
Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges



Abstract
:Simple Summary
Abstract
1. Introduction
2. Recent Advancements in ctDNA-Based Genomic Profiling Assays
2.1. ctDNA Reflects the General Genomic Landscape of Tumors
2.2. Promising Clinical Outcomes following ctDNA Profiling for Treatment Selection
2.3. Shorter Turnaround Time (TAT) with Improved Clinical Trial Enrolment Rate
2.4. FDA Approval of Multigene ctDNA NGS Tests for CGP and as In Vitro Companion Diagnostics
3. Limitations and Challenges for the Routine Use of ctDNA-Based CGP for Treatment Selection
3.1. Technical Limitations Leading to False Positive and Negative Results
3.2. Biological Limitations: Low Tumor Shedding and Non-Tumoral Origin of cfDNA
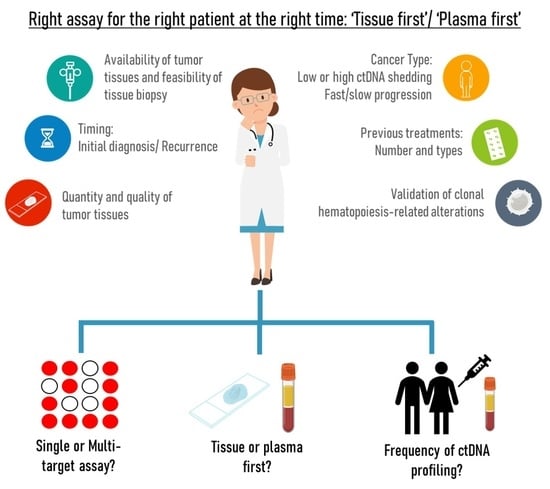
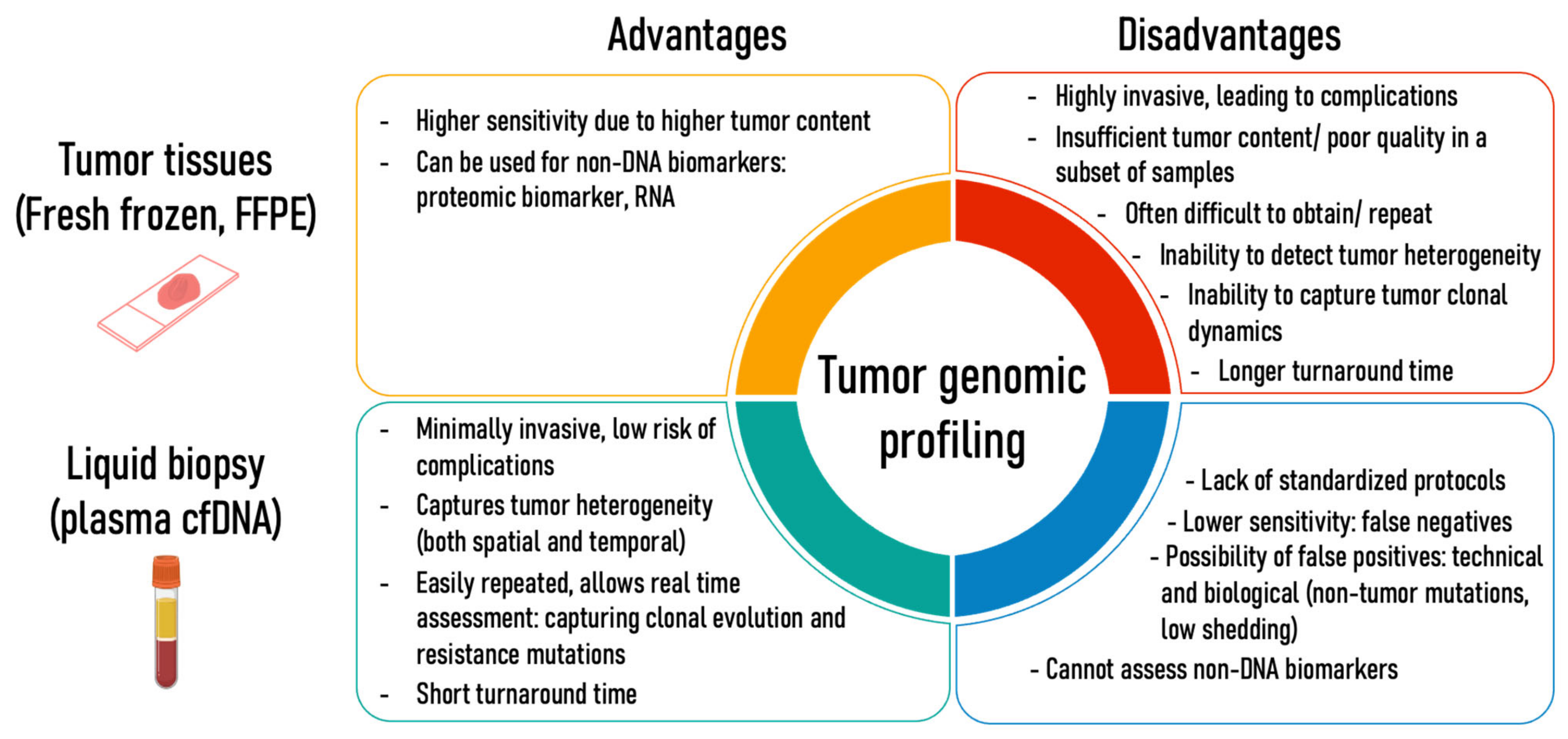
3.3. Lack of Standardized Evidence-Based Guidelines for Tissue or Plasma First Approach
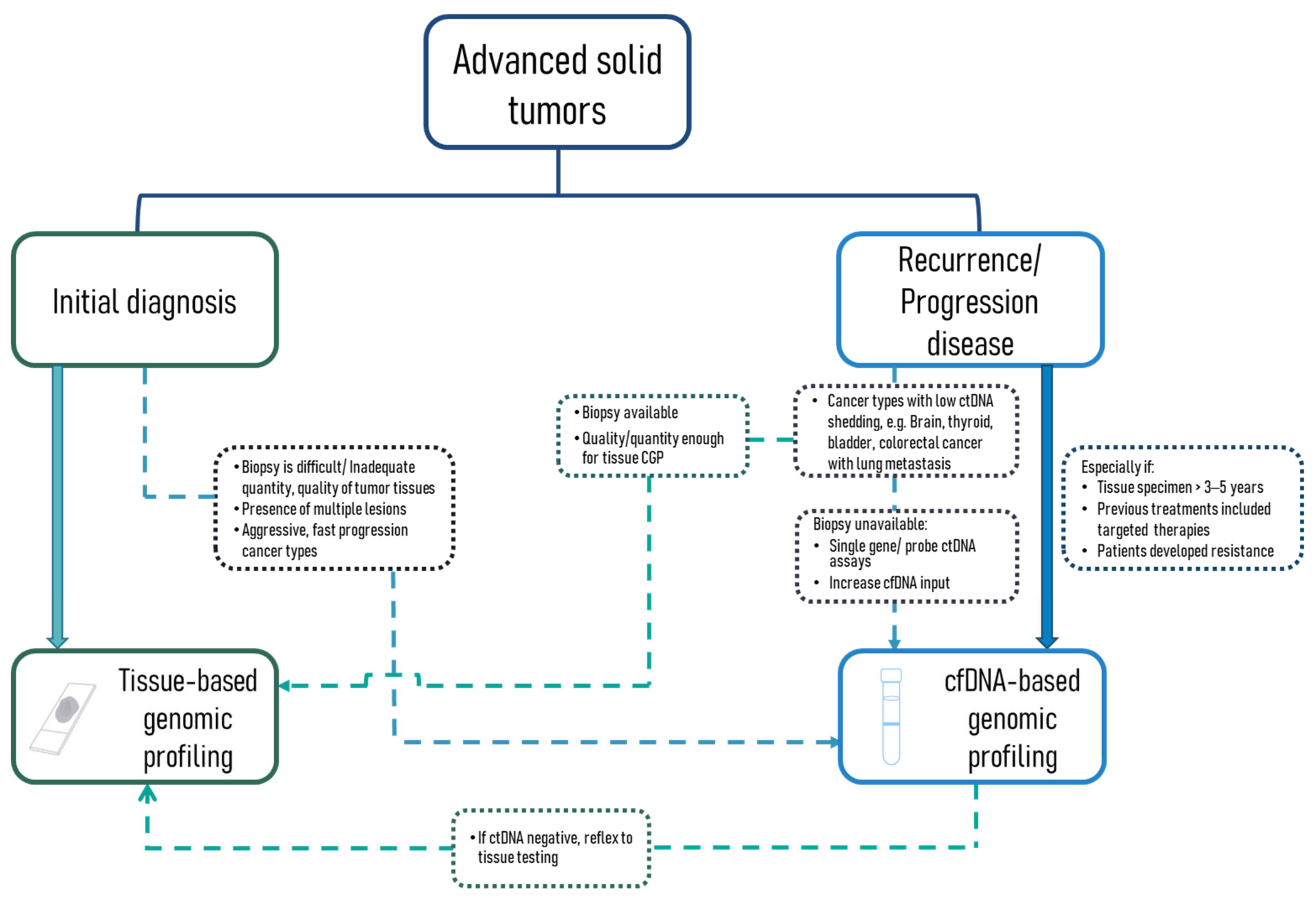
3.3.1. Availability of Excision Tumor Tissue, Quality and Quantity of Biopsy; the Presence of Multiple Lesions; Cancer Types
3.3.2. Timing: Initial Diagnosis or Recurrence/Progression Disease
4. Future Perspectives of ctDNA-Based CGP to Maximize Its Utilities in Personalizing Oncology Management
4.1. Standardizing Methods to Exclude CH Mutations
4.2. Establishing the VAF Threshold for Treatment Initiation
4.3. Frequency of ctDNA CGP for Treatment Optimization
4.4. ctDNA Biomarkers for Immunotherapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chakravarty, D.; Solit, D.B. Clinical cancer genomic profiling. Nat. Rev. Genet. 2021, 22, 483–501. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Al-Kateb, H.; Nguyen, T.T.; Steger-May, K.; Pfeifer, J.D. Identification of major factors associated with failed clinical molecular oncology testing performed by next generation sequencing (NGS). Mol. Oncol. 2015, 9, 1737–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Hovelson, D.H.; Suga, J.M.; Anderson, D.M.; Koh, H.A.; Dees, E.C.; McNulty, B.; Burkard, M.E.; Guarino, M.; Khatri, J.; et al. Real-World Performance of a Comprehensive Genomic Profiling Test Optimized for Small Tumor Samples. JCO Precis. Oncol. 2021, 5, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Uozu, S.; Imaizumi, K.; Yamaguchi, T.; Goto, Y.; Kawada, K.; Minezawa, T.; Okamura, T.; Akao, K.; Hayashi, M.; Isogai, S.; et al. Feasibility of tissue re-biopsy in non-small cell lung cancers resistant to previous epidermal growth factor receptor tyrosine kinase inhibitor therapies. BMC Pulm. Med. 2017, 17, 175. [Google Scholar] [CrossRef] [Green Version]
- Chouaid, C.; Dujon, C.; Do, P.; Monnet, I.; Madroszyk, A.; Le Caer, H.; Auliac, J.B.; Berard, H.; Thomas, P.; Lena, H.; et al. Feasibility and clinical impact of re-biopsy in advanced non small-cell lung cancer: A prospective multicenter study in a real-world setting (GFPC study 12-01). Lung Cancer 2014, 86, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Modak, J.; Kopetz, S.; Murthy, R.; Yao, J.C.; Hicks, M.E.; Abbruzzese, J.L.; Tam, A.L. Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J. Clin. Oncol. 2013, 31, 17–22. [Google Scholar] [CrossRef]
- Kelly, R.J.; Turner, R.; Chen, Y.-W.; Rigas, J.R.; Fernandes, A.W.; Karve, S. Complications and Economic Burden Associated with Obtaining Tissue for Diagnosis and Molecular Analysis in Patients with Non–Small-Cell Lung Cancer in the United States. J. Oncol. Pract. 2019, 15, e717–e727. [Google Scholar] [CrossRef]
- Wu, C.C.; Maher, M.M.; Shepard, J.-A.O. Complications of CT-Guided Percutaneous Needle Biopsy of the Chest: Prevention and Management. Am. J. Roentgenol. 2011, 196, W678–W682. [Google Scholar] [CrossRef]
- Nam, B.D.; Yoon, S.H.; Hong, H.; Hwang, J.H.; Goo, J.M.; Park, S. Tissue Adequacy and Safety of Percutaneous Transthoracic Needle Biopsy for Molecular Analysis in Non-Small Cell Lung Cancer: A Systematic Review and Meta-analysis. Korean J. Radiol. 2021, 22, 2082–2093. [Google Scholar] [CrossRef]
- Jekunen, A.P. Role of Rebiopsy in Relapsed Non-Small Cell Lung Cancer for Directing Oncology Treatments. J. Oncol. 2015, 2015, 809835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C. The future of liquid biopsy. Nature 2020, 579, S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocetto, F.; Barone, B.; Ferro, M.; Busetto, G.M.; La Civita, E.; Buonerba, C.; Di Lorenzo, G.; Terracciano, D.; Schalken, J.A. Liquid biopsy in bladder cancer: State of the art and future perspectives. Crit. Rev. Oncol. 2022, 170, 103577. [Google Scholar] [CrossRef]
