
The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
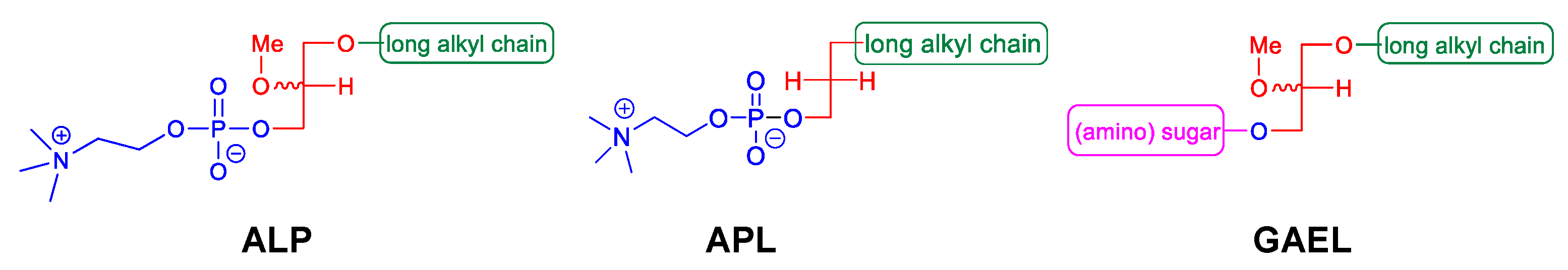
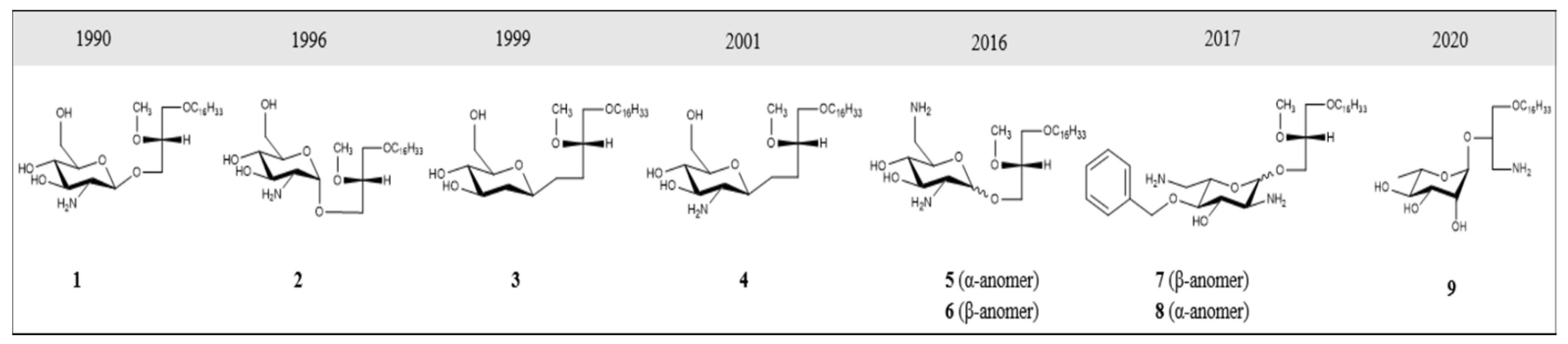
2. GAELs: How Did We Get Here?
3. GAEL Structure–Activity Relationship
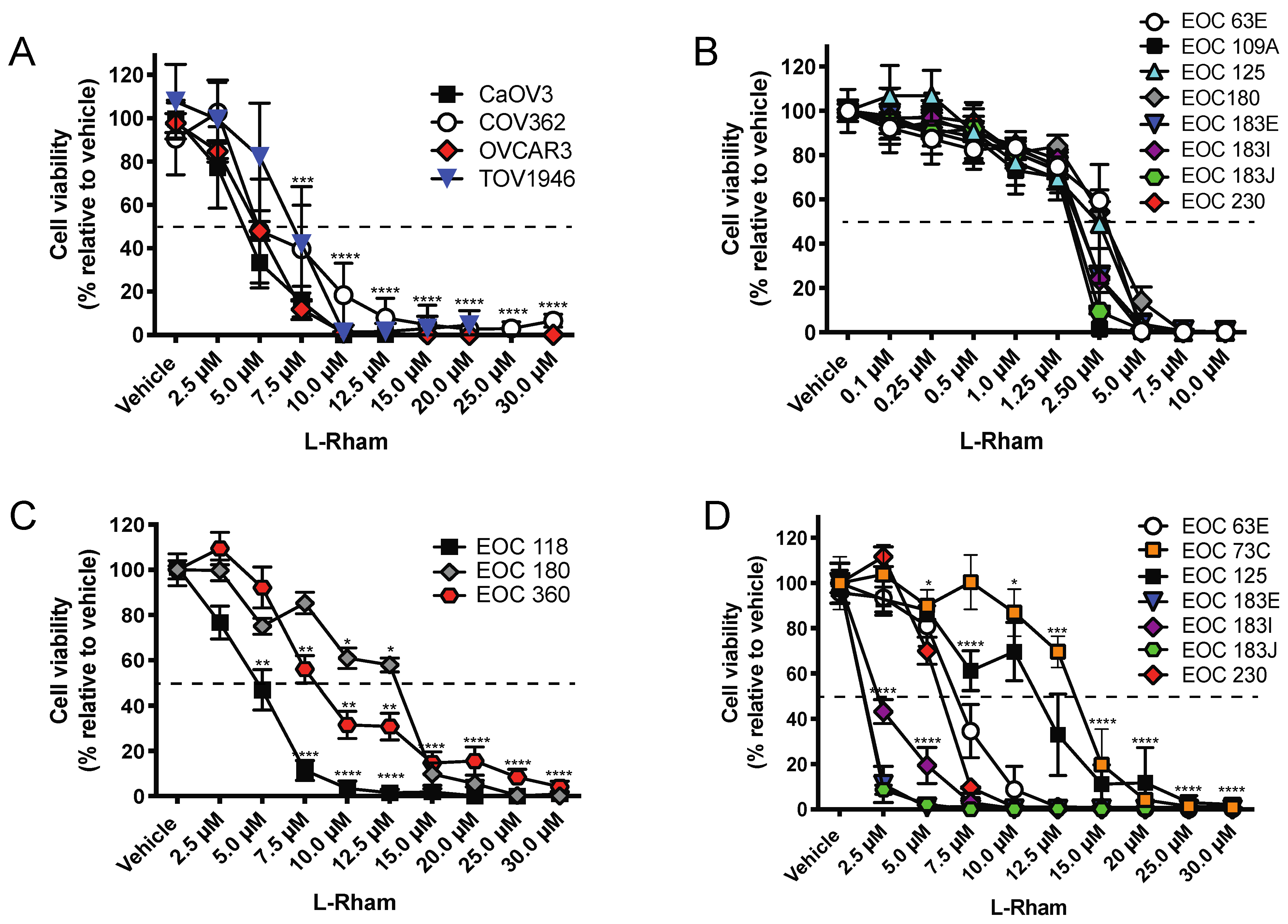
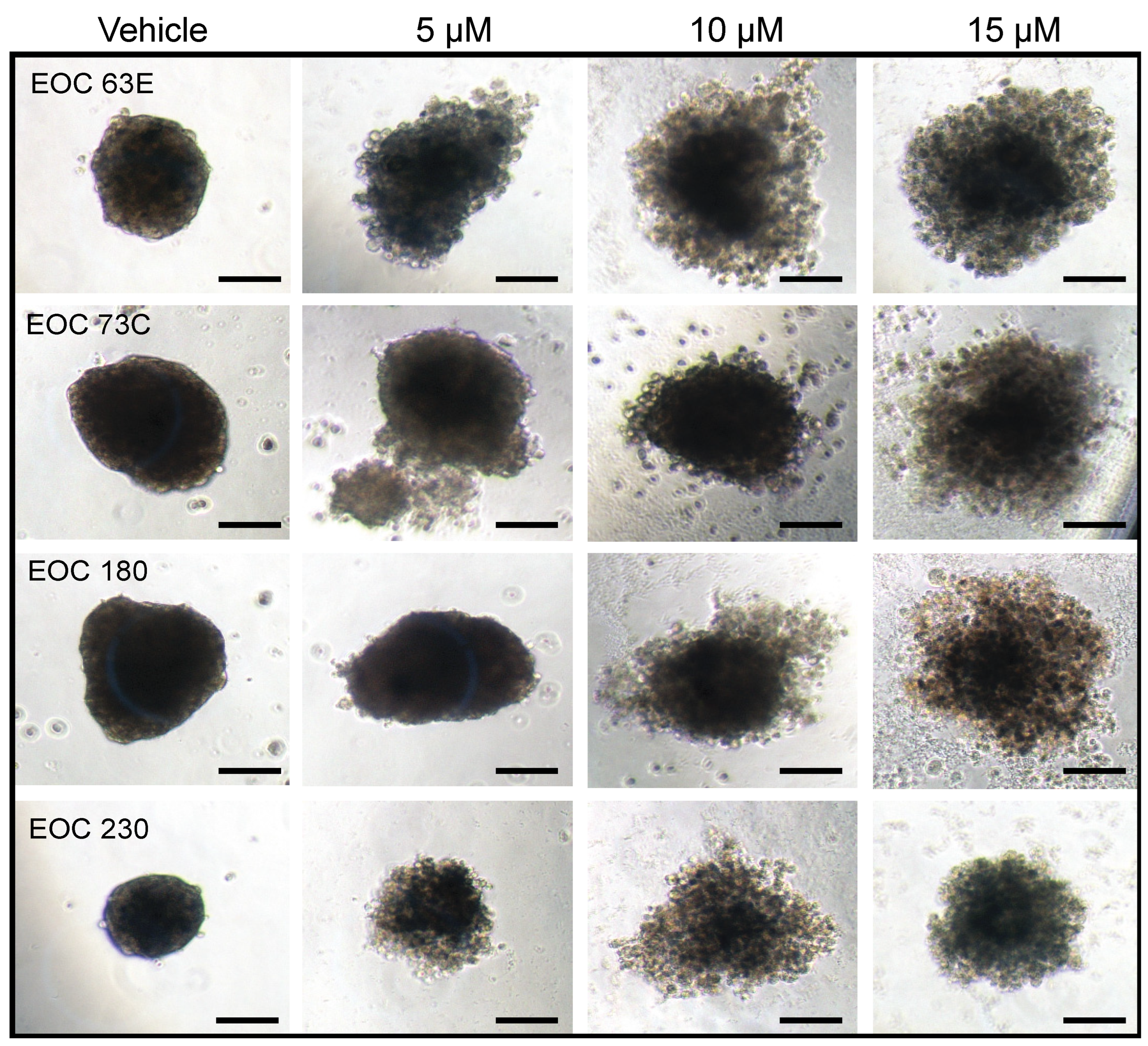
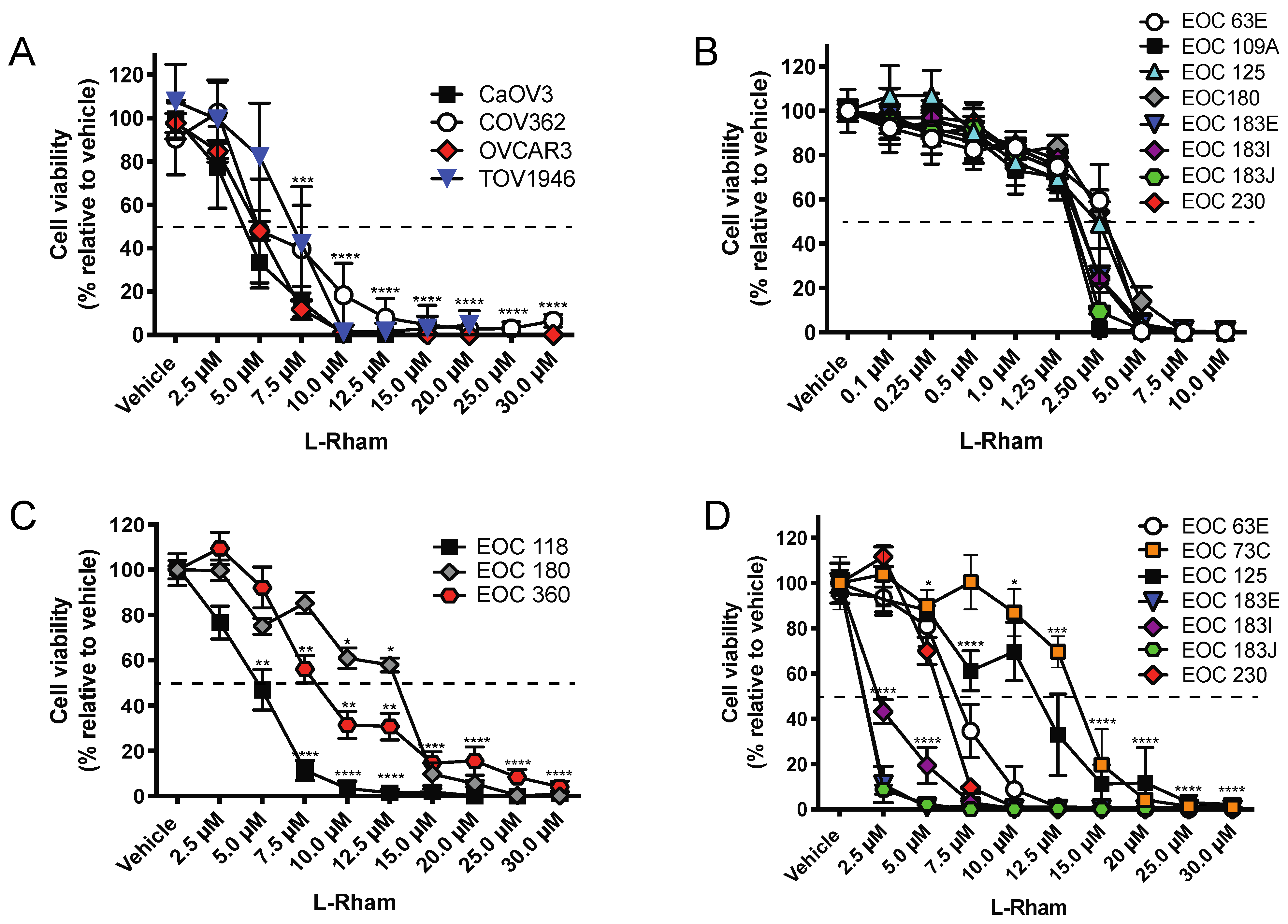
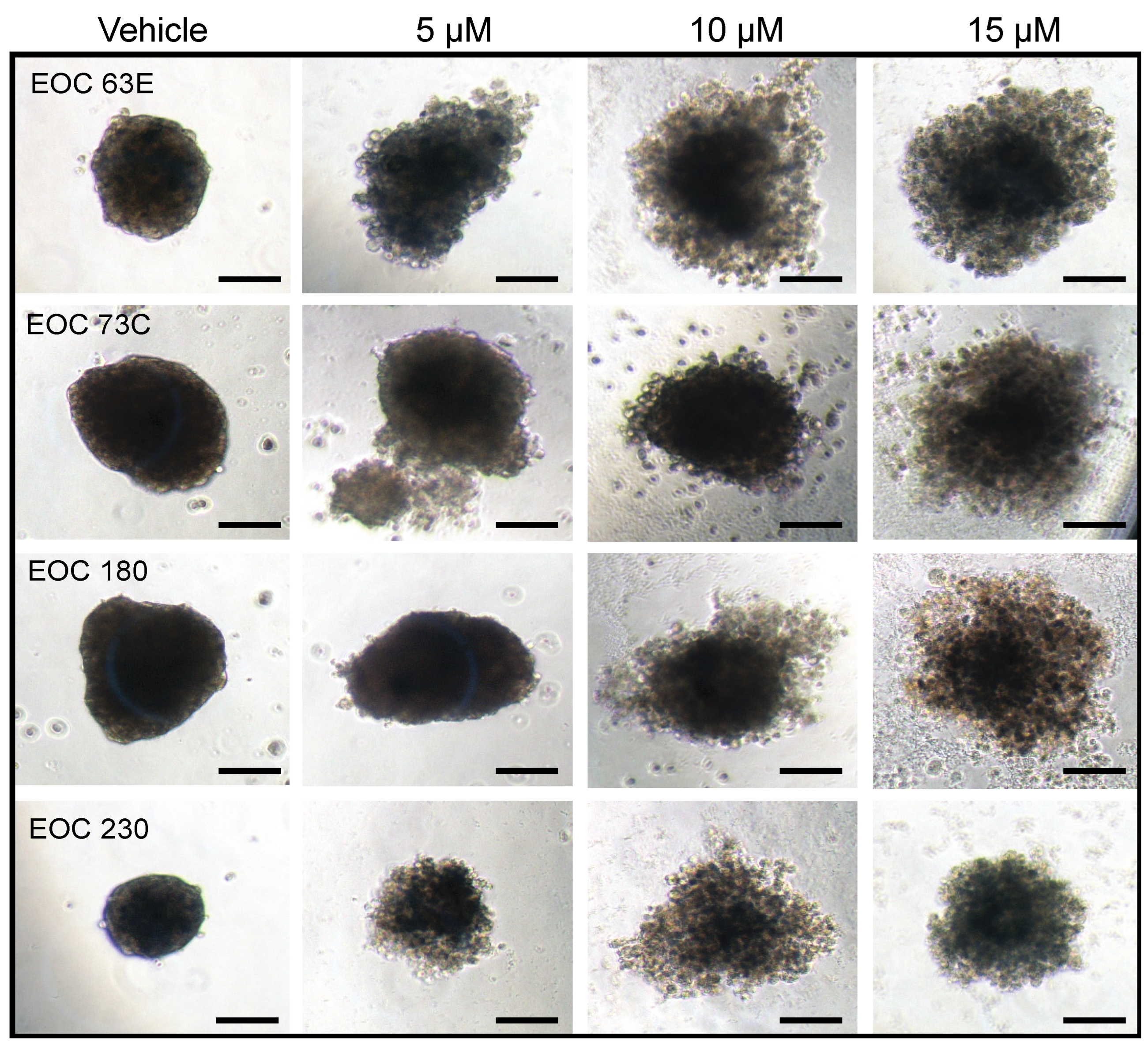
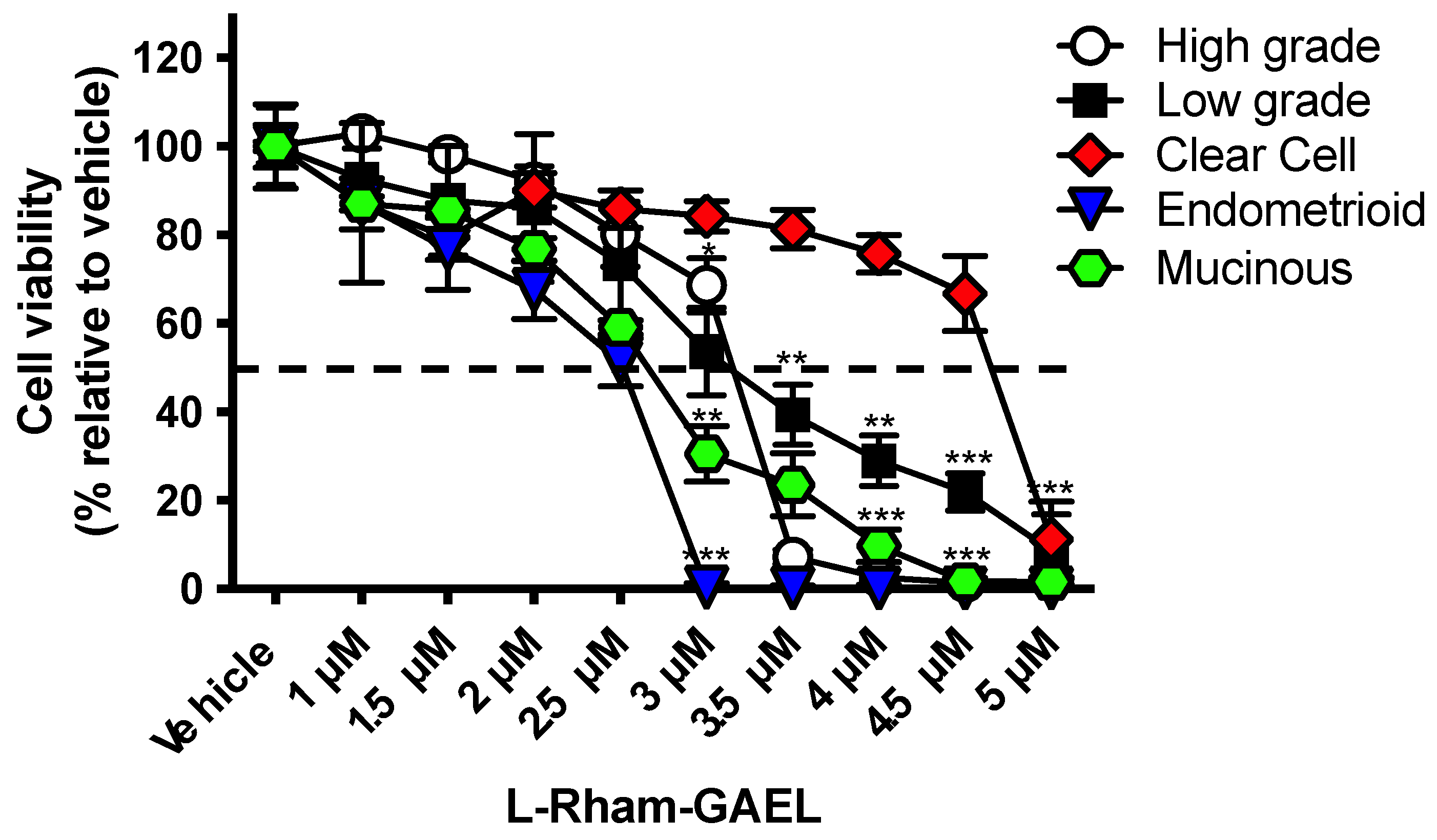
4. GAEL Cell-Killing Activity against Epithelial Ovarian Cancer
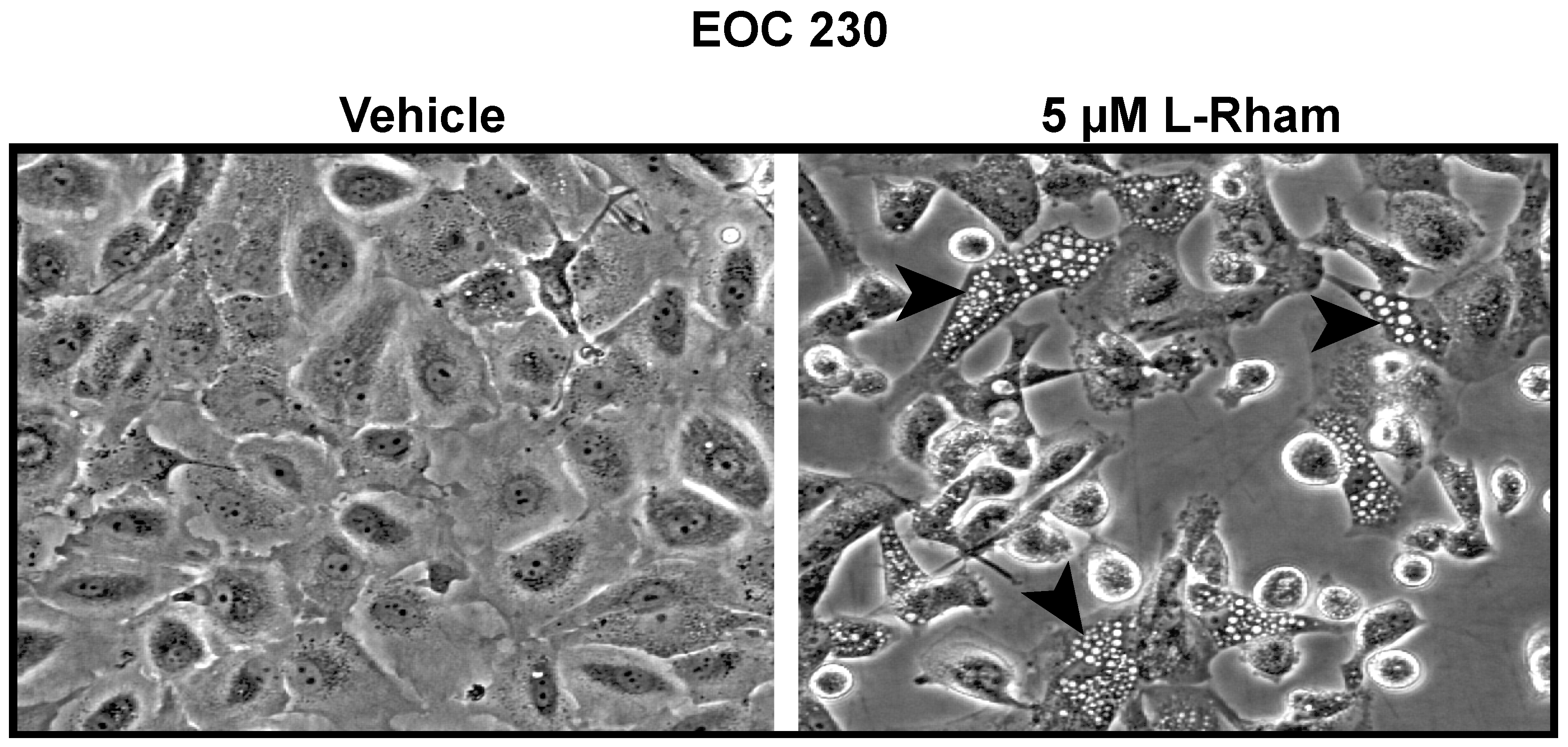
5. GAEL Mechanism of Action
6. Conclusions: Utility of GAELs and Methuosis-Inducing Compounds for EOC Treatment
Author Contributions
Funding
Conflicts of Interest
References
- CCS. Canadian Cancer Statistics 2021. 2021. Available online: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf?rev=2b9d2be7a2d34c1dab6a01c6b0a6a32d&hash=01DE85401DBF0217F8B64F2B7DF43986 (accessed on 14 June 2022).
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C. Effect of screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Basso, O.; Sampalis, J.; Karp, I.; Martins, C.; Feng, J. Assessment of symptomatic women for early diagnosis of ovarian cancer: Results from the prospective DOvE pilot project. Lancet Oncol. 2012, 13, 285–291. [Google Scholar] [CrossRef]
- Goff, B.A.; Mandel, L.S.; Drescher, C.W.; Urban, N.; Gough, S.; Schurman, K.M. Development of an ovarian cancer symptom index: Possibilities for earlier detection. Cancer 2007, 109, 221–227. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.H.; Skates, S.; Hernandez, M.A.; Bedi, D.; Bevers, T.; Leeds, L. A 2-stage ovarian cancer screening strategy using the Risk of Ovarian Cancer Algorithm (ROCA) identifies early-stage incident cancers and demonstrates high positive predictive value. Cancer 2013, 119, 3454–3461. [Google Scholar] [CrossRef]
- Altman, A.D.; Lambert, P.; Love, A.J.; Turner, D.; Lotocki, R.; Dean, E. Examining the Effects of Time to Diagnosis, Income, Symptoms, and Incidental Detection on Overall Survival in Epithelial Ovarian Cancer: Manitoba Ovarian Cancer Outcomes (MOCO) Study Group. Int. J. Gynecol. Cancer 2017, 27, 1637–1644. [Google Scholar] [CrossRef]
- Nagle, C.M.; Francis, J.E.; Nelson, A.E.; Zorbas, H.; Luxford, K.; de Fazio, A. Reducing time to diagnosis does not improve outcomes for women with symptomatic ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2011, 29, 2253–2258. [Google Scholar] [CrossRef]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: Three-year results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [Green Version]
