
Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy


Abstract
:Simple Summary
Abstract
1. Tumour Heterogeneity: From Historical Perspectives to Novel Insights
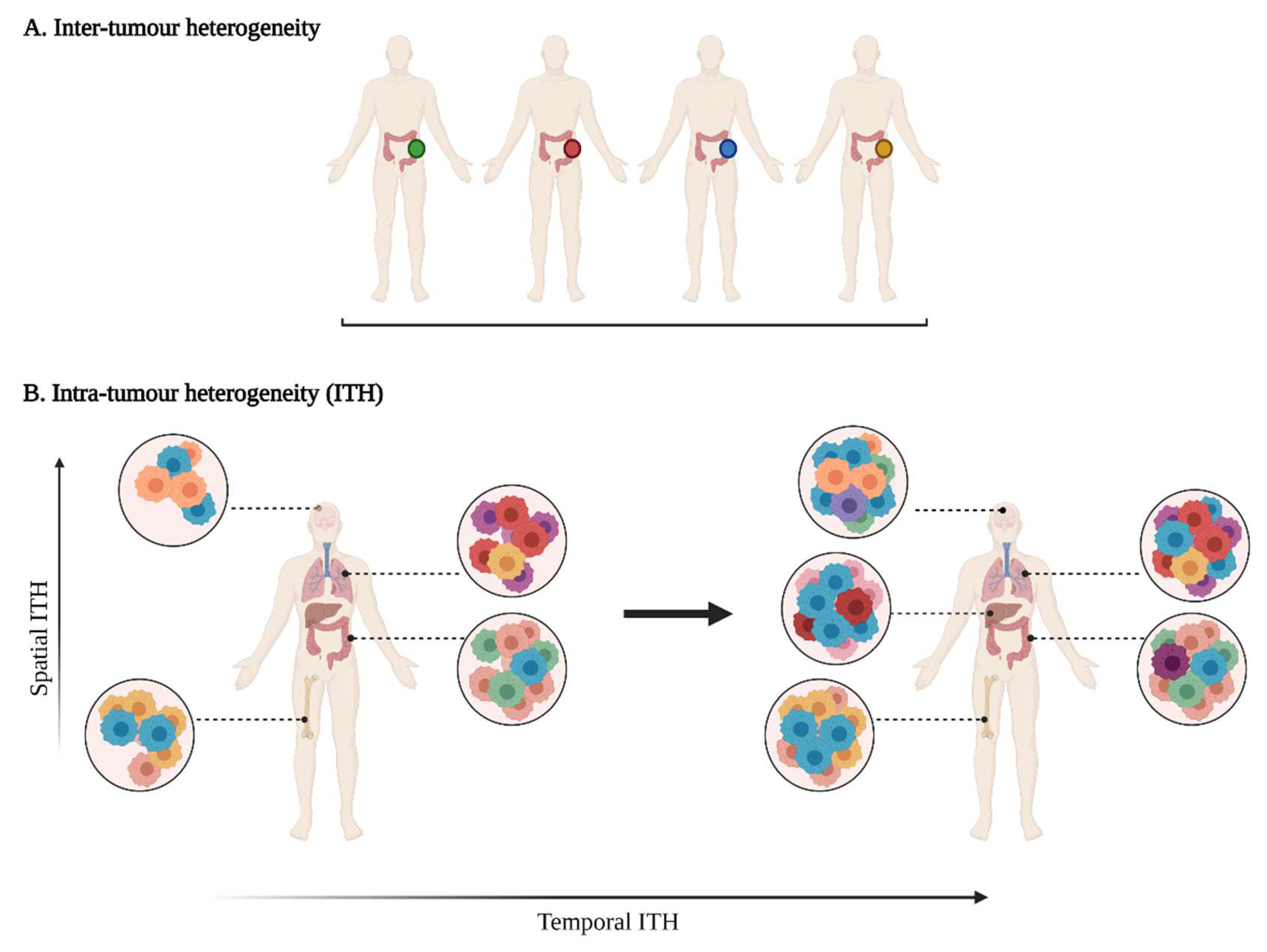
1.1. Varying Degrees of Tumour Heterogeneity
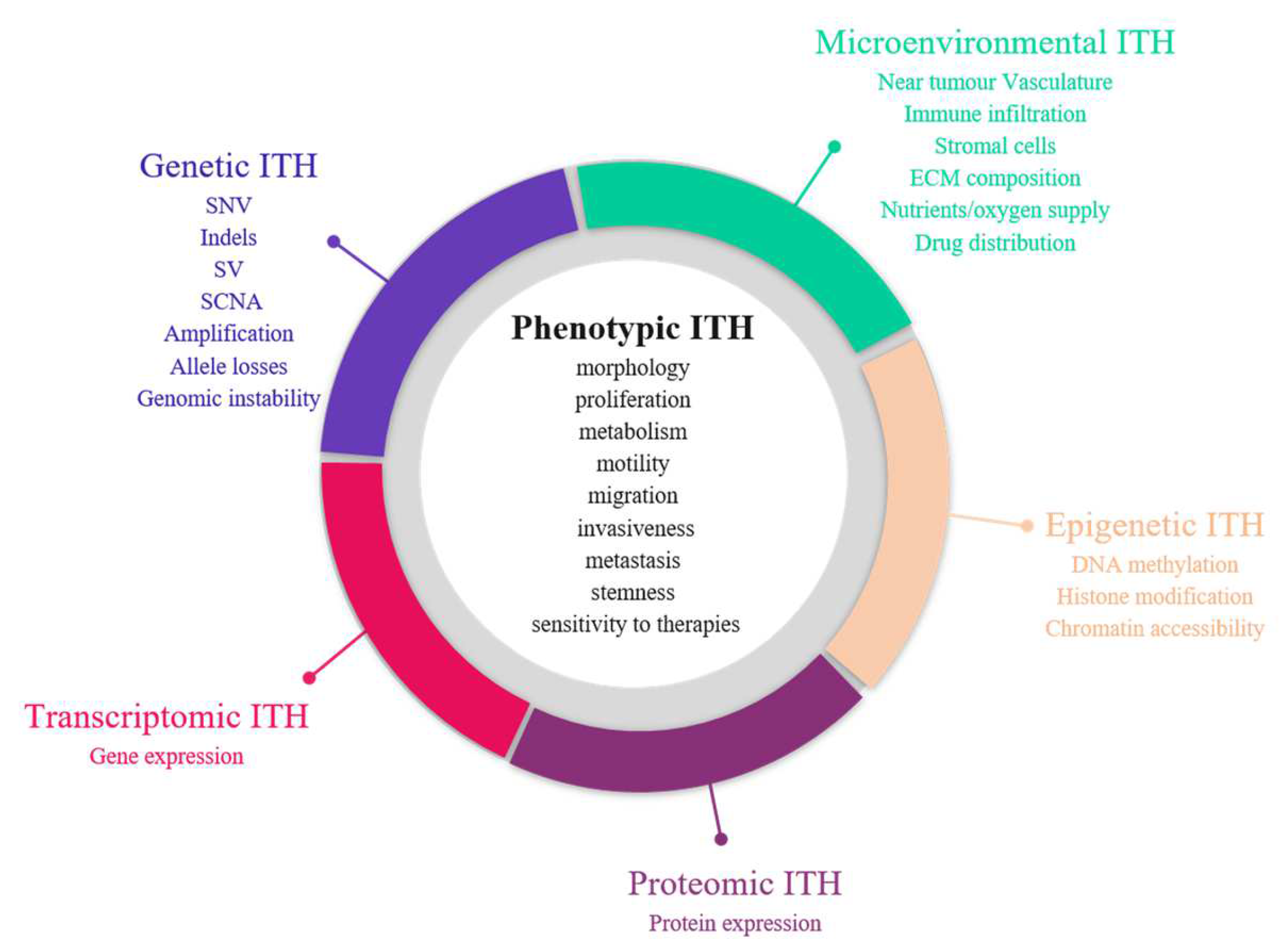
1.1.1. Phenotypic Heterogeneity
1.1.2. Molecular Heterogeneity
1.1.3. Tumour Micro-Environment (TME) Heterogeneity
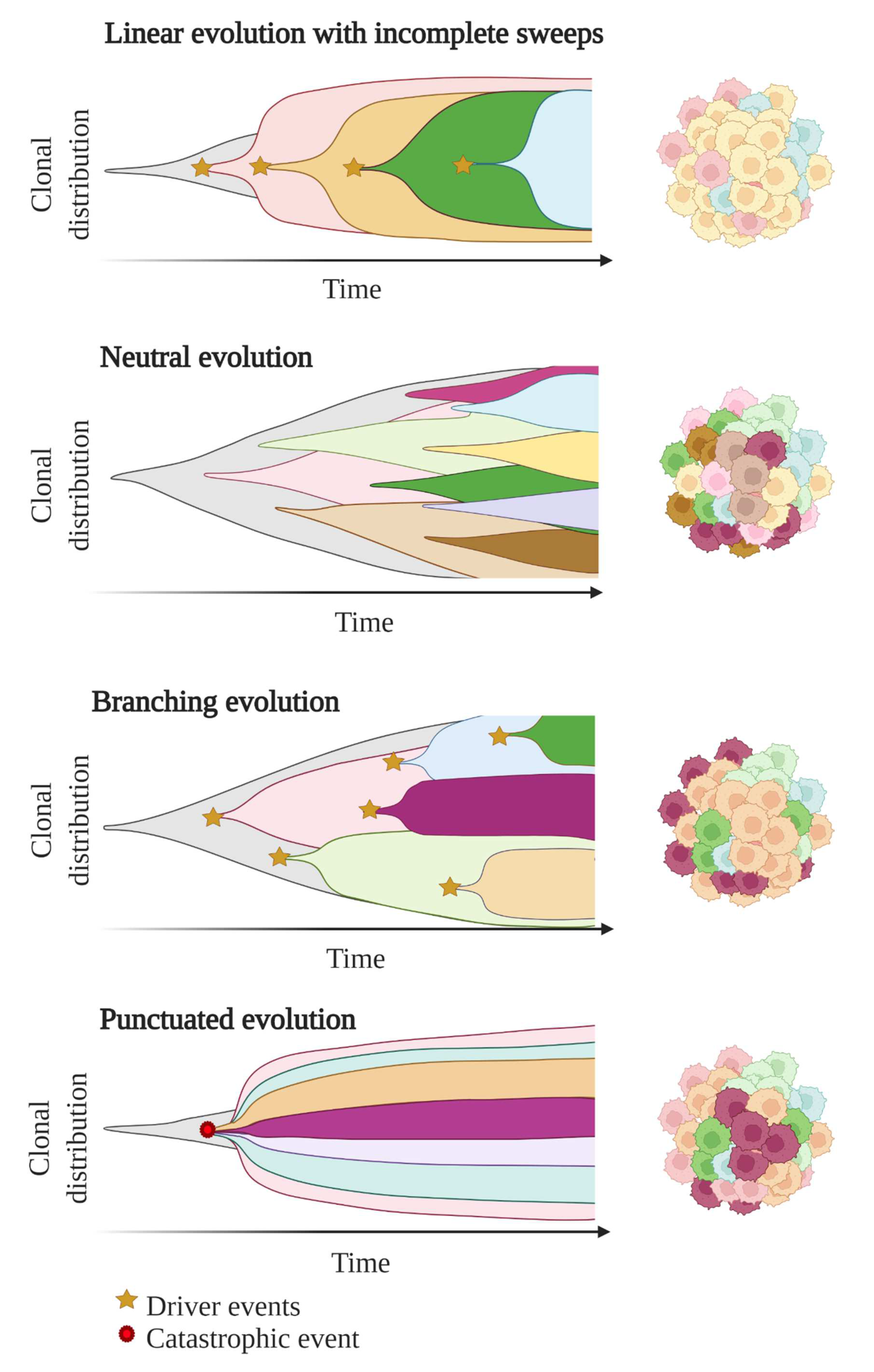
1.2. Unravelling Evolutionary Processes behind Tumour Heterogeneity
2. Clinical Consequences of Tumour Heterogeneity
2.1. Impact on Diagnosis, Prognosis and Therapeutic Predictions
2.1.1. Tumour Sampling Bias Due to Spatial ITH
2.1.2. Tumour Sampling Bias Due to Temporal ITH
2.1.3. Determining ITH to Decipher the Identity of the Tumour or of Specific Regions of the Tumour
2.1.4. ITH Is Associated with Poorer Clinical Outcomes
2.1.5. High Amounts of Biomarkers to Analyse in Order to Fully Decipher ITH
2.2. Impact on Therapeutic Strategies
3. Emerging Approaches to Evaluate ITH
3.1. Bulk-Cell Versus Single-Cell Approaches
3.2. Tissue Biopsy Sampling Approaches
3.3. Post-Mortem Samples
3.4. Liquid Biopsy-Based Approaches: A Better Reflection of ITH?
3.4.1. CTCs
3.4.2. ctDNA
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational Implications of Tumor Heterogeneity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosman, F.T. Tumor Heterogeneity: Will It Change What Pathologists Do. Pathobiology 2018, 85, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Bignold, L.P.; Coghlan, B.L.D.; Jersmann, H.P.A. (Eds.) Hansemann’s Ideas of the Nature of Cancer: Description and Analysis. In David Paul von Hansemann: Contributions to Oncology: Context, Comments and Translations; Birkhäuser: Basel, Switzerland, 2007; pp. 75–90. ISBN 978-3-7643-7769-4. [Google Scholar]
- Müller, J. On the Nature and Structural Characteristics of Cancer: General Observations on the Minute Structure of Morbid Growths. CA. Cancer J. Clin. 1973, 23, 307–312. [Google Scholar] [CrossRef]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and Annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef]
- Parquet, R.A. Rudolf Carl Virchow. Acta Gastroenterol. Latinoam. 2014, 44, 202. [Google Scholar]
- Stanta, G.; Jahn, S.W.; Bonin, S.; Hoefler, G. Tumour Heterogeneity: Principles and Practical Consequences. Virchows Arch. Int. J. Pathol. 2016, 469, 371–384. [Google Scholar] [CrossRef]
- Ramón, Y.; Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical Implications of Intratumor Heterogeneity: Challenges and Opportunities. J. Mol. Med. Berl. Ger. 2020, 98, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Polyak, K. Tumor Heterogeneity: Causes and Consequences. Biochim. Biophys. Acta 2010, 1805, 105. [Google Scholar] [CrossRef] [Green Version]
- Heppner, G.H. Tumor Heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar]
- Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers 2019, 11, 1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in Understanding Cancer Genomes through Second-Generation Sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing Genetic Intra-Tumor Heterogeneity across 2658 Human Cancer Genomes. Cell 2021, 184, 2239–2254.e39. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-Cancer Analysis of the Extent and Consequences of Intratumor Heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational Signatures: The Patterns of Somatic Mutations Hidden in Cancer Genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. 2018, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Dietz, S.; Harms, A.; Endris, V.; Eichhorn, F.; Kriegsmann, M.; Longuespée, R.; Stenzinger, A.; Sültmann, H.; Warth, A.; Kazdal, D. Spatial Distribution of EGFR and KRAS Mutation Frequencies Correlates with Histological Growth Patterns of Lung Adenocarcinomas. Int. J. Cancer 2017, 141, 1841–1848. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Brocks, D.; Gerhäuser, C. Intratumor Heterogeneity in Epigenetic Patterns. Semin. Cancer Biol. 2018, 51, 12–21. [Google Scholar] [CrossRef]
- McQuerry, J.A.; Chang, J.T.; Bowtell, D.D.L.; Cohen, A.; Bild, A.H. Mechanisms and Clinical Implications of Tumor Heterogeneity and Convergence on Recurrent Phenotypes. J. Mol. Med. Berl. Ger. 2017, 95, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic Heterogeneity in Cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining Epigenetic Drugs with Other Therapies for Solid Tumours—Past Lessons and Future Promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Beyes, S.; Bediaga, N.G.; Zippo, A. An Epigenetic Perspective on Intra-Tumour Heterogeneity: Novel Insights and New Challenges from Multiple Fields. Cancers 2021, 13, 4969. [Google Scholar] [CrossRef]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- van ’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene Expression Profiling Predicts Clinical Outcome of Breast Cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, R.K. Design and Multiseries Validation of a Web-Based Gene Expression Assay for Predicting Breast Cancer Recurrence and Patient Survival. J. Mol. Diagn. 2011, 13, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Daza, J.; Itzel, T.; Betge, J.; Zhan, T.; Marmé, F.; Teufel, A. Prognostic Cancer Gene Expression Signatures: Current Status and Challenges. Cells 2021, 10, 648. [Google Scholar] [CrossRef]
- Bateman, N.W.; Conrads, T.P. Recent Advances and Opportunities in Proteomic Analyses of Tumour Heterogeneity. J. Pathol. 2018, 244, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-Tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. Poznan Pol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Ricketts, C.J. The Cancer Genome Atlas of Renal Cell Carcinoma: Findings and Clinical Implications. Nat. Rev. Urol. 2019, 16, 539–552. [Google Scholar] [CrossRef]
- ICGC/TCGA. Pan-Cancer Analysis of Whole Genomes Consortium Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Cancer Genome Atlas Research Network Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues That Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [Green Version]