- Crocetto, F.; Cimmino, A.; Ferro, M.; Terracciano, D. Circulating tumor cells in bladder cancer: A new horizon of liquid biopsy for precision medicine. J. Basic Clin. Physiol. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating Tumor DNA as a Liquid Biopsy for Cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2020, 124, 345–358. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Dennis, Y.M. Circulating DNA in plasma and serum: Biology, preanalytical issues and diagnostic applications. Clin. Chem. Lab. Med. 2002, 40, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar] [PubMed]
- Lui, Y.Y.N.; Chik, K.-W.; Chiu, R.W.K.; Ho, C.-Y.; Lam, C.W.K.; Lo, Y.M.D. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.K.J.; Gai, W.; Sun, K.; Wong, R.S.; Chan, R.W.; Jiang, P.; Chan, N.P.; Hui, W.I.; Chan, A.; Szeto, C.-C.; et al. DNA of Erythroid Origin Is Present in Human Plasma and Informs the Types of Anemia. Clin. Chem. 2017, 63, 1614–1623. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Bellosillo, B.; Montagut, C. High-accuracy liquid biopsies. Nat. Med. 2019, 25, 1820–1821. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, K.C.S.; Ramos, I.B.; Silva, J.M.C.; Barra, W.F.; Riggins, G.J.; Palande, V.; Pinho, C.T.; Frenkel-Morgenstern, M.; Santos, S.E.; Assumpcao, P.P.; et al. Current Perspectives on Circulating Tumor DNA, Precision Medicine, and Personalized Clinical Management of Cancer. Mol. Cancer Res. 2020, 18, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendrell, J.A.; Quantin, X.; Aussel, A.; Solassol, I.; Serre, I.; Solassol, J. EGFR-dependent mechanisms of resistance to osimertinib determined by ctDNA NGS analysis identify patients with better outcome. Transl. Lung Cancer Res. 2021, 10, 4084–4094. [Google Scholar] [CrossRef]
- Nakajima, H.; Kotani, D.; Bando, H.; Kato, T.; Oki, E.; Shinozaki, E.; Sunakawa, Y.; Yamazaki, K.; Yuki, S.; Nakamura, Y.; et al. REMARRY and PURSUIT trials: Liquid biopsy-guided rechallenge with anti-epidermal growth factor receptor (EGFR) therapy with panitumumab plus irinotecan for patients with plasma RAS wild-type metastatic colorectal cancer. BMC Cancer 2021, 21, 674. [Google Scholar] [CrossRef] [PubMed]
- Van Helden, E.J.; Angus, L.; Menke-van der Houven van Oordt, C.W.; Heideman, D.V.M.; Boon, E.; van Es, S.C.; Radema, S.A.; van Herpen, C.M.L.; de Groot, D.J.A.; de Vries, E.G.E.; et al. RAS and BRAF mutations in cell-free DNA are predictive for outcome of cetuximab monotherapy in patients with tissue-tested RAS wild-type advanced colorectal cancer. Mol. Oncol. 2019, 13, 2361–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Ye, Y.; Liu, X.; Gong, Y.; Xu, M.; Song, L.; Liu, H. Intra-Tumor Heterogeneity of Colorectal Cancer Necessitates the Multi-Regional Sequencing for Comprehensive Mutational Profiling. Cancer Manag. Res. 2021, 13, 9209–9223. [Google Scholar] [CrossRef] [PubMed]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L. Analysis of Circulating Tumor DNA to Confirm Somatic KRAS Mutations. JNCI J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat. Biotechnol. 2021, 39, 1115–1128. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 2021, 12, 11. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.-K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotani, K.; Fukunaga, Y.; Ueno, M.; Nakamura, Y.; Low, S. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar] [CrossRef]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.-J.; Jardin, F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals 2021, 14, 596. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor–Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.A.; Mulder, H.L.; Edmonson, M.N.; Liu, Y.; Chen, X.; Newman, S.; Nakitandwe, J.; et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef] [Green Version]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Dono, M. The Opportunities and Challenges of Molecular Tagging Next-Generation Sequencing in Liquid Biopsy. Mol. Diagn. Ther. 2021, 25, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Davis, A.A.; Jacob, S.; Gerratana, L.; Shah, A.N.; Wehbe, F.; Katam, N.; Zhang, Q.; Flaum, L.; Siziopikou, K.P.; Platanias, L.C.; et al. Landscape of circulating tumour DNA in metastatic breast cancer. eBioMedicine 2020, 58, 102914. [Google Scholar] [CrossRef]