- International Collaborative Ovarian Neoplasm Group. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: The ICON3 randomised trial. Lancet 2002, 360, 505–515. [Google Scholar] [CrossRef]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Becker, K.G.; Wood, W.H., III; Zhang, Y.; Morin, P.J. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, D.H.; Kim, M.K.; No, J.H.; Chung, H.H.; Song, Y.S. Metabolic approaches to overcoming chemoresistance in ovarian cancer. Ann. N. Y. Acad. Sci. 2011, 1229, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- May, T.; Altman, A.; McGee, J.; Lu, L.; Xu, W.; Lane, K. Examining Survival Outcomes of 852 Women with Advanced Ovarian Cancer: A Multi-institutional Cohort Study. Int. J. Gynecol. Cancer 2018, 28, 925–931. [Google Scholar] [CrossRef]
- Vergote, I.; Trope, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Katsumata, N.; Yasuda, M.; Takahashi, F.; Isonishi, S.; Jobo, T.; Aoki, D. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: A phase 3, open-label, randomised controlled trial. Lancet 2009, 374, 1331–1338. [Google Scholar] [CrossRef]
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 396–405. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Van Driel, W.J.; Koole, S.N.; Sikorska, K.; Schagen van Leeuwen, J.H.; Schreuder, H.W.R.; Hermans, R.H.M. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N. Engl. J. Med. 2018, 378, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.K.; Brady, M.F.; Penson, R.T.; Huang, H.; Birrer, M.J.; Walker, J.L. Weekly vs. Every-3-Week Paclitaxel and Carboplatin for Ovarian Cancer. N. Engl. J. Med. 2016, 374, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Walker, J.L.; Brady, M.F.; Wenzel, L.; Fleming, G.F.; Huang, H.Q.; DiSilvestro, P.A. Randomized Trial of Intravenous Versus Intraperitoneal Chemotherapy Plus Bevacizumab in Advanced Ovarian Carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. J. Clin. Oncol. 2019, 37, 1380–1390. [Google Scholar] [CrossRef]
- DiSilvestro, P.; Colombo, N.; Harter, P.; Gonzalez-Martin, A.; Ray-Coquard, I.; Coleman, R.L. Maintenance Treatment of Newly Diagnosed Advanced Ovarian Cancer: Time for a Paradigm Shift? Cancers 2021, 13, 5756. [Google Scholar] [CrossRef]
- Markman, M.; Liu, P.Y.; Wilczynski, S.; Monk, B.; Copeland, L.J.; Alvarez, R.D. Phase III randomized trial of 12 versus 3 months of maintenance paclitaxel in patients with advanced ovarian cancer after complete response to platinum and paclitaxel-based chemotherapy: A Southwest Oncology Group and Gynecologic Oncology Group trial. J. Clin. Oncol. 2003, 21, 2460–2465. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e116–e131. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Leary, A.; Tan, D.; Ledermann, J. Immune checkpoint inhibitors in ovarian cancer: Where do we stand? Ther. Adv. Med. Oncol. 2021, 13, 17588359211039899. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.A. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995-2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Cabasag, C.J.; Butler, J.; Arnold, M.; Rutherford, M.; Bardot, A.; Ferlay, J. Exploring variations in ovarian cancer survival by age and stage (ICBP SurvMark-2): A population-based study. Gynecol. Oncol. 2020, 157, 234–244. [Google Scholar] [CrossRef]
- Coleman, M.P.; Forman, D.; Bryant, H.; Butler, J.; Rachet, B.; Maringe, C. Cancer survival in Australia, Canada, Denmark, Norway, Sweden, and the UK, 1995–2007 (the International Cancer Benchmarking Partnership): An analysis of population-based cancer registry data. Lancet 2011, 377, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kim, S.; Kim, Y.T.; Lim, M.C.; Lee, B.; Jung, K.W. Changes in ovarian cancer survival during the 20 years before the era of targeted therapy. BMC Cancer 2018, 18, 601. [Google Scholar] [CrossRef]
- Lin, J.J.; Egorova, N.; Franco, R.; Prasad-Hayes, M.; Bickell, N.A. Ovarian Cancer Treatment and Survival Trends Among Women Older Than 65 Years of Age in the United States, 1995–2008. Obstet. Gynecol. 2016, 127, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Norell, C.H.; Butler, J.; Farrell, R.; Altman, A.; Bentley, J.; Cabasag, C.J. Exploring international differences in ovarian cancer treatment: A comparison of clinical practice guidelines and patterns of care. Int. J. Gynecol. Cancer 2020, 30, 1748–1756. [Google Scholar] [CrossRef]
- Vogler, W.R.; Liu, J.; Volpert, O.; Ades, E.W.; Bouck, N. The anticancer drug edelfosine is a potent inhibitor of neovascularization in vivo. Cancer Investig. 1998, 16, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Engel, J. Miltefosine—Discovery of the antileishmanial activity of phospholipid derivatives. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Erdlenbruch, B.; Jendrossek, V.; Marx, M.; Hunold, A.; Eibl, H.; Lakomek, M. Antitumor effects of erucylphosphocholine on brain tumor cells in vitro and in vivo. Anticancer Res. 1998, 18, 2551–2557. [Google Scholar] [PubMed]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Arthur, G.; Schweizer, F.; Ogunsina, M. Synthetic glycosylated ether glycerolipids as anticancer agents. In Carbohydrates in Drug Design and Discovery; Jiménez-Barbero, J., Cañada, F.J., Martin-Santamaria, S., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Jahreiss, L.; Renna, M.; Bittman, R.; Arthur, G.; Rubinsztein, D.C. 