoff, C. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel–Lindau renal cell carcinoma. Oncogene 2004, 24, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Capony, F.; Rougeot, C.; Montcourrier, P.; Cavailles, V.; Salazar, G.; Rochefort, H. Increased secretion, altered processing, and glycosylation of pro-cathepsin D in human mammary cancer cells. Cancer Res. 1989, 49, 3904–3909. [Google Scholar]
- Brouillet, J.P.; Dufour, F.; Lemamy, G.; Garcia, M.; Schlup, N.; Grenier, J.; Mani, J.C.; Rochefort, H. Increased cathepsin D level in the serum of patients with metastatic breast carcinoma detected with a specific pro-cathepsin D immunoassay. Cancer 1997, 79, 2132–2136. [Google Scholar] [CrossRef]
- Jarosz, D.; Hamer, P.; Tenney, D.; Zabrecky, J. Elevated levels of pro-cathepsin-d in the plasma of breast-cancer patients. Int. J. Oncol. 1995, 6, 859–865. [Google Scholar] [CrossRef]
- Graham, C.H.; Forsdike, J.; Fitzgerald, C.J.; Macdonald-Goodfellow, S. Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int. J. Cancer 1999, 80, 617–623. [Google Scholar] [CrossRef]
- Büchler, P.; Reber, H.A.; Tomlinson, J.S.; Hankinson, O.; Kallifatidis, G.; Friess, H.; Herr, I.; Hines, O.J. Transcriptional Regulation of Urokinase-type Plasminogen Activator Receptor by Hypoxia-Inducible Factor 1 Is Crucial for Invasion of Pancreatic and Liver Cancer. Neoplasia 2009, 11, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Giannoni, E.; Taddei, L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013, 12, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrech, D.S.; Longaker, M.T.; Mehrara, B.J.; Saadeh, P.; Chin, G.S.; Gerrets, R.P.; Chau, D.C.; Rowe, N.M.; Gittes, G.K. Fibroblast Response to Hypoxia: The Relationship between Angiogenesis and Matrix Regulation. J. Surg. Res. 1999, 84, 127–133. [Google Scholar] [CrossRef]
- Zhao, B.; Guan, H.; Liu, J.-Q.; Zheng, Z.; Zhou, Q.; Zhang, J.; Su, L.-L.; Hu, D.-H. Hypoxia drives the transition of human dermal fibroblasts to a myofibroblast-like phenotype via the TGF-β1/Smad3 pathway. Int. J. Mol. Med. 2016, 39, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Falanga, V.; Martin, T.A.; Takagi, H.; Kirsner, R.S.; Helfman, T.; Pardes, J.; Ochoa, M.S. Low oxygen tension increases mRNA levels of alpha 1 (I) procollagen in human dermal fibroblasts. J. Cell. Physiol. 1993, 157, 408–412. [Google Scholar] [CrossRef]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmouliere, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Ammirante, M.; Shalapour, S.; Kang, Y.; Jamieson, C.A.M.; Karin, M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci. USA 2014, 111, 14776–14781. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Long, Q.; Zhang, L.; Shi, Y.; Liu, X.; Li, X.; Guan, B.; Tian, Y.; Wang, X.; Li, L.; et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1α signaling. Int. J. Oncol. 2015, 47, 2064–2072. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Ziani, L.; Buart, S.; Chouaib, S.; Thiery, J. Hypoxia increases melanoma-associated fibroblasts immunosuppressive potential and inhibitory effect on T cell-mediated cytotoxicity. OncoImmunology 2021, 10, 1950953. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.-Y.; Yang, J.-T.; Huang, T.-H.; Liu, H.-W. Crosstalk with Cancer-Associated Fibroblasts Increases the Growth and Radiation Survival of Cervical Cancer Cells. Radiat. Res. 2014, 181, 540–547. [Google Scholar] [CrossRef]