- Li, B.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.; Shen, R.; Isbell, J.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Tukachinsky, H.; Madison, R.W.; Chung, J.H.; Gjoerup, O.V.; Severson, E.A.; Dennis, L.; Fendler, B.J.; Morley, S.; Zhong, L.; Graf, R.P.; et al. Genomic Analysis of Circulating Tumor DNA in 3,334 Patients with Advanced Prostate Cancer Identifies Targetable BRCA Alterations and AR Resistance Mechanisms. Clin. Cancer Res. 2021, 27, 3094–3105. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef]
- Litchfield, K.; Turajlic, S.; Swanton, C. The GENIE Is Out of the Bottle: Landmark Cancer Genomics Dataset Released. Cancer Discov. 2017, 7, 796–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Brannon, A.R.; Jayakumaran, G.; Diosdado, M.; Patel, J.; Razumova, A.; Hu, Y.; Meng, F.; Haque, M.; Sadowska, J.; Murphy, B.J.; et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat. Commun. 2021, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Bieg-Bourne, C.C.; Okamura, R.; Kurzrock, R. Concordance between TP53 alterations in blood and tissue: Impact of time interval, biopsy site, cancer type and circulating tumor DNA burden. Mol. Oncol. 2020, 14, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.T.; Lam, V.K.; Elamin, Y.Y.; Hong, L.; Colen, R.; Elshafeey, N.A.; Hassan, I.S.A.; Altan, M.; Blumenschein, G.R.; Rinsurongkawong, W.; et al. Clinical Outcomes in Non–Small-Cell Lung Cancer Patients Treated With EGFR-Tyrosine Kinase Inhibitors and Other Targeted Therapies Based on Tumor Versus Plasma Genomic Profiling. JCO Precis. Oncol. 2021, 5, 1241–1249. [Google Scholar] [CrossRef]
- Park, S.; Olsen, S.; Ku, B.M.; Lee, M.; Jung, H.; Sun, J.; Lee, S.; Ahn, J.S.; Park, K.; Choi, Y.; et al. High concordance of actionable genomic alterations identified between circulating tumor DNA–based and tissue-based next-generation sequencing testing in advanced non–small cell lung cancer: The Korean Lung Liquid Versus Invasive Biopsy Program. Cancer 2021, 127, 3019–3028. [Google Scholar] [CrossRef]
- Madison, R.; Schrock, A.B.; Castellanos, E.; Gregg, J.P.; Snider, J.; Ali, S.M.; Miller, V.A.; Singal, G.; Alexander, B.M.; Venstrom, J.M.; et al. Retrospective analysis of real-world data to determine clinical outcomes of patients with advanced non-small cell lung cancer following cell-free circulating tumor DNA genomic profiling. Lung Cancer 2020, 148, 69–78. [Google Scholar] [CrossRef]
- Mack, P.C.; Ms, K.C.B.; Ms, C.R.E.; Ms, R.A.B.; Zill, O.A.; Ms, C.E.L.; Riess, J.W.; Mortimer, S.A.; Talasaz, A.; Lanman, R.B.; et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non–small cell lung cancer: Analysis of over 8000 cases. Cancer 2020, 126, 3219–3228. [Google Scholar] [CrossRef]
- Sabari, J.K.; Offin, M.; Stephens, D.; Ni, A.; Lee, A.; Pavlakis, N.; Clarke, S.; I Diakos, C.; Datta, S.; Tandon, N.; et al. A Prospective Study of Circulating Tumor DNA to Guide Matched Targeted Therapy in Lung Cancers. JNCI J. Natl. Cancer Inst. 2018, 111, 575–583. [Google Scholar] [CrossRef]
- Bujak, A.Z.; Weng, C.-F.; Silva, M.J.; Yeung, M.; Lo, L.; Ftouni, S.; Litchfield, C.; Ko, Y.-A.; Kuykhoven, K.; Van Geelen, C.; et al. Circulating tumour DNA in metastatic breast cancer to guide clinical trial enrolment and precision oncology: A cohort study. PLoS Med. 2020, 17, e1003363. [Google Scholar] [CrossRef]
- Zhang, M.; Qi, C.; Wang, Z.; Chen, H.; Zhao, X.; Zhang, X.; Zhou, Y.; Gao, C.; Bai, Y.; Jia, S.; et al. Molecular characterization of ctDNA from Chinese patients with advanced gastric adenocarcinoma reveals actionable alterations for targeted and immune therapy. Klin. Wochenschr. 2021, 99, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Olsen, S.; Zhang, N.; Liao, J.; Yoshino, T. Comprehensive Genomic Profiling of Circulating Tumor DNA in Patients with Previously Treated Metastatic Colorectal Cancer: Analysis of a Real-World Healthcare Claims Database. Curr. Oncol. 2022, 29, 3433–3448. [Google Scholar] [CrossRef] [PubMed]
- Botrus, G.; Kosirorek, H.; Sonbol, M.B.; Kusne, Y.; Uson, P.L.S.; Borad, M.J.; Ahn, D.H.; Kasi, P.M.; Drusbosky, L.M.; Dada, H.; et al. Circulating Tumor DNA-Based Testing and Actionable Findings in Patients with Advanced and Metastatic Pancreatic Adenocarcinoma. Oncologist 2021, 26, 569–578. [Google Scholar] [CrossRef]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.-S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Matsubara, N.; de Bono, J.S.; Olmos, D.; Procopio, G.; Kawakami, S.; Urun, Y.; van Alphen, R.J.; Flechon, A.; Carducci, M.A.; Choi, Y.D.; et al. Olaparib efficacy in patients with metastatic castration-resistant prostate cancer (mCRPC) carrying circulating tumor (ct) DNA alterations in BRCA1, BRCA2 or ATM: Results from the PROfound study. J. Clin. Oncol. 2021, 39, 27. [Google Scholar] [CrossRef]