1-O-Hexadecyl-2-O-methyl-3-O-(2′-acetamido-2′-deoxy-beta-D-glucopyranosyl)-sn-gly cerol (Gln) induces cell death with more autophagosomes which is autophagy-independent. Autophagy 2009, 5, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Moraya, A.I.; Ali, J.L.; Samadder, P.; Liang, L.; Morrison, L.C.; Werbowetski-Ogilvie, T.E. Novel glycolipid agents for killing cisplatin-resistant human epithelial ovarian cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 67. [Google Scholar] [CrossRef] [Green Version]
- Nachtigal, M.W.; Musaphir, P.; Dhiman, S.; Altman, A.D.; Schweizer, F.; Arthur, G. Cytotoxic capacity of a novel glycosylated antitumor ether lipid in chemotherapy-resistant high grade serous ovarian cancer in vitro and in vivo. Transl. Oncol. 2021, 14, 101203. [Google Scholar] [CrossRef]
- Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Design, synthesis and evaluation of cytotoxic properties of bisamino glucosylated antitumor ether lipids against cancer cells and cancer stem cells. Med. Chem. Comm. 2016, 7, 2100–2110. [Google Scholar] [CrossRef]
- Samadder, P.; Bittman, R.; Byun, H.S.; Arthur, G. A glycosylated antitumor ether lipid kills cells via paraptosis-like cell death. Biochem. Cell Biol. 2009, 87, 401–414. [Google Scholar] [CrossRef] [Green Version]
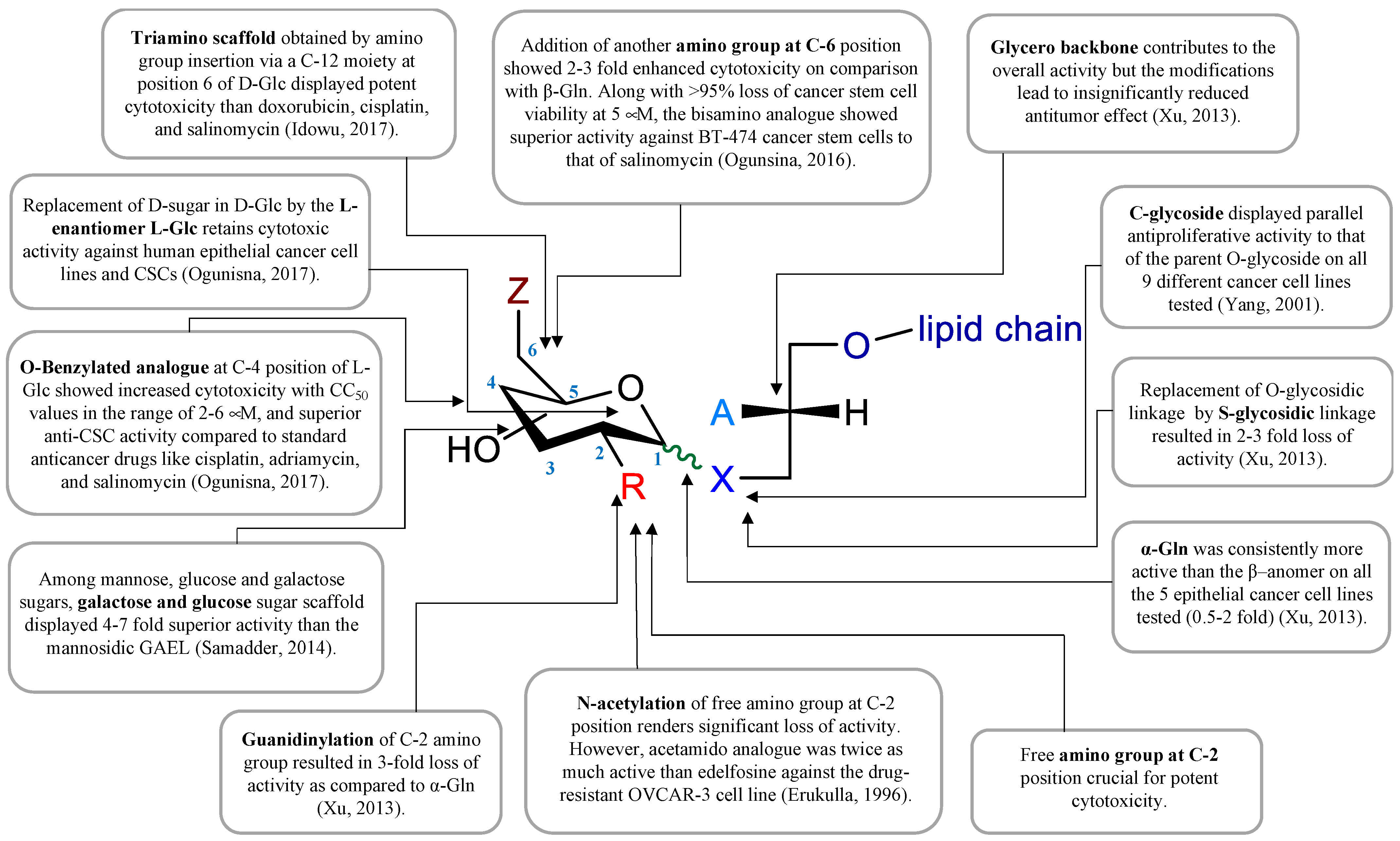
- Samadder, P.; Xu, Y.Z.; Schweizer, F.; Arthur, G. Cytotoxic properties of D-gluco-, D-galacto- and D-manno-configured 2-amino-2-deoxy-glycerolipids against epithelial cancer cell lines and BT-474 breast cancer stem cells. Eur. J. Med. Chem. 2014, 78, 225–235. [Google Scholar] [CrossRef]
- Burdzy, K.; Munder, P.G.; Fischer, H.; Westphal, O. Increase in the Phagocytosis of Peritoneal Macrophages by Lysolecithin. Z. Naturforsch. B. 1964, 19, 1118–1120. [Google Scholar] [CrossRef]
- Fischer, H. Lysolecithin and the Action of Complement. Ann. N. Y. Acad. Sci. 1964, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Munder, P.G.; Modolell, M.; Ferber, E.; Fischer, H. Phospholipids in quartz-damaged macrophages. Biochem. Z. 1966, 344, 310–313. [Google Scholar] [PubMed]
- Arnold, D.; Weltzien, H.U.; Westphal, O. Concerning the synthesis of lysolecithin and its ether analogs. Justus Liebigs Ann. Chem. 1967, 709, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Eibl, H.; Westphal, O. Palmitoyl-propandiol-(1.3)-phosphorylcholine (2-desoxylysolecithin) and omega.omega’-alkanediol-analogs. Justus Liebigs Ann. Chem. 1967, 709, 244–245. [Google Scholar] [CrossRef]
- Weltzien, H.U.; Westphal, O. O-methylated and O-acetylated lysolecithin. Justus Liebigs Ann. Chem. 1967, 709, 240–243. [Google Scholar] [CrossRef]