- Garattini, S.; Fuso Nerini, I.; D’Incalci, M. Not Only Tumor but also Therapy Heterogeneity. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 13–19. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Wei, W.-F.; Wu, H.-Z.; Fan, L.-S.; Wang, W. Cancer-Associated Fibroblast Heterogeneity: A Factor That Cannot Be Ignored in Immune Microenvironment Remodeling. Front. Immunol. 2021, 12, 2760. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2019, 112, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhu, B.; Zhang, M.; Wang, X. Roles of Immune Microenvironment Heterogeneity in Therapy-Associated Biomarkers in Lung Cancer. Semin. Cell Dev. Biol. 2017, 64, 90–97. [Google Scholar] [CrossRef]
- Fico, F.; Santamaria-Martínez, A. The Tumor Microenvironment as a Driving Force of Breast Cancer Stem Cell Plasticity. Cancers 2020, 12, 3863. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Ohga, N.; Akiyama, K.; Maishi, N.; Hida, Y. Heterogeneity of Tumor Endothelial Cells. Cancer Sci. 2013, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers 2019, 11, 1511. [Google Scholar] [CrossRef] [Green Version]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of Tumor Endothelial Cells: Comparison between Tumor Endothelial Cells Isolated from High- and Low-Metastatic Tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; He, S. Multi-Faceted Role of Cancer-Associated Adipocytes in the Tumor Microenvironment. Mol. Med. Rep. 2021, 24, 866. [Google Scholar] [CrossRef]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K.; Senthebane, D.A.; Thomford, N.E.; Rowe, A.; Dandara, C.; Parker, M.I. Not Everyone Fits the Mold: Intratumor and Intertumor Heterogeneity and Innovative Cancer Drug Design and Development. Omics J. Integr. Biol. 2018, 22, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Zhao, W.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; Zhang, T.; Zhu, B.; Wang, M.; Jones, K.; Hicks, B.; et al. Genetic and Epigenetic Intratumor Heterogeneity Impacts Prognosis of Lung Adenocarcinoma. Nat. Commun. 2020, 11, 2459. [Google Scholar] [CrossRef]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer Stem Cell Hypothesis for Therapeutic Innovation in Clinical Oncology? Taking the Root Out, Not Chopping the Leaf. Omics J. Integr. Biol. 2016, 20, 681–691. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity—A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-Tumor Heterogeneity from a Cancer Stem Cell Perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- van Niekerk, G.; Davids, L.M.; Hattingh, S.M.; Engelbrecht, A.-M. Cancer Stem Cells: A Product of Clonal Evolution? Int. J. Cancer 2017, 140, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer Evolution: Darwin and Beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Hu, Z.; Yang, Z.; Yang, F.; Li, Y.; Lin, P.; Chen, K.; Dong, L.; Cao, L.; Tao, Y.; et al. Extremely High Genetic Diversity in a Single Tumor Points to Prevalence of Non-Darwinian Cell Evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Swanton, C. Cancer Evolution: The Final Frontier of Precision Medicine? Ann. Oncol. 2014, 25, 549–551. [Google Scholar] [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor Evolution: Linear, Branching, Neutral or Punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Janku, F. Tumor Heterogeneity in the Clinic: Is It a Real Problem? Ther. Adv. Med. Oncol. 