- Sharma, P.; Abramson, V.G.; O’Dea, A.P.; Nye, L.; Mayer, I.A.; Pathak, H.B.; Hoffmann, M.; Stecklein, S.R.; Elia, M.; Lewis, S.; et al. Clinical and Biomarker Results from Phase I/II Study of PI3K Inhibitor Alpelisib plus Nab-paclitaxel in HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 3896–3904. [Google Scholar] [CrossRef]
- Rothé, F.; Silva, M.J.; Venet, D.; Campbell, C.; Bradburry, I.; Rouas, G.; de Azambuja, E.; Maetens, M.; Fumagalli, D.; Rodrik-Outmezguine, V.; et al. Circulating Tumor DNA in HER2-Amplified Breast Cancer: A Translational Research Substudy of the NeoALTTO Phase III Trial. Clin. Cancer Res. 2019, 25, 3581–3588. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Weipert, C.; Gumas, S.; Okamura, R.; Lee, S.; Sicklick, J.K.; Saam, J.; Kurzrock, R. Therapeutic Actionability of Circulating Cell-Free DNA Alterations in Carcinoma of Unknown Primary. JCO Precis. Oncol. 2021, 5, 1687–1698. [Google Scholar] [CrossRef]
- Kolling, S.; Ventre, F.; Geuna, E.; Milan, M.; Pisacane, A.; Boccaccio, C.; Sapino, A.; Montemurro, F. “Metastatic Cancer of Unknown Primary” or “Primary Metastatic Cancer”? Front. Oncol. 2019, 9, 1546. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Mok, T.; Peters, S.; Han, J.-Y.; Alatorre-Alexander, J.; Leighl, N.; Sriuranpong, V.; Pérol, M.; Junior, G.D.C.; Nadal, E.; et al. Blood First Assay Screening Trial (BFAST) in Treatment-Naive Advanced or Metastatic NSCLC: Initial Results of the Phase 2 ALK-Positive Cohort. J. Thorac. Oncol. 2021, 16, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Clark, E.W.; Milan, M.S.; Champagne, C.; Michael, K.S.; Awad, M.M.; Barbie, D.A.; Cheng, M.L.; Kehl, K.L.; Marcoux, J.P.; et al. Turnaround Time of Plasma Next-Generation Sequencing in Thoracic Oncology Patients: A Quality Improvement Analysis. JCO Precis. Oncol. 2020, 4, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Chattopadhyay, R.; Kato, S.; Fanta, P.T.; Banks, K.C.; Choi, I.S.; Piccioni, D.E.; Ikeda, S.; Talasaz, A.; Lanman, R.B.; et al. Genomic Alterations in Circulating Tumor DNA from Diverse Cancer Patients Identified by Next-Generation Sequencing. Cancer Res. 2017, 77, 5419–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, Y.; Tan, M.-H.; Shi, J.L.; Tee, A.; Ngeow, K.C.; Poh, J.; Goh, R.R.; Mong, J. Complementing Tissue Testing with Plasma Mutation Profiling Improves Therapeutic Decision-Making for Patients with Lung Cancer. Front. Med. 2022, 9, 758464. [Google Scholar] [CrossRef]
- FoundationMedicine. FoundationOne Liquid CDx. 2022. Available online: https://www.foundationmedicine.com/test/foundationone-liquid-cdx#:~:text=FoundationOne%20Liquid%20CDx%20is%20an,liquid%20biopsy%20on%20the%20market (accessed on 16 February 2022).
- GuardantHealth. Guardant360 CDx. 2022. Available online: https://guardant360cdx.com/ (accessed on 16 February 2022).
- Low, S.-K.K.; Uchibori, K.; Hayashi, R.; Chan, H.T.; Ariyasu, R.; Kitazono, S.; Yanagitani, N.; Nishio, M.; Nakamura, Y. Evaluation of Genexus system that automates specimen-to-report for cancer genomic profiling within a day using liquid biopsy. J. Clin. Oncol. 2020, 38, 3538. [Google Scholar] [CrossRef]
- Low, S.-K.; Ariyasu, R.; Uchibori, K.; Hayashi, R.; Chan, H.T.; Chin, Y.M.; Akita, T.; Harutani, Y.; Kiritani, A.; Tsugitomi, R.; et al. Rapid genomic profiling of circulating tumor DNA in non-small cell lung cancer using Oncomine Precision Assay with GenexusTM integrated sequencer. Transl. Lung Cancer Res. 2021, 11, 711–721. [Google Scholar] [CrossRef]
- Al Zoughbi, W.; Fox, J.; Beg, S.; Papp, E.; Hissong, E.; Ohara, K.; Keefer, L.; Sigouros, M.; Kane, T.; Bockelman, D.; et al. Validation of a Circulating Tumor DNA-Based Next-Generation Sequencing Assay in a Cohort of Patients with Solid tumors: A Proposed Solution for Decentralized Plasma Testing. Oncologist 2021, 26, e1971–e1981. [Google Scholar] [CrossRef]
- FDA. Cobas EGFR Mutation Test v2. 2016. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/cobas-egfr-mutation-test-v2 (accessed on 17 February 2022).