- Munder, P.G.; Modolell, M.; Andreesen, R.; Weltzien, H.U.; Westphal, O. Lysophosphatidylcholine (Lysolecithin) and its Synthetic Analogues. Immunomodulating and Other Biologic Effects. In Immunostimulation; Chedid, L., Miescher, P.A., Mueller-Eberhard, H.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar]
- Andreesen, R.; Modolell, M.; Weltzien, H.U.; Eibl, H.; Common, H.H.; Lohr, G.W. Selective destruction of human leukemic cells by alkyl-lysophospholipids. Cancer Res. 1978, 38, 3894–3899. [Google Scholar]
- Modolell, M.; Andreesen, R.; Pahlke, W.; Brugger, U.; Munder, P.G. Disturbance of phospholipid metabolism during the selective destruction of tumor cells induced by alkyl-lysophospholipids. Cancer Res. 1979, 39, 4681–4686. [Google Scholar]
- Tarnowski, G.S.; Mountain, I.M.; Stock, C.C.; Munder, P.G.; Weltzien, H.U.; Westphal, O. Effect of lysolecithin and analogs on mouse ascites tumors. Cancer Res. 1978, 38, 339–344. [Google Scholar]
- Berdel, W.E.; Bausert, W.R.; Weltzien, H.U.; Modolell, M.L.; Widmann, K.H.; Munder, P.G. The influence of alkyl-lysophospholipids and lysophospholipid-activated macrophages on the development of metastasis of 3-Lewis lung carcinoma. Eur. J. Cancer 1980, 16, 1199–1204. [Google Scholar] [CrossRef]
- Berger, M.R.; Munder, P.G.; Schmahl, D.; Westphal, O. Influence of the alkyllysophospholipid ET-18-OCH3 on methylnitrosourea-induced rat mammary carcinomas. Oncology 1984, 41, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Helfman, D.M.; Barnes, K.C.; Kinkade, J.M.; Vogler, W.R.; Shoji, M.; Kuo, J.F. Phospholipid-Sensitive Ca-2+-Dependent Protein-Phosphorylation System in Various Types of Leukemic-Cells from Human Patients and in Human-Leukemic Cell Line-Hl60 and Line-K562, and Its Inhibition by Alkyl-Lysophospholipid. Cancer Res. 1983, 43, 2955–2961. [Google Scholar] [PubMed]
- Parker, J.; Daniel, L.W.; Waite, M. Evidence of Protein-Kinase-C Involvement in Phorbol Diester-Stimulated Arachidonic-Acid Release and Prostaglandin Synthesis. J. Biol. Chem. 1987, 262, 5385–5393. [Google Scholar] [CrossRef]
- Daniel, L.W.; Etkin, L.A.; Morrison, B.T.; Parker, J.; Morris-Natschke, S.; Surles, J.R. Ether lipids inhibit the effects of phorbol diester tumor promoters. Lipids 1987, 22, 851–855. [Google Scholar] [CrossRef]
- Vogler, W.R.; Whigham, E.; Bennett, W.D.; Olson, A.C. Effect of alkyl-lysophospholipids on phosphatidylcholine biosynthesis in leukemic cell lines. Exp. Hematol. 1985, 13, 629–633. [Google Scholar] [PubMed]
- Berdel, W.E.; Fromm, M.; Fink, U.; Pahlke, W.; Bicker, U.; Reichert, A. Cytotoxicity of thioether-lysophospholipids in leukemias and tumors of human origin. Cancer Res. 1983, 43, 5538–5543. [Google Scholar]