- Tsai, K.K.; Chuang, E.Y.-Y.; Little, J.B.; Yuan, Z.-M. Cellular Mechanisms for Low-Dose Ionizing Radiation–Induced Perturbation of the Breast Tissue Microenvironment. Cancer Res. 2005, 65, 6734–6744. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.E.; Paget, J.T.E.; Khan, A.; Harrington, K. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. 2019, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Chiavarina, B.; Martinez-Outschoorn, U.; Whitaker-Menezes, D.; Howell, A.; Tanowitz, H.B.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Metabolic reprogramming and two-compartment tumor metabolism. Cell Cycle 2012, 11, 3280–3289. [Google Scholar] [CrossRef] [Green Version]
- Del Rey, M.J.; Valín, A.; Usategui, A.; Garcia, F.J.B.; Sánchez-Aragó, M.; Cuezva, J.M.; Galindo, M.; Bravo, B.; Cañete, J.D.; Blanco, F.J.; et al. Hif-1α Knockdown Reduces Glycolytic Metabolism and Induces Cell Death of Human Synovial Fibroblasts Under Normoxic Conditions. Sci. Rep. 2017, 7, 3644. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, A.M.; Lone, A.; Betts, D.H.; Cumming, R.C. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 2020, 10, 8388. [Google Scholar] [CrossRef]
- Sun, K.; Tang, S.; Hou, Y.; Xi, L.; Chen, Y.; Yin, J.; Peng, M.; Zhao, M.; Cui, X.; Liu, M. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EbioMedicine 2019, 41, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.; Peng, M.; Liu, S.; Liu, Y.; Wan, X.; Hou, Y.; Qin, Y.; Yang, L.; Chen, S.; Zeng, H.; et al. Hypoxia-stimulated ATM activation regulates autophagy-associated exosome release from cancer-associated fibroblasts to promote cancer cell invasion. J. Extracell. Vesicles 2021, 10, e12146. [Google Scholar] [CrossRef]
- Becker, L.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Jena, B.C.; Das, C.K.; Banerjee, I.; Bharadwaj, D.; Majumder, R.; Das, S.; Biswas, A.; Kundu, M.; Roy, P.K.; Kundu, C.N.; et al. TGF-β1 induced autophagy in cancer associated fibroblasts during hypoxia contributes EMT and glycolysis via MCT4 upregulation. Exp. Cell Res. 2022, 417, 113195. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [Green Version]
- Chiavarina, B.; Whitaker-Menezes, D.; Migneco, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Tanowitz, H.B.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. HIF1-alpha functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells. Cell Cycle 2010, 9, 3534–3551. [Google Scholar] [CrossRef] [Green Version]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Kudo, M. Signaling Pathways Governing Tumor Angiogenesis. Oncology 2011, 81, 24–29. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Evans, C.; Weidemann, A.; Takeda, N.; Lee, Y.S.; Stockmann, C.; Branco-Price, C.; Brandberg, F.; Leone, G.; Ostrowski, M.C.; et al. Loss of Fibroblast HIF-1α Accelerates Tumorigenesis. Cancer Res. 2012, 72, 3187–3195. [Google Scholar] [CrossRef] [Green Version]
- Kugeratski, F.G.; Atkinson, S.J.; Neilson, L.J.; Lilla, S.; Knight, J.R.P.; Serneels, J.; Juin, A.; Ismail, S.; Bryant, D.M.; Markert, E.K.; et al. Hypoxic cancer–associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci. Signal. 2019, 12, eaan8247. [Google Scholar] [CrossRef] [Green Version]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H.M. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Chen, D.; Hurwitz, H. Combinations of Bevacizumab with Cancer Immunotherapy. Cancer J. 2018, 24, 193–204. [Google Scholar] [CrossRef]
- Kuchnio, A.; Moens, S.; Bruning, U.; Kuchnio, K.; Cruys, B.; Thienpont, B.; Broux, M.; Ungureanu, A.A.; de Oliveira, R.L.; Bruyère, F.; et al. The Cancer Cell Oxygen Sensor PHD2 Promotes Metastasis via Activation of Cancer-Associated Fibroblasts. Cell Rep. 2015, 12, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.; Pilkington, R.; Munoz, A.G.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; von Kriegsheim, A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Kang, J.; Gee, H.Y.; Park, J.-W. A novel HIF1AN substrate KANK3 plays a tumor-suppressive role in hepatocellular carcinoma. Cell Biol. Int. 2017, 42, 303–312. [Google Scholar] [CrossRef]