- Roche. Roche Receives FDA Approval for the cobas EGFR Mutation Test v2 as the First Companion Diagnostic Test for Expanded EGFR TKI Therapies in Patients with Non-Small Cell Lung Cancer. 2020. Available online: https://diagnostics.roche.com/global/en/news-listing/2020/fda-class-claim-for-cobas-egfr-mutation-test-v2.html#:~:text=Roche%20receives%20FDA%20approval%20for,non%2Dsmall%20cell%20lung%20cancer (accessed on 17 February 2022).
- Heeke, S.; Benzaquen, J.; Hofman, V.; Ilié, M.; Allegra, M.; Long-Mira, E.; Lassalle, S.; Tanga, V.; Salacroup, C.; Bonnetaud, C.; et al. Critical Assessment in Routine Clinical Practice of Liquid Biopsy for EGFR Status Testing in Non–Small-Cell Lung Cancer: A Single-Laboratory Experience (LPCE, Nice, France). Clin. Lung Cancer 2020, 21, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Shin, S.; Lee, K.-A. A Comparative Study for Detection of EGFR Mutations in Plasma Cell-Free DNA in Korean Clinical Diagnostic Laboratories. BioMed Res. Int. 2018, 2018, 7392419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mountzios, G.; Koumarianou, A.; Bokas, A.; Mavroudis, D.; Samantas, E.; Fergadis, E.; Linardou, H.; Katsaounis, P.; Athanasiadis, E.; Karamouzis, M.; et al. A Real-World, Observational, Prospective Study to Assess the Molecular Epidemiology of Epidermal Growth Factor Receptor (EGFR) Mutations upon Progression on or after First-Line Therapy with a First- or Second-Generation EGFR Tyrosine Kinase Inhibitor in EGFR Mutation-Positive Locally Advanced or Metastatic Non-Small Cell Lung Cancer: The ‘LUNGFUL’ Study. Cancers 2021, 13, 3172. [Google Scholar] [CrossRef] [PubMed]
- FDA. FoundationOne Liquid CDx—P190032. 2020. Available online: https://www.fda.gov/medical-devices/recently-approved-devices/foundationone-liquid-cdx-p190032 (accessed on 17 February 2022).
- FDA. Guardant360 CDx—P200010. 2020. Available online: https://www.fda.gov/medical-devices/recently-approved-devices/guardant360-cdx-p200010 (accessed on 17 February 2022).
- Guardant Health, Guardant360 CDx Technical Information. 2021. Available online: https://guardant360cdx.com/wp-content/uploads/guardant360-cdx-technical-information.pdf (accessed on 31 May 2022).
- Foundation Medicine, Foundation One Liquid CDx Technical Information. 2021. Available online: https://info.foundationmedicine.com/hubfs/FMI%20Labels/FoundationOne_Liquid_CDx_Label_Technical_Info.pdf (accessed on 31 May 2022).
- Larribère, L.; Martens, U.M. Advantages and Challenges of Using ctDNA NGS to Assess the Presence of Minimal Residual Disease (MRD) in Solid Tumors. Cancers 2021, 13, 5698. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J. Characterizing the Cancer Genome in Blood. Cold Spring Harb. Perspect. Med. 2018, 9, a026880. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Liu, X.; Zheng, B.; Ke, R.; Tzeng, C.-M. Liquid Biopsy, ctDNA Diagnosis through NGS. Life 2021, 11, 890. [Google Scholar] [CrossRef]
- Ross, M.G.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and measuring bias in sequence data. Genome Biol. 2013, 14, R51-20. [Google Scholar] [CrossRef] [Green Version]
- Willey, J.C.; Morrison, T.B.; Austermiller, B.; Crawford, E.L.; Craig, D.J.; Blomquist, T.M.; Jones, W.D.; Wali, A.; Lococo, J.S.; Haseley, N.; et al. Advancing NGS quality control to enable measurement of actionable mutations in circulating tumor DNA. Cell Rep. Methods 2021, 1, 100106. [Google Scholar] [CrossRef]
- Stetson, D.; Ahmed, A.; Xu, X.; Nuttall, B.; Lubinski, T.J.; Johnson, J.H.; Barrett, J.C.; Dougherty, B.A. Orthogonal Comparison of Four Plasma NGS Tests with Tumor Suggests Technical Factors are a Major Source of Assay Discordance. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- El Achi, H.; Khoury, J.D.; Loghavi, S. Liquid Biopsy by Next-Generation Sequencing: A Multimodality Test for Management of Cancer. Curr. Hematol. Malign. Rep. 2019, 14, 358–367. [Google Scholar] [CrossRef]
- Shu, Y.; Wu, X.; Tong, X.; Wang, X.; Chang, Z.; Mao, Y.; Chen, X.; Sun, J.; Wang, Z.; Hong, Z.; et al. Circulating Tumor DNA Mutation Profiling by Targeted Next Generation Sequencing Provides Guidance for Personalized Treatments in Multiple Cancer Types. Sci. Rep. 2017, 7, 583. [Google Scholar] [CrossRef]
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical impact of subclonal EGFR T790M mutations in advanced-stage EGFR-mutant non-small-cell lung cancers. Nat. Commun. 2021, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Kohrn, B.F.; Loubet-Senear, K.J.; Dunn, Y.J.; Ahn, E.H.; O’Sullivan, J.N.; Salk, J.J.; Bronner, M.P.; Beckman, R.A. Extensive subclonal mutational diversity in human colorectal cancer and its significance. Proc. Natl. Acad. Sci. USA 2019, 116, 26863–26872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2015, 13, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical Relevance of KRAS-Mutated Subclones Detected with Picodroplet Digital PCR in Advanced Colorectal Cancer Treated with Anti-EGFR Therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Francesco Marass, K.H. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Takano, S.; Maekawa, S.; Yamaguchi, T.; Yoshida, T.; Kobayashi, S.; Iwamoto, F.; Kuno, T.; Hayakawa, H.; Matsuda, S.; et al. Fractionated small cell-free DNA increases possibility to detect cancer-related gene mutations in advanced colorectal cancer. JGH Open 2020, 4, 978–986. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Heider, K.; Gale, D.; Murphy, S.; Fisher, E.; Mouliere, F.; Ruiz-Valdepenas, A.; Santonja, A.; Morris, J.; Chandrananda, D.; et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. 2020, 12, eaaz8084. [Google Scholar] [CrossRef]
- Esagian, S.M.; Grigoriadou, G.I.; Nikas, I.P.; Boikou, V.; Sadow, P.M.; Won, J.-K.; Economopoulos, K.P. Comparison of liquid-based to tissue-based biopsy analysis by targeted next generation sequencing in advanced non-small cell lung cancer: A comprehensive systematic review. J. Cancer Res. Clin. Oncol. 2020, 146, 2051–2066. [Google Scholar] [CrossRef]