- Storme, G.A.; Berdel, W.E.; van Blitterswijk, W.J.; Bruyneel, E.A.; De Bruyne, G.K.; Mareel, M.M. Antiinvasive effect of racemic 1-O-octadecyl-2-O-methylglycero-3-phosphocholine on MO4 mouse fibrosarcoma cells in vitro. Cancer Res. 1985, 45, 351–357. [Google Scholar]
- Houlihan, W.J.; Lohmeyer, M.; Workman, P.; Cheon, S.H. Phospholipid antitumor agents. Med. Res. Rev. 1995, 15, 157–223. [Google Scholar] [CrossRef]
- Smets, L.A.; Van Rooij, H.; Salomons, G.S. Signalling steps in apoptosis by ether lipids. Apoptosis 1999, 4, 419–427. [Google Scholar] [CrossRef]
- Berdel, W.E.; Fink, U.; Rastetter, J. Clinical phase I pilot study of the alkyl lysophospholipid derivative ET-18-OCH3. Lipids 1987, 22, 967–969. [Google Scholar] [CrossRef]
- Drings, P.; Günther, I.; Gatzemeier, U.; Ulbrich, F.; Khanavkar, B.; Schreml, W. Final Evaluation of a Phase II Study on the Effect of Edelfosine (an Ether Lipid) in Advanced Non-Small-Cell Bronchogenic Carcinoma. Onkologie 1992, 15, 375–382. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH(3) (edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab. 2002, 3, 491–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, D.B.; Neumann, H.A.; Berdel, W.E.; Heim, M.E.; Fromm, M.; Boerner, D. Phase I trial of the thioether phospholipid analogue BM 41.440 in cancer patients. Lipids 1987, 22, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Hilgard, P.; Stekar, J.; Voegeli, R.; Harleman, J.H. Experimental therapeutic studies with miltefosine in rats and mice. Prog. Exp. Tumor Res. 1992, 34, 116–130. [Google Scholar] [CrossRef]
- Unger, C.; Eibl, H. Hexadecylphosphocholine: Preclinical and the first clinical results of a new antitumor drug. Lipids 1991, 26, 1412–1417. [Google Scholar] [CrossRef]
- Jendrossek, V.; Erdlenbruch, B.; Hunold, A.; Kugler, W.; Eibl, H.; Lakomek, M. Erucylphosphocholine, a novel antineoplastic ether lipid, blocks growth and induces apoptosis in brain tumor cell lines in vitro. Int. J. Oncol. 1999, 14, 15–22. [Google Scholar] [CrossRef]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Van Blitterswijk, W.J.; Verheij, M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 2013, 1831, 663–674. [Google Scholar] [CrossRef]
- Keane, N.A.; Glavey, S.V.; Krawczyk, J.; O’Dwyer, M. AKT as a therapeutic target in multiple myeloma. Expert Opin. Ther. Targets 2014, 18, 897–915. [Google Scholar] [CrossRef]
- Zentaris, A. Assessment of Efficacy and Safety of Perifosine, Bortezomib and Dexamethasone in Multiple Myeloma Patients. 2013. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01002248 (accessed on 14 June 2022).