- Kim, I.; Shin, S.-H.; Lee, J.E.; Park, J.-W. Oxygen sensor FIH inhibits HACE1-dependent ubiquitination of Rac1 to enhance metastatic potential in breast cancer cells. Oncogene 2019, 38, 3651–3666. [Google Scholar] [CrossRef]
- Kim, I.; Park, J.-W. Hypoxia-driven epigenetic regulation in cancer progression: A focus on histone methylation and its modifying enzymes. Cancer Lett. 2020, 489, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Lippl, K.; Tian, Y.-M.; Pegg, H.B.; Figg, W.D.J.; Abboud, M.I.; Heilig, R.; Fischer, R.; Myllyharju, J.; Schofield, C.J.; et al. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. eLife 2019, 8, e46490. [Google Scholar] [CrossRef]
- Manresa, M.C.; Tambuwala, M.M.; Radhakrishnan, P.; Harnoss, J.M.; Brown, E.; Cavadas, M.A.M.A.; Keogh, C.E.C.E.; Cheong, A.; Barrett, K.E.K.E.; Cummins, E.P.; et al. Hydroxylase inhibition regulates inflammation-induced intestinal fibrosis through the suppression of ERK-mediated TGF-beta 1 signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G405. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.D.; Pedersen, J.T.; Venning, F.A.; Singh, L.B.; Moeendarbary, E.; Charras, G.; Cox, T.; Sahai, E.; Erler, J.T. Hypoxia and loss of PHD 2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015, 16, 1394–1408. [Google Scholar] [CrossRef] [PubMed]
- Janbandhu, V.; Tallapragada, V.; Patrick, R.; Li, Y.; Abeygunawardena, D.; Humphreys, D.T.; Martin, E.M.; Ward, A.O.; Contreras, O.; Farbehi, N.; et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell 2021, 29, 281–297.e12. [Google Scholar] [CrossRef]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Koga, Y.; Miyazaki, K. Tumor–stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int. J. Cancer 2006, 119, 2750–2759. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, J.; Bao, C. Exosomal circEIF3K from cancer-associated fibroblast promotes colorectal cancer (CRC) progression via miR-214/PD-L1 axis. BMC Cancer 2021, 21, 933. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer Associated Fibroblasts Exploit Reactive Oxygen Species Through a Proinflammatory Signature Leading to Epithelial Mesenchymal Transition and Stemness. Antioxid. Redox Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Mironchik, Y.; Jacob, D.; Goggins, E.; Kakkad, S.; Ofori, F.; Dore-Savard, L.; Bharti, S.K.; Wildes, F.; Penet, M.-F.; et al. Hypoxia theranostics of a human prostate cancer xenograft and the resulting effects on the tumor microenvironment. Neoplasia 2020, 22, 679–688. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts—Heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Ohshio, Y.; Hanaoka, J.; Kontani, K.; Teramoto, K. Tranilast Inhibits the Function of Cancer-Associated Fibroblasts Responsible for the Induction of Immune Suppressor Cell Types. Scand. J. Immunol. 2014, 80, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashiro, M.; Murahashi, K.; Matsuoka, T.; Nakazawa, K.; Tanaka, H.; Osaka, H.; Koyama, T.; Ohira, M.; Chung, K.H. Tranilast (N-3,4-dimethoxycinamoyl anthranilic acid): A novel inhibitor of invasion-stimulating interaction between gastric cancer cells and orthotopic fibroblasts. Anticancer Res. 2003, 23, 3899–3904. [Google Scholar] [PubMed]