- Bruno, R.; Fontanini, G. Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Lee, J.K.; Hazar-Rethinam, M.; Decker, B.; Gjoerup, O.; Madison, R.W.; Lieber, D.S.; Chung, J.H.; Schrock, A.B.; Creeden, J.; Venstrom, J.M.; et al. The Pan-Tumor Landscape of Targetable Kinase Fusions in Circulating Tumor DNA. Clin. Cancer Res. 2021, 28, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Woodhouse, S.; Howarth, K.; Lensing, S.; Smith, M.; Epstein, M.; Madi, M.; Smalley, S.; Leroy, C.; Hinton, J.; et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE 2018, 13, e0193802. [Google Scholar] [CrossRef] [PubMed]
- Supplee, J.G.; Milan, M.S.; Lim, L.P.; Potts, K.T.; Sholl, L.M.; Oxnard, G.R.; Paweletz, C.P. Sensitivity of next-generation sequencing assays detecting oncogenic fusions in plasma cell-free DNA. Lung Cancer 2019, 134, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, N.; Kohsaka, S.; Kurokawa, K.; Shinno, Y.; Nakamura, I.T.; Ueno, T.; Kojima, S.; Kawazu, M.; Suehara, Y.; Ishijima, M.; et al. Highly sensitive fusion detection using plasma cell-free RNA in non-small-cell lung cancers. Cancer Sci. 2021, 112, 4393–4403. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Tsoulos, N.; Tsantikidi, K.; Metaxa-Mariatou, V.; Stamou, P.E.; Kladi-Skandali, A.; Kapeni, E.; Tsaousis, G.; Pentheroudakis, G.; Petrakis, D.; et al. Clinical feasibility of NGS liquid biopsy analysis in NSCLC patients. PLoS ONE 2019, 14, e0226853. [Google Scholar] [CrossRef] [Green Version]
- Jahangiri, L.; Hurst, T. Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers 2019, 11, 1938. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef] [Green Version]
- Underhill, H.R. Leveraging the Fragment Length of Circulating Tumour DNA to Improve Molecular Profiling of Solid Tumour Malignancies with Next-Generation Sequencing: A Pathway to Advanced Non-invasive Diagnostics in Precision Oncology? Mol. Diagn. Ther. 2021, 25, 389–408. [Google Scholar] [CrossRef]
- Weber, Z.T.; Collier, K.A.; Tallman, D.; Forman, J.; Shukla, S.; Asad, S.; Rhoades, J.; Freeman, S.; Parsons, H.A.; Williams, N.O.; et al. Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer. Genome Med. 2021, 13, 89. [Google Scholar] [CrossRef]
- Lee, S.; Park, Y.-S.; Chang, W.-J.; Choi, J.Y.; Lim, A.; Kim, B.; Lee, S.-B.; Lee, J.-W.; Kim, S.-H.; Kim, J.; et al. Clinical Implication of Liquid Biopsy in Colorectal Cancer Patients Treated with Metastasectomy. Cancers 2021, 13, 2231. [Google Scholar] [CrossRef]
- Bando, H.; Kagawa, Y.; Kato, T.; Akagi, K.; Denda, T.; Nishina, T.; Komatsu, Y.; Oki, E.; Kudo, T.; Kumamoto, H.; et al. Correction: A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br. J. Cancer 2020, 122, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, Y.; Elez, E.; García-Foncillas, J.; Bando, H.; Taniguchi, H.; Vivancos, A.; Akagi, K.; García, A.; Denda, T.; Ros, J.; et al. Combined Analysis of Concordance between Liquid and Tumor Tissue Biopsies for RAS Mutations in Colorectal Cancer with a Single Metastasis Site: The METABEAM Study. Clin. Cancer Res. 2021, 27, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.W.Y.; Cheng, M.L.; Shady, M.; Yang, J.L.; Stephens, D.; Won, H.; Srinivasan, P.; Huberman, K.; Meng, F.; Jing, X.; et al. Tumor fraction-guided cell-free DNA profiling in metastatic solid tumor patients. Genome Med. 2021, 13, 96. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Leal, A.; Van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Bacon, J.V.; Annala, M.; Soleimani, M.; Lavoie, J.-M.; So, A.; Gleave, M.E.; Fazli, L.; Wang, G.; Chi, K.N.; Kollmannsberger, C.K.; et al. Plasma Circulating Tumor DNA and Clonal Hematopoiesis in Metastatic Renal Cell Carcinoma. Clin. Genitourin. Cancer 2020, 18, 322–331. [Google Scholar] [CrossRef]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Burstein, H.J.; Somerfield, M.R.; Barton, D.L.; Dorris, A.; Fallowfield, L.J.; Jain, D.; Johnston, S.R.D.; Korde, L.A.; Litton, J.K.; Macrae, E.R.; et al. Endocrine Treatment and Targeted Therapy for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 3959–3977. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology—Breast Cancer. Version 5.2020; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2020. [Google Scholar]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement from the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Kalemkerian, G.P.; Narula, N.; Kennedy, E.B. Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment with Targeted Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement Summary of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J. Oncol. Pract. 2018, 14, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non–Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef] [PubMed]
- Sunami, K.; Bando, H.; Yatabe, Y.; Naito, Y.; Takahashi, H.; Tsuchihara, K.; Toyooka, S.; Mimori, K.; Kohsaka, S.; Uetake, H.; et al. Appropriate use of cancer comprehensive genome profiling assay using circulating tumor DNA. Cancer Sci. 2021, 112, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chen, C.T.; Goyal, L.; Walmsley, C.; Pinto, C.J.; Baiev, I.; Allen, R.; Henderson, L.; Saha, S.; Reyes, S.; et al. Cell-free DNA captures tumor heterogeneity and driver alterations in rapid autopsies with pre-treated metastatic cancer. Nat. Commun. 2021, 12, 3199. [Google Scholar] [CrossRef]
- Moreno, F.; Gayarre, J.; López-Tarruella, S.; del Monte-Millán, M.; Picornell, A.C.; Álvarez, E.; García-Saenz, J.; Jerez, Y.; Márquez-Rodas, I.; Echavarría, I.; et al. Concordance of Genomic Variants in Matched Primary Breast Cancer, Metastatic Tumor, and Circulating Tumor DNA: The MIRROR Study. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Liebs, S.; Eder, T.; Klauschen, F.; Schütte, M.; Yaspo, M.-L.; Keilholz, U.; Tinhofer, I.; Kidess-Sigal, E.; Braunholz, D. Applicability of liquid biopsies to represent the mutational profile of tumor tissue from different cancer entities. Oncogene 2021, 40, 5204–5212. [Google Scholar] [CrossRef]