2014, 6, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic Architecture and Evolution of Clear Cell Renal Cell Carcinomas Defined by Multiregion Sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor Cells Can Follow Distinct Evolutionary Paths to Become Resistant to Epidermal Growth Factor Receptor Inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary Dynamics of Carcinogenesis and Why Targeted Therapy Does Not Work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.T.; Riaz, N.; Desrichard, A.; Şenbabaoğlu, Y.; Hakimi, A.A.; Makarov, V.; Reis-Filho, J.S.; Chan, T.A. Pan-Cancer Analysis of Intratumor Heterogeneity as a Prognostic Determinant of Survival. Oncotarget 2016, 7, 10051–10063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell, D.R.; Swanton, C. The Role of Tumour Heterogeneity and Clonal Cooperativity in Metastasis, Immune Evasion and Clinical Outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA Methylation Heterogeneity Defines a Disease Spectrum in Ewing Sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Jiang, Y.; Boi, M.; Tabbò, F.; Redmond, D.; Nie, K.; Ladetto, M.; Chiappella, A.; Cerchietti, L.; Shaknovich, R.; et al. Epigenomic Evolution in Diffuse Large B-Cell Lymphomas. Nat. Commun. 2015, 6, 6921. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct Evolution and Dynamics of Epigenetic and Genetic Heterogeneity in Acute Myeloid Leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Su, A.T.; Hajiabadi, H.; Tran, M.; Nguyen, Q. Applying Machine Learning for Integration of Multi-Modal Genomics Data and Imaging Data to Quantify Heterogeneity in Tumour Tissues. Methods Mol. Biol. Clifton NJ 2021, 2190, 209–228. [Google Scholar] [CrossRef]
- Laurinavicius, A.; Rasmusson, A.; Plancoulaine, B.; Shribak, M.; Levenson, R. Machine-Learning–Based Evaluation of Intratumoral Heterogeneity and Tumor-Stroma Interface for Clinical Guidance. Am. J. Pathol. 2021, 191, 1724–1731. [Google Scholar] [CrossRef]
- West, J.; You, L.; Zhang, J.; Gatenby, R.A.; Brown, J.S.; Newton, P.K.; Anderson, A.R.A. Towards Multidrug Adaptive Therapy. Cancer Res. 2020, 80, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating Evolutionary Dynamics into Treatment of Metastatic Castrate-Resistant Prostate Cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. Cancer Genomics: One Cell at a Time. Genome Biol. 2014, 15, 452. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, A.; Tarabichi, M.; Espiritu, S.M.G.; Deshwar, A.G.; David, M.; Wilson, N.M.; Dentro, S.; Wintersinger, J.A.; Liu, L.Y.; Ko, M.; et al. A Community Effort to Create Standards for Evaluating Tumor Subclonal Reconstruction. Nat. Biotechnol. 2020, 38, 97–107. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-Cell Profiling of Tumor Heterogeneity and the Microenvironment in Advanced Non-Small Cell Lung Cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Bao, X.; Shi, R.; Zhao, T.; Wang, Y.; Anastasov, N.; Rosemann, M.; Fang, W. Integrated Analysis of Single-Cell RNA-Seq and Bulk RNA-Seq Unravels Tumour Heterogeneity plus M2-like Tumour-Associated Macrophage Infiltration and Aggressiveness in TNBC. Cancer Immunol. Immunother. 2021, 70, 189–202. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.A.; Poirion, O.; Zhu, X.; Huang, S.; Wolfgruber, T.K.; Sebra, R.; Garmire, L.X. Using Single-Cell Multiple Omics Approaches to Resolve Tumor Heterogeneity. Clin. Transl. Med. 2017, 6, 46. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A Highly Reproducible and Sensitive Single-Cell RNA Sequencing Method, Reveals Non-Genetic Gene-Expression Heterogeneity. Genome Biol. 2013, 14, 3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef]