- Weber, N.; Benning, H. Synthesis of Ether Glyceroglycolipids. Chem. Phys. Lipids 1986, 41, 93–100. [Google Scholar] [CrossRef]
- Guivisdalsky, P.N.; Bittman, R.; Smith, Z.; Blank, M.L.; Snyder, F.; Howard, S. Synthesis and antineoplastic properties of ether-linked thioglycolipids. J. Med. Chem. 1990, 33, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Rengan, K.; Bittman, R.; Arthur, G. The Alpha-Anomers and Beta-Anomers of 1-O-Hexadecyl-2-O-Methyl-3-S-Thioglucosyl-Sn-Glycerol Inhibit the Proliferation of Epithelial Cancer Cell-Lines. Oncol. Rep. 1994, 1, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Erukulla, R.K.; Zhou, X.; Samadder, P.; Arthur, G.; Bittman, R. Synthesis and evaluation of the antiproliferative effects of 1-O-hexadecyl-2-O-methyl-3-O-(2′-acetamido-2′-deoxy-beta-D-glucopyranosyl)-sn-glycerol and 1-O-hexadecyl-2-O-methyl-3-0-(2′-amino-2′-deoxy-beta-D-glucopyranosyl)-sn-glycerol on epithelial cancer cell growth. J. Med. Chem. 1996, 39, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F. Design, synthesis and antitumor properties of glycosylated antitumor ether lipid (GAEL)- chlorambucil-hybrids. Chem. Phys. Lipids 2016, 194, 139–148. [Google Scholar] [CrossRef]
- Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Replacing D-Glucosamine with Its L-Enantiomer in Glycosylated Antitumor Ether Lipids (GAELs) Retains Cytotoxic Effects against Epithelial Cancer Cells and Cancer Stem Cells. J. Med. Chem. 2017, 60, 2142–2147. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Ogunsina, M.; Samadder, P.; Arthur, G.; Schweizer, F. StructureActivity Relationships of Glucosamine-Derived Glycerolipids: The Role of the Anomeric Linkage, the Cationic Charge and the Glycero Moiety on the Antitumor Activity. ChemMedChem 2013, 8, 511–520. [Google Scholar] [CrossRef]
- Yang, G.L.; Franck, R.W.; Bittman, R.; Samadder, P.; Arthur, G. Synthesis and growth inhibitory properties of glucosamine-derived glycerolipids. Org. Lett. 2001, 3, 197–200. [Google Scholar] [CrossRef]
- Yang, G.L.; Franck, R.W.; Byun, H.S.; Bittman, R.; Samadder, P.; Arthur, G. Convergent C-glycolipid synthesis via the Ramberg-Backlund reaction: Active antiproliferative glycolipids. Org. Lett. 1999, 1, 2149–2151. [Google Scholar] [CrossRef]
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F. Amphiphilic Modulation of Glycosylated Antitumor Ether Lipids Results in a Potent Triamino Scaffold against Epithelial Cancer Cell Lines and BT474 Cancer Stem Cells. J. Med. Chem. 2017, 60, 9724–9738. [Google Scholar] [CrossRef]
- Ogunsina, M.; Samadder, P.; Idowu, T.; Nachtigal, M.W.; Schweizer, F.; Arthur, G. Syntheses of L-Rhamnose linked amino glycerolipids and their cytotoxic activities against human cancer cells. Molecules 2020, 25, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, T.G.; Theriault, B.L.; Campbell, E.J.; Nachtigal, M.W. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat. Protoc. 2006, 1, 2643–2649. [Google Scholar] [CrossRef] [PubMed]
- Theriault, B.L.; Portelance, L.; Mes-Masson, A.M.; Nachtigal, M.W. Establishment of primary cultures from ovarian tumor tissue and ascites fluid. Methods Mol. Biol. 2013, 1049, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Samadder, P.; Byun, H.S.; Bittman, R.; Arthur, G. An active endocytosis pathway is required for the cytotoxic effects of glycosylated antitumor ether lipids. Anticancer Res. 2011, 31, 3809–3818. [Google Scholar]