- Barefoot, M.E.; Loyfer, N.; Kiliti, A.J.; McDeed, A.P.I.; Kaplan, T.; Wellstein, A. Detection of Cell Types Contributing to Cancer from Circulating, Cell-Free Methylated DNA. Front. Genet. 2021, 12, 671057. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Han, D.S.C.; Jiang, P.; Chiu, R.W.K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Zhang, X.; Zhang, M.; Wang, Q.; Chen, X.; Yu, R.; Bao, H.; Liu, J.; Wu, X.; Shao, Y.; et al. Real-world circulating tumor DNA analysis depicts resistance mechanism and clonal evolution in ALK inhibitor-treated lung adenocarcinoma patients. ESMO Open 2022, 7, 100337. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Stenehjem, D.; Nussenzveig, R.; Carroll, E.; Bailey, E.; Batten, J.; Maughan, B.L.; Agarwal, N. Evolution of the genomic landscape of circulating tumor DNA (ctDNA) in metastatic prostate cancer over treatment and time. Cancer Treat. Res. Commun. 2019, 19, 100120. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Othman, T.; Chen, C.; Sandhu, J.; Ouyang, C.; Fakih, M. Guardant360 Circulating Tumor DNA Assay Is Concordant with FoundationOne Next-Generation Sequencing in Detecting Actionable Driver Mutations in Anti-EGFR Naive Metastatic Colorectal Cancer. Oncol. 2019, 25, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Abbosh, C.; Swanton, C.; Birkbak, N. Clonal haematopoiesis: A source of biological noise in cell-free DNA analyses. Ann. Oncol. 2019, 30, 358–359. [Google Scholar] [CrossRef]
- Marass, F.; Stephens, D.; Ptashkin, R.; Zehir, A.; Berger, M.F.; Solit, D.B.; A Diaz, L.; Tsui, D.W.Y. Fragment Size Analysis May Distinguish Clonal Hematopoiesis from Tumor-Derived Mutations in Cell-Free DNA. Clin. Chem. 2020, 66, 616–618. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Ai, X.; Cui, J.; Zhang, J.; Chen, R.; Lin, W.; Xie, C.; Liu, A.; Zhang, J.; Yang, W.; Hu, X.; et al. Clonal Architecture of EGFR Mutation Predicts the Efficacy of EGFR-Tyrosine Kinase Inhibitors in Advanced NSCLC: A Prospective Multicenter Study (NCT03059641). Clin. Cancer Res. 2021, 27, 704–712. [Google Scholar] [CrossRef]
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients with RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol. 2021, 7, 1529–1535. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Fenocchio, E.; Amatu, A.; Corallo, S.; Manai, C.; Tosi, F.; et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: The CHRONOS trial. J. Clin. Oncol. 2021, 39, 3506. [Google Scholar] [CrossRef]
- Parseghian, C.; Loree, J.; Morris, V.; Liu, X.; Clifton, K.; Napolitano, S.; Henry, J.; Pereira, A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siena, S.; Sartore-Bianchi, A.; Garcia-Carbonero, R.; Karthaus, M.; Smith, D.; Tabernero, J.; Van Cutsem, E.; Guan, X.; Boedigheimer, M.; Ang, A.; et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 29, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.M.; Shibayama, T.; Chan, H.T.; Otaki, M.; Hara, F.; Kobayashi, T.; Kobayashi, K.; Hosonaga, M.; Fukada, I.; Inagaki, L.; et al. Serial circulating tumor DNA monitoring of CDK4/6 inhibitors response in metastatic breast cancer. Cancer Sci. 2022, 113, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Hardy-Bessard, A.-C.; Bachelot, T.; Pierga, J.-Y.; Canon, J.-L.; Clatot, F.; Andre, F.; De La Motte Rouge, T.; Pistilli, B.; Dalenc, F.; et al. Fulvestrant-palbociclib vs. continuing AI-palbociclib upon detection of circulating ESR1 mutation in HR+ HER2–metastatic breast cancer patients: Results of PADA-1, a UCBG-GINECO randomized phase 3 trial. In Proceedings of the 2021 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 7–10 December 2021. [Google Scholar]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, M.; Mehnert, J.; Silk, A.; Jabbour, S.; Ganesan, S.; Popli, P.; Riedlinger, G.; Stephenson, R.; de Meritens, A.; Leiser, A.; et al. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open 2021, 7, 100336. [Google Scholar] [CrossRef]
- Snyder, A.; Morrissey, M.P.; Hellmann, M.D. Use of Circulating Tumor DNA for Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 6909–6915. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients with Non–Small Cell Lung Cancer with Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Ahn, M.-J.; Luft, A.; van den Heuvel, M.; Cobo, M.; Smolin, A.; Vicente, D.; et al. Durvalumab With or Without Tremelimumab vs. Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Si, H.; Kuziora, M.; Quinn, K.J.; Helman, E.; Ye, J.; Liu, F.; Scheuring, U.; Peters, S.; Rizvi, N.A.; Brohawn, P.Z.; et al. A Blood-based Assay for Assessment of Tumor Mutational Burden in First-line Metastatic NSCLC Treatment: Results from the MYSTIC Study. Clin. Cancer Res. 2020, 27, 1631–1640. [Google Scholar] [CrossRef]
- Stadler, J.-C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Ba, H.; Liu, L.; Peng, Q.; Chen, J.; Zhu, Y.-D. The relationship between blood-based tumor mutation burden level and efficacy of PD-1/PD-L1 inhibitors in advanced non-small cell lung cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, X.; Gao, C.; Gong, J.; Wang, X.; Gao, J.; Li, Z.; Wang, J.; Yang, B.; Wang, L.; et al. Plasma-based microsatellite instability detection strategy to guide immune checkpoint blockade treatment. J. Immunother. Cancer 2020, 8, e001297. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, A.; Durham, J.N.; Keefer, L.A.; Bartlett, B.R.; Zielonka, M.; Murphy, D.; White, J.R.; Lu, S.; Verner, E.L.; Ruan, F.; et al. Noninvasive Detection of Microsatellite Instability and High Tumor Mutation Burden in Cancer Patients Treated with PD-1 Blockade. Clin. Cancer Res. 2019, 25, 7024–7034. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Method | Name | Example | Number of Targets | LOD |
|---|---|---|---|---|
| PCR-based | qPCR | COLD-PCR | 1 | 0.1–1% |
| dPCR | BEAMing | 1–20 targets | 0.01–0.1% | |
| dPCR | ddPCR | up to 5 targets | 0.01–0.1% | |
| MassSpec PCR | UltraSeek | Multigenes | 0.1–1% | |
| NGS-based | Amplicon-based | IonTorrent-Oncomine | Multigenes | 0.1% |
| Safe-SeqS; Plasma-SeqSensei | Multigenes | 0.04–0.2% | ||
| Hybrid capture | Avenio, TruSight 500 | Multigenes | 0.5% | |
| CAPP-Seq; Guardant360; FoundationOne Liquid | Multigenes | 0.02% |