- Kim, T.H.; Zhou, X.; Chen, M. Demystifying “Drop-Outs” in Single-Cell UMI Data. Genome Biol. 2020, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-Cell Triple Omics Sequencing Reveals Genetic, Epigenetic, and Transcriptomic Heterogeneity in Hepatocellular Carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Goldman, S.L.; MacKay, M.; Afshinnekoo, E.; Melnick, A.M.; Wu, S.; Mason, C.E. The Impact of Heterogeneity on Single-Cell Sequencing. Front. Genet. 2019, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, K.; Stanislaw, S.; Spain, L.; Gallegos, L.L.; Rowan, A.; Schnidrig, D.; Rosenbaum, H.; Harle, A.; Au, L.; Hill, S.M.; et al. Representative Sequencing: Unbiased Sampling of Solid Tumor Tissue. Cell Rep. 2020, 31, 107550. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; et al. Intratumor Heterogeneity in Localized Lung Adenocarcinomas Delineated by Multiregion Sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, B.; Traulsen, A.; Sottoriva, A.; Dingli, D. Detecting Truly Clonal Alterations from Multi-Region Profiling of Tumours. Sci. Rep. 2017, 7, 44991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e12. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610.e11. [Google Scholar] [CrossRef] [Green Version]
- Masoodi, T.; Siraj, S.; Siraj, A.K.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Tulbah, A.; Al-Dayel, F.; AlHusaini, H.; AlOmar, O.; et al. Genetic Heterogeneity and Evolutionary History of High-Grade Ovarian Carcinoma and Matched Distant Metastases. Br. J. Cancer 2020, 122, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Harbst, K.; Lauss, M.; Cirenajwis, H.; Isaksson, K.; Rosengren, F.; Törngren, T.; Kvist, A.; Johansson, M.C.; Vallon-Christersson, J.; Baldetorp, B.; et al. Multiregion Whole-Exome Sequencing Uncovers the Genetic Evolution and Mutational Heterogeneity of Early-Stage Metastatic Melanoma. Cancer Res. 2016, 76, 4765–4774. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Niu, X.; Wang, Z.; Song, C.-L.; Huang, Z.; Chen, K.-N.; Duan, J.; Bai, H.; Xu, J.; Zhao, J.; et al. Multiregion Sequencing Reveals the Genetic Heterogeneity and Evolutionary History of Osteosarcoma and Matched Pulmonary Metastases. Cancer Res. 2019, 79, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Heide, T.; Maurer, A.; Eipel, M.; Knoll, K.; Geelvink, M.; Veeck, J.; Knuechel, R.; van Essen, J.; Stoehr, R.; Hartmann, A.; et al. Multiregion Human Bladder Cancer Sequencing Reveals Tumour Evolution, Bladder Cancer Phenotypes and Implications for Targeted Therapy. J. Pathol. 2019, 248, 230–242. [Google Scholar] [CrossRef]