- Zhang, W.; Ma, J.; Wang, S.; Huang, T.; Xia, M. Tranilast attenuates neuropathic pain during type-2 diabetes by inhibiting hypoxia-induced pro-inflammatory cytokines in Zucker diabetic fatty rat model. Arch. Physiol. Biochem. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Funaki, S.; Fukui, E.; Kimura, K.; Kanou, T.; Ose, N.; Minami, M.; Shintani, Y. Effects of pirfenidone targeting the tumor microenvironment and tumor-stroma interaction as a novel treatment for non-small cell lung cancer. Sci. Rep. 2020, 10, 10900. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwitz, S.; Turkowski, K.; Nitschkowski, D.; Weigert, A.; Brandenburg, J.; Reiling, N.; Thomas, M.; Reck, M.; Drömann, D.; Seeger, W.; et al. The Multi-Modal Effect of the Anti-fibrotic Drug Pirfenidone on NSCLC. Front. Oncol. 2020, 9, 1550. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone Inhibits Pancreatic Cancer Desmoplasia by Regulating Stellate Cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yin, Z.; Qin, W.; Ma, X.; Zhang, Y.; Liu, E.; Chu, Y. Pirfenidone Inhibits Hypoxic Pulmonary Hypertension through the NADPH/ROS/p38 Pathway in Adventitial Fibroblasts in the Pulmonary Artery. Mediat. Inflamm. 2020, 2020, 2604967. [Google Scholar] [CrossRef]
- Dauer, P.; Zhao, X.; Gupta, V.K.; Sharma, N.; Kesh, K.; Gnamlin, P.; Dudeja, V.; Vickers, S.M.; Banerjee, S.; Saluja, A. Inactivation of Cancer-Associated-Fibroblasts Disrupts Oncogenic Signaling in Pancreatic Cancer Cells and Promotes Its Regression. Cancer Res. 2018, 78, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Modi, S.; McGinn, O.; Zhao, X.; Dudeja, V.; Ramakrishnan, S.; Saluja, A.K. Impaired Synthesis of Stromal Components in Response to Minnelide Improves Vascular Function, Drug Delivery, and Survival in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 415–425. [Google Scholar] [CrossRef] [Green Version]
- McGinn, O.; Gupta, V.K.; Dauer, P.; Arora, N.; Sharma, N.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 2017, 7, 7872. [Google Scholar] [CrossRef] [PubMed]
- Skorupan, N.; Ahmad, M.I.; Steinberg, S.M.; Trepel, J.B.; Cridebring, D.; Han, H.; Von Hoff, D.D.; Alewine, C. Abstract PO-054: A phase II trial of the super-enhancer inhibitor Minnelide in advanced refractory adenosquamous carcinoma of the pancreas (ASCP). Cancer Res. 2021, 81, 37–38. [Google Scholar] [CrossRef]
- Tang, Y.-A.; Chen, Y.-F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.-C.; Su, C.-H.; Liu, C.-Y.; Chen, T.-H.; Chen, C.-P.; Wang, H.-S. Transforming growth factor-β induces CD44 cleavage that promotes migration of MDA-MB-435s cells through the up-regulation of membrane type 1-matrix metalloproteinase. Int. J. Cancer 2009, 124, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.C.; Yin, L.; Yang, J.J.; Lee, H.-W.; Ivanov, I.; Yan, C.; Yang, H.; Grossniklaus, H.E.; Wang, S.; Ma, C.; et al. Rational design of a protein that binds integrin αvβ3 outside the ligand binding site. Nat. Commun. 2016, 7, 11675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turaga, R.C.; Sharma, M.; Mishra, F.; Krasinskas, A.; Yuan, Y.; Yang, J.J.; Wang, S.; Liu, C.; Li, S.; Liu, Z.-R. Modulation of Cancer-Associated Fibrotic Stroma by An Integrin αvβ3 Targeting Protein for Pancreatic Cancer Treatment. Cell. Mol. Gastroenterol. Hepatol. 2020, 11, 161–179. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27, 2040206619829382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2015, 35, 816–826. [Google Scholar] [CrossRef]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X.-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer—Spotlight on Cancer-Associated Fibroblasts. Cancers 2020, 12, 3650. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, Y.; Matsui, T.; Takakura, N. CD44 Expressed on Cancer-Associated Fibroblasts Is a Functional Molecule Supporting the Stemness and Drug Resistance of Malignant Cancer Cells in the Tumor Microenvironment. Stem Cells 2013, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, F.; Okoye, I.; Esmaily, M.; Chaleshtari, M.G.; Masjedi, A.; Azizi, G.; Irandoust, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F. Hypoxia inducible factors in the tumor microenvironment as therapeutic targets of cancer stem cells. Life Sci. 2019, 237, 116952. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel–Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Romero, D. Belzutifan is active in VHL-related cancers. Nat. Rev. Clin. Oncol. 2021, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gong, K. Belzutifan: A novel therapy for von Hippel–Lindau disease. Nat. Rev. Nephrol. 2022, 18, 205–206. [Google Scholar] [CrossRef]
- Dahl, K.D.C.; Robertson, S.E.; Weaver, V.M.; Simon, M.C. Hypoxia-inducible factor regulates alpha v ss 3 integrin cell surface expression. Mol. Biol. Cell 2005, 16, 1901–1912. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. αv Integrins in Angiogenesis and Cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Xiao, L.; Joo, K.-I.; Liu, Y.; Zhang, C.; Liu, S.; Conti, P.S.; Li, Z.; Wang, P. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int. J. Cancer 2015, 138, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Investig. 2009, 119, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolás-Boluda, A.; Vaquero, J.; Laurent, G.; Renault, G.; Bazzi, R.; Donnadieu, E.; Roux, S.; Fouassier, L.; Gazeau, F. Photothermal Depletion of Cancer-Associated Fibroblasts Normalizes Tumor Stiffness in Desmoplastic Cholangiocarcinoma. ACS Nano 2020, 14, 5738–5753. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Drugs | Mechanisms | Effects | Cancer Models | Status | References |
|---|---|---|---|---|---|
| Tranilast | TGF-β inhibition | Inhibits CAF-mediated fibrosis by reducing pro-inflammatory cytokines | Lymphoma, Lewis lung carcinoma, gastric cancer | Preclinical | [111,112,113] |
| Pirfenidone | TGF-β inhibition | Inhibits CAF activation and proliferation | Non-small-cell lung carcinoma, pancreatic cancer | Preclinical | [114,115,116,117,118] |
| Minnelide | TGF-β and HIF inhibition | Induces CAF inactivation | Pancreatic cancer | Phase I-II | [119,120,121,122] |
| SD208 | TGF-β inhibition | Reduces CAF-induced chemoresistance | Colorectal cancer | Preclinical | [123] |
| GANT61 | GLI inhibition | Reduces CAF-induced chemoresistance | Colorectal cancer | Preclinical | [123] |
| PD98059 | ERK1/2 inhibition | Inhibits CAF signaling | Melanoma | Preclinical | [124] |
| LY294002 | PI3K inhibition | Inhibits CAF signaling | Melanoma | Preclinical | [124] |
| ProAgio | αvβ3 inhibition | Induces apoptosis | Pancreatic cancer | Phase I | [125,126] |
| AMD3100 | CXCR4 inhibition | Inhibits CAF-mediated immune evasion | Multiple myeloma, non-Hodgkin’s lymphoma, pancreatic ductal adenocarcinoma | Approved | [127,128,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, I.; Choi, S.; Yoo, S.; Lee, M.; Kim, I.-S. Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers 2022, 14, 3321. https://doi.org/10.3390/cancers14143321
Kim I, Choi S, Yoo S, Lee M, Kim I-S. Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers. 2022; 14(14):3321. https://doi.org/10.3390/cancers14143321
Chicago/Turabian StyleKim, Iljin, Sanga Choi, Seongkyeong Yoo, Mingyu Lee, and In-San Kim. 2022. "Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment" Cancers 14, no. 14: 3321. https://doi.org/10.3390/cancers14143321
APA StyleKim, I., Choi, S., Yoo, S., Lee, M., & Kim, I.-S. (2022). Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers, 14(14), 3321. https://doi.org/10.3390/cancers14143321