| Cancer Type | Sample Size | Method | Number of Genes | Types of Variants | Detection Rate | Detection of Actionable Mutations from ctDNA | CH Elimination | Tissue Plasma Concordance * | Comments | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Pan Cancer | 11,525 | Customized hybridization capture NGS | 1021 | SNVs, Indels, CNVs, Fusion, bTMB | 73.50% | 41.2% | Yes | N.D | [40] | |
| Pan Cancer | 681 | MSK-ACCESS hybridization capture NGS | 129 | SNVs, Indels, CNVs, Fusion, bTMB | 73% | 56.0% | Yes | 59.0% | Variable collection interval between tissue and plasma | [63] |
| Pan Cancer | 433 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 37% | N.D. | No | 45.0% | Only examined TP53; variable collection interval between tissue and plasma | [65] |
| Pan Cancer | 161 | Customized hybridization capture NGS (GRAIL) | 508 | SNVs, Indels, CNVs, Fusion | 84% | N.D. | Yes | 72.0% | [42] | |
| Pan Cancer | 10,593 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 86% | 72.0% | No | 92.0% | Concordance based on 543 patients, in 7 genes | [57] |
| Lung | 1971 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 87.30% | 26.70% | No | N.D. | [66] | |
| Lung | 262 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 60.40% | 60.40% | No | 67.70% | Initial diagnosis/treatment naïve; only examined 6 genes (EGFR, ALK, MET, ROS1, RET, KRAS) | [67] |
| Lung | 934 | FoundationLiquid/FoundationACT NGS | 62/70 | SNVs, Indels, CNVs, Fusion | 90.00% | 20.00% | No | N.D. | ctDNA: 937 patients; Tissue: 5582 patients | [68] |
| Lung | 8388 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 86.00% | 48.00% | No | N.D. | [69] | |
| Lung | 282 | Guardant 360 hybridization capture NGS/ddPCR | 73 | SNVs, Indels, CNVs, Fusion | - | 27.30% | No | 80.0% | [53] | |
| Lung | 127 | Customized hybridization capture NGS (GRAIL) | 37 | SNVs, Indels, CNVs, Fusion | - | - | Yes | 75.0% | [56] | |
| Lung | 210 | ResBio ctDx-Lung amplicon-based NGS | 21 | SNVs, Indels, CNVs, Fusion | 64.30% | 21.90% | No | 60.6% | A subset of patients subjected to treatment at the time of plasma collection | [70] |
| Lung | 323 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | - | 33.00% | No | N.D. | [64] | |
| Breast | 162 | Customized amplicon-based NGS | 39 | SNVs | 92.50% | 39.00% | No | N.D. | [71] | |
| Breast | 1044 | Guardant 360 hybridization capture NGS/ddPCR | ddPCR: 4; NGS: 73 | SNVs, Indels, CNVs, Fusion | 51.10% | 34.50% | No | 93% | Concordance is based on 77 patients in 4 genes (AKT1, HER2, ESR1, PIK3CA). Negative concordance was included. | [54] |
| Breast | 255 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 89.00% | 26.00% | No | 79–91% | Actionable alterations in PIK3CA, ESR1, ERBB2 | [55] |
| Gastrointestinal | 1687 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 91.40% | 57.30% | No | 8.3–80.3% (<0.3 vs. >0.3 clonality) | Concordance is based on 287 patients | [59] |
| Gastrointestinal | 200 | Customized amplicon-based NGS | 150 | SNVs, Indels, CNVs, Fusion, bTMB, bMSI | 84.05% | 45.50% | No | N.D. | [72] | |
| Gastrointestinal | 1064 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 93.70% | 47.70% | No | N.D. | Only included metastatic colorectal cancer patients | [73] |
| Gastrointestinal | 282 | Guardant 360 hybridization capture NGS | 73 | SNVs, Indels, CNVs, Fusion | 75.00% | 48.00% | No | 50–86% | [74] | |
| Prostate | 3334 | FoundationLiquid/FoundationACT NGS | 62/70 | SNVs, Indels, CNVs, Fusion | 79.50% | 30% (DDR gene alteration) | Yes | 75.3% (SNVs); 70.3% (rearrangements); 27.5% (CNVs) | DDR alterations: BRCA1/2; CDK12; MSI-H | [58] |
| Approved Diagnostic Tool | Technology | Number of Genes | Input (ng) | Disease | Drug | Biomarker | LOD |
|---|---|---|---|---|---|---|---|
| Cobas EGFR | RT-PCR | 1 | Undefined; (2 mL of plasma) | NSCLC | Erlotinib & Gefitinib | EGFR Exon 19 deletions; L858R | Exon 19 deletions: 0.1–0.5%; L858R: 0.4–0.8% |
| Osimertinib | EGFR T790M | Exon 19 deletions: 0.1–0.5%; L858R: 0.4–0.8%; T790M: 0.4–0.8% | |||||
| Therascreen | RQT-PCR | 1 | Undefined; (2 mL of plasma) | Breast | Alpelisib | PIK3CA (C420R, E542K, E545A, E545D [1635G > T only], E545G, E545K, Q546E, Q546R; and H1047L, H1047R, and H1047Y) | 1.82–7.07% |
| FoundationOne Liquid CDx | NGS-hybridization enrichment | 324 (311 FDA approved) | 20 | NSCLC | Alectinib | ALK rearrangements: ALK-EML4 | ALK-EML4: 0.24% |
| Osimertinib & Erlotinib | EGFR Exon 19 deletions; L858R | Exon 19 deletions: 0.27%; L858R: 0.34% | |||||
| Capmatinib | MET SNVs & Indels that lead to MET exon 14 skipping | Substitutions: 0.4%; Indels: 0.28% | |||||
| Prostate | Olaparib | BRAC1 | Substitutions: 0.34%; Indels: 0.38% | ||||
| BRCA2 | Substitutions: 0.37%; Indels: 0.36%; | ||||||
| ATM alterations | Indels: 0.51% | ||||||
| Rucaparib | BRCA1 | Substitutions: 0.34%; Indels: 0.38% | |||||
| BRCA2 | Substitutions: 0.37%; Indels: 0.36% | ||||||
| Ovarian | Rucaparib | BRCA1 | Substitutions: 0.34%; Indels: 0.38% | ||||
| BRCA2 | Substitutions: 0.37%; Indels: 0.36% | ||||||
| Breast | Alpelisib | PIK3CA (C420R, E542K, E545A, E545D [1635G > T only], E545G, E545K, Q546E, Q546R; and H1047L, H1047R, and H1047Y) | Substitutions: 0.34% | ||||
| Guardant360 CDx | NGS- hybridization enrichment | 74 (55 FDA approved) | 30 | NSCLC | Osimertinib | EGFR Exon 19 deletions; L858R; T790M | 0.20% |
| Amivantamab-vmjw | EGFR exon 20 insertions | 0.30% | |||||
| Sotorasib | KRAS G12C | 0.50% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, H.T.; Chin, Y.M.; Low, S.-K. Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers 2022, 14, 3275. https://doi.org/10.3390/cancers14133275
Chan HT, Chin YM, Low S-K. Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers. 2022; 14(13):3275. https://doi.org/10.3390/cancers14133275
Chicago/Turabian StyleChan, Hiu Ting, Yoon Ming Chin, and Siew-Kee Low. 2022. "Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges" Cancers 14, no. 13: 3275. https://doi.org/10.3390/cancers14133275
APA StyleChan, H. T., Chin, Y. M., & Low, S.-K. (2022). Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers, 14(13), 3275. https://doi.org/10.3390/cancers14133275