- Yan, T.; Cui, H.; Zhou, Y.; Yang, B.; Kong, P.; Zhang, Y.; Liu, Y.; Wang, B.; Cheng, Y.; Li, J.; et al. Multi-Region Sequencing Unveils Novel Actionable Targets and Spatial Heterogeneity in Esophageal Squamous Cell Carcinoma. Nat. Commun. 2019, 10, 1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duregon, E.; Schneider, J.; DeMarzo, A.M.; Hooper, J.E. Rapid Research Autopsy Is a Stealthy but Growing Contributor to Cancer Research. Cancer 2019, 125, 2915–2919. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Z.; Bonneville, R.; Paruchuri, A.; Reeser, J.W.; Wing, M.R.; Samorodnitsky, E.; Krook, M.A.; Smith, A.M.; Dao, T.; Miya, J.; et al. Genomic and Transcriptomic Characterization of Relapsed SCLC Through Rapid Research Autopsy. JTO Clin. Res. Rep. 2021, 2, 100164. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qiao, Y.; Brady, S.W.; Factor, R.E.; Downs-Kelly, E.; Farrell, A.; McQuerry, J.A.; Shrestha, G.; Jenkins, D.; Johnson, W.E.; et al. Novel Temporal and Spatial Patterns of Metastatic Colonization from Breast Cancer Rapid-Autopsy Tumor Biopsies. Genome Med. 2021, 13, 170. [Google Scholar] [CrossRef]
- Krook, M.A.; Bonneville, R.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Martin, D.M.; Smith, A.M.; Dao, T.; Samorodnitsky, E.; Paruchuri, A.; et al. Tumor Heterogeneity and Acquired Drug Resistance in FGFR2-Fusion-Positive Cholangiocarcinoma through Rapid Research Autopsy. Mol. Case Stud. 2019, 5, a004002. [Google Scholar] [CrossRef] [Green Version]
- Makishima, K.; Suehara, Y.; Abe, Y.; Hattori, K.; Kusakabe, M.; Matsuoka, R.; Chiba, S.; Sakata-Yanagimoto, M. Intratumor Heterogeneity of Lymphoma Identified by Multiregion Sequencing of Autopsy Samples. Cancer Sci. 2022, 113, 362–364. [Google Scholar] [CrossRef]
- Haffner, M.C.; De Marzo, A.M.; Yegnasubramanian, S.; Epstein, J.I.; Carter, H.B. Diagnostic Challenges of Clonal Heterogeneity in Prostate Cancer. J. Clin. Oncol. 2015, 33, e38–e40. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-Invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How Liquid Biopsies Can Change Clinical Practice in Oncology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattox, A.K.; Bettegowda, C.; Zhou, S.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Applications of Liquid Biopsies for Cancer. Sci. Transl. Med. 2019, 11, eaay1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Gabriel, M.; Heymann, M.-F.; Heymann, D. Circulating Tumor Cells as a Tool for Assessing Tumor Heterogeneity. Theranostics 2019, 9, 4580–4594. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid Biopsy and Tumor Heterogeneity in Metastatic Solid Tumors: The Potentiality of Blood Samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]
- Brown, H.K.; Tellez-Gabriel, M.; Cartron, P.-F.; Vallette, F.M.; Heymann, M.-F.; Heymann, D. Characterization of Circulating Tumor Cells as a Reflection of the Tumor Heterogeneity: Myth or Reality? Drug Discov. Today 2019, 24, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Bidard, F.-C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical Validity of Circulating Tumour Cells in Patients with Metastatic Breast Cancer: A Pooled Analysis of Individual Patient Data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Trapp, E.; Janni, W.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; de Gregorio, A.; Alunni-Fabbroni, M.; Tzschaschel, M.; Polasik, A.; Koch, J.G.; et al. Presence of Circulating Tumor Cells in High-Risk Early Breast Cancer During Follow-Up and Prognosis. J. Natl. Cancer Inst. 2019, 111, 380–387. [Google Scholar] [CrossRef]