- Maltese, W.A.; Overmeyer, J.H. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front. Physiol. 2015, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Geno, E.; Patoor, M.; Reid, A.; McDonald, R.; Hild, M. Indolyl-Pyridinyl-Propenone-Induced Methuosis through the Inhibition of PIKFYVE. ACS Omega 2018, 3, 6097–6103. [Google Scholar] [CrossRef]
- Huang, W.; Sun, X.; Li, Y.; He, Z.; Li, L.; Deng, Z. Discovery and Identification of Small Molecules as Methuosis Inducers with in Vivo Antitumor Activities. J. Med. Chem. 2018, 61, 5424–5434. [Google Scholar] [CrossRef]
- Li, Z.; Mbah, N.E.; Overmeyer, J.H.; Sarver, J.G.; George, S.; Trabbic, C.J. The JNK signaling pathway plays a key role in methuosis (non-apoptotic cell death) induced by MOMIPP in glioblastoma. BMC Cancer 2019, 19, 77. [Google Scholar] [CrossRef]
- Overmeyer, J.H.; Young, A.M.; Bhanot, H.; Maltese, W.A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Nara, A.; Aki, T.; Funakoshi, T.; Unuma, K.; Uemura, K. Hyperstimulation of macropinocytosis leads to lysosomal dysfunction during exposure to methamphetamine in SH-SY5Y cells. Brain Res. 2012, 1466, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, B.; Su, X.; Chen, G.; Li, Y.; Yu, L. An Ursolic Acid Derived Small Molecule Triggers Cancer Cell Death through Hyperstimulation of Macropinocytosis. J. Med. Chem. 2017, 60, 6638–6648. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, F.; Simbari, F.; Abad, J.L.; Casasampere, M.; Fabrias, G.; Futerman, A.H. Jaspine B induces nonapoptotic cell death in gastric cancer cells independently of its inhibition of ceramide synthase. J. Lipid Res. 2017, 58, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minna, E.; Romeo, P.; De Cecco, L.; Dugo, M.; Cassinelli, G.; Pilotti, S. miR-199a-3p displays tumor suppressor functions in papillary thyroid carcinoma. Oncotarget 2014, 5, 2513–2528. [Google Scholar] [CrossRef] [Green Version]

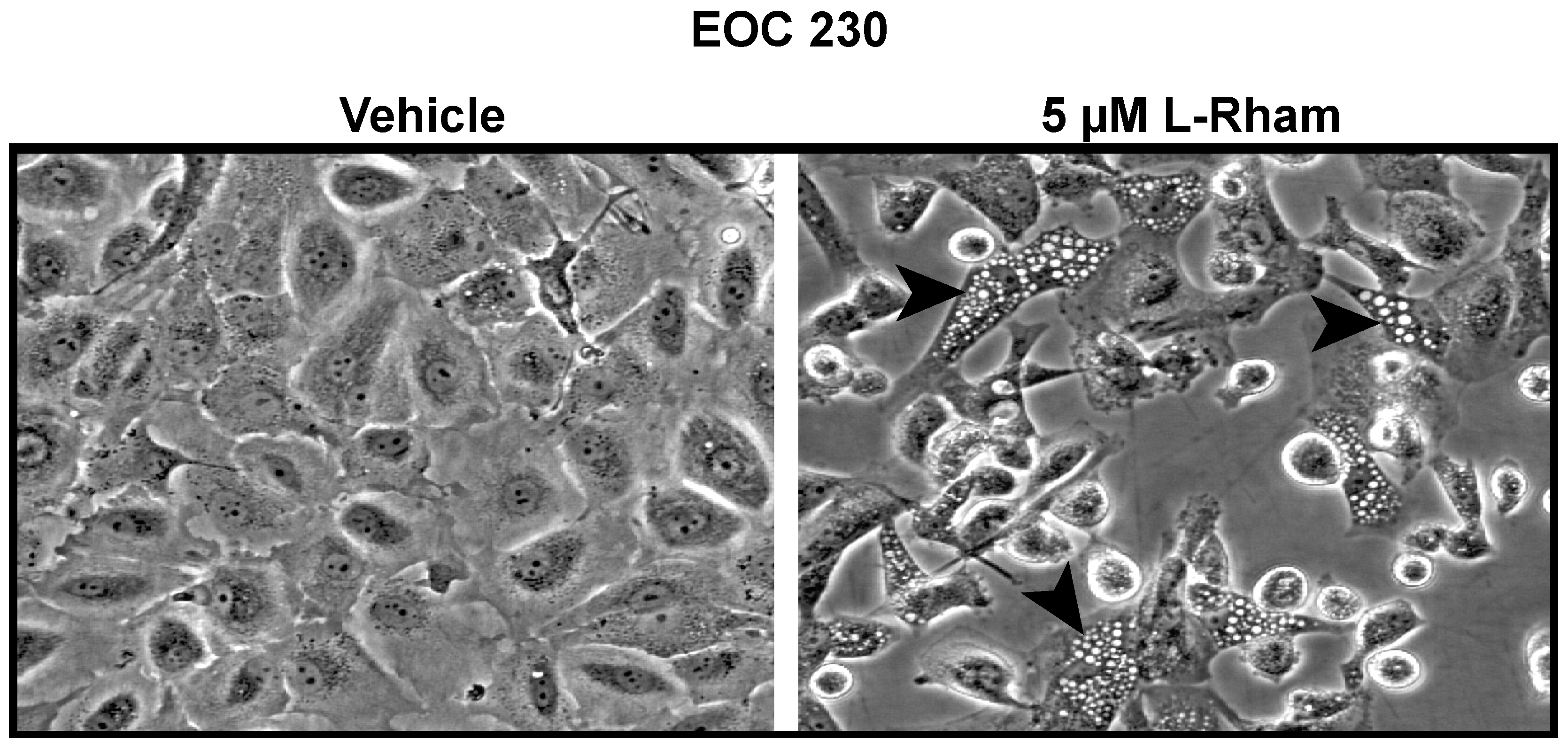


{kind=link}
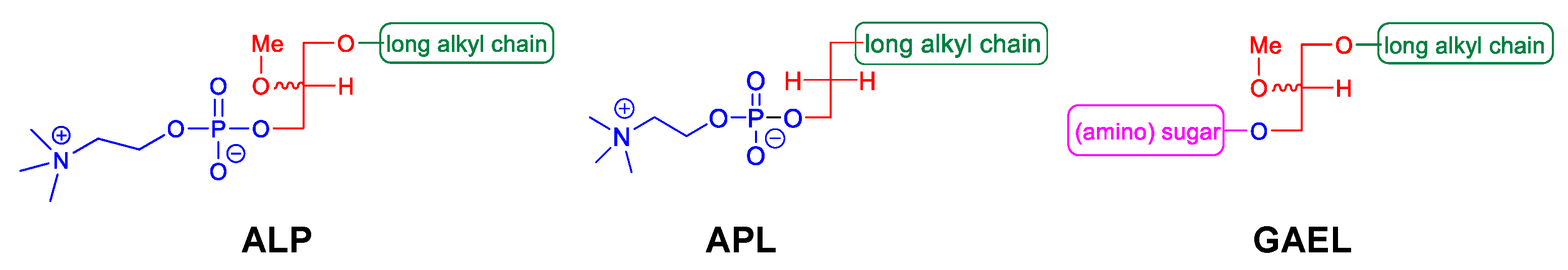
{kind=link}
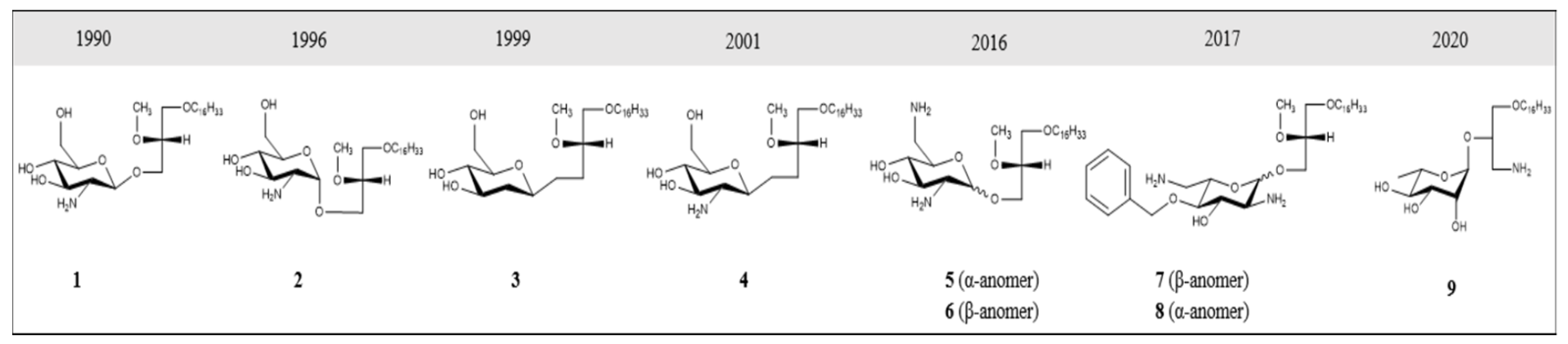
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Group Name | Dose Schedule/Termination | Mice with Solid Tumors | Ascites (%) |
|---|---|---|---|---|
| 2 * | Vehicle—2 | Q4Dx8 Termination: day 74 *** | 100% (12/12) @ | 92% (11/12) |
| 4 * | L-Rham—low 2 | Q4Dx8 Termination: day 74 *** | 33% (2/6) | 0% (0/6) |
| 5 ** | L-Rham—high | Q4Dx8 Termination: day 74 *** | 100% (6/6) | 0% (0/6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nachtigal, M.W.; Altman, A.D.; Arora, R.; Schweizer, F.; Arthur, G. The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer. Cancers 2022, 14, 3318. https://doi.org/10.3390/cancers14143318
Nachtigal MW, Altman AD, Arora R, Schweizer F, Arthur G. The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer. Cancers. 2022; 14(14):3318. https://doi.org/10.3390/cancers14143318
Chicago/Turabian StyleNachtigal, Mark W., Alon D. Altman, Rajat Arora, Frank Schweizer, and Gilbert Arthur. 2022. "The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer" Cancers 14, no. 14: 3318. https://doi.org/10.3390/cancers14143318
APA StyleNachtigal, M. W., Altman, A. D., Arora, R., Schweizer, F., & Arthur, G. (2022). The Potential of Novel Lipid Agents for the Treatment of Chemotherapy-Resistant Human Epithelial Ovarian Cancer. Cancers, 14(14), 3318. https://doi.org/10.3390/cancers14143318