- Franken, A.; Honisch, E.; Reinhardt, F.; Meier-Stiegen, F.; Yang, L.; Jaschinski, S.; Esposito, I.; Alberter, B.; Polzer, B.; Huebner, H.; et al. Detection of ESR1 Mutations in Single Circulating Tumor Cells on Estrogen Deprivation Therapy but Not in Primary Tumors from Metastatic Luminal Breast Cancer Patients. J. Mol. Diagn. 2020, 22, 111–121. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Lee, R.J.; Stott, S.L.; Ting, D.T.; Wittner, B.S.; Ulman, M.; Smas, M.E.; Lord, J.B.; Brannigan, B.W.; Trautwein, J.; et al. Androgen Receptor Signaling in Circulating Tumor Cells as a Marker of Hormonally Responsive Prostate Cancer. Cancer Discov. 2012, 2, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Jendrisak, A.; Wang, Y.; Lee, J.; Greene, S.; Krupa, R.; Lu, D.; et al. Phenotypic Heterogeneity of Circulating Tumor Cells Informs Clinical Decisions between AR Signaling Inhibitors and Taxanes in Metastatic Prostate Cancer. Cancer Res. 2017, 77, 5687–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.-T.; Cui, X.; Chen, Q.; Li, Y.-F.; Cui, Y.-H.; Wang, Y.; Jiang, J. Circulating Tumor Cell Status Monitors the Treatment Responses in Breast Cancer Patients: A Meta-Analysis. Sci. Rep. 2017, 7, 43464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudineh, M.; Sargent, E.H.; Pantel, K.; Kelley, S.O. Profiling Circulating Tumour Cells and Other Biomarkers of Invasive Cancers. Nat. Biomed. Eng. 2018, 2, 72–84. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Challenges in Circulating Tumour Cell Research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Riethdorf, S.; O’Flaherty, L.; Hille, C.; Pantel, K. Clinical Applications of the CellSearch Platform in Cancer Patients. Adv. Drug Deliv. Rev. 2018, 125, 102–121. [Google Scholar] [CrossRef]
- Pantel, K.; Denève, E.; Nocca, D.; Coffy, A.; Vendrell, J.-P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabières, C. Circulating Epithelial Cells in Patients with Benign Colon Diseases. Clin. Chem. 2012, 58, 936–940. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, Structures, and Functions of Circulating DNA in Oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic CtDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.; Stein, S. Circulating Cell-Free Tumour DNA in the Management of Cancer. Int. J. Mol. Sci. 2015, 16, 14122–14142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, P. Enrichment and analysis of CtDNA. In Tumor Liquid Biopsies; Schaffner, F., Merlin, J.L., von Bubnoff, N., Eds.; Springer: Cham, Switzerland, 2020; Volume 215, pp. 181–211. [Google Scholar] [CrossRef]
- Underhill, H.R. Leveraging the Fragment Length of Circulating Tumour DNA to Improve Molecular Profiling of Solid Tumour Malignancies with Next-Generation Sequencing: A Pathway to Advanced Non-Invasive Diagnostics in Precision Oncology? Mol. Diagn. Ther. 2021, 25, 389–408. [Google Scholar] [CrossRef]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS Mutation Analysis in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: The AGEO RASANC Prospective Multicenter Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Wang, J.; Zhou, S.; Wang, C.L.; Ye, M.Z.; Wang, X.Y.; Song, Y.; Wang, Y.Q.; Zhang, L.T.; et al. Biological Background of the Genomic Variations of Cf-DNA in Healthy Individuals. Ann. Oncol. 2019, 30, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Danlos, F.-X.; Papo, M.; Micol, J.-B. Clonal haematopoiesis: A concise review. Rev. Med. Interne 2019, 40, 684–692. [Google Scholar] [CrossRef]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.-K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer Therapy Shapes the Fitness Landscape of Clonal Hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TME Cell Types | TME Heterogeneity | Consequences on Tumour Development | References |
|---|---|---|---|
| Cancer-associated fibroblasts (CAFs) | CAFs differ by their origins (cellular precursors and cellular locations) and their marker expression profiles. CAFs subgroups are differentially expressed depending on the cancer types. | CAF subsets display opposite functions in cancers: some favour cancer development through the promotion of angiogenesis, metastasis and drug resistance, while others exhibit tumour-suppressor properties by contributing to growth inhibition, immune surveillance of the tumour and drug sensitivity. | [52] |
| Immune cells (macrophages, dendritic cells, mast cells, natural killer cells, B and T lymphocytes) | Variable levels of immune infiltration are observed in tumours depending on cancer types and subtypes. The immune cell composition (innate/adaptive immune cells, immune cell types) also differs between tumours. | Numerous studies report the interest of tumour-infiltrating lymphocytes (TILs) as a major prognostic marker in diverse cancers. High density of CD8+ T cells in tumours is strongly correlated with good prognosis, while high regulatory T-cell (Tregs) infiltration was associated with early recurrence and poor outcomes. In the same way, high density of NK cells in tumours was shown to predict good patient survival. Tumour-associated macrophages and neutrophils promote tumour cell plasticity and cancer stem cell phenotype, notably though the secretion of specific cytokines. | [53,54,55] |
| Tumour endothelial cells (TECs) | TECs show differences in terms of origins, morphology, structure, functions and marker expression. TECs derived from highly metastatic tumours harbour more cytogenic abnormalities and proangiogenic properties than those from tumours with low metastasis. TECs from tumours with high metastatic potential display a stem cell-like phenotype with the remarkable capacity to form spheres. | The overexpression of adhesion molecules in TECs allows cancer cell extravasation and metastasis spreading. TECs can secrete angiocrine factors at various levels that contribute to cancer cell proliferation, migration, invasion and angiogenesis. TECs also contribute to the emergence of drug resistance by increasing the expression of ATP-binding cassette transporters or helping tumour cells to switch to resistant phenotypes. TECs also modulate cancer immune surveillance by secreting growth factors that inhibit immune cells homing and induce the apoptosis of activated CD8+ T cells. TECs expressing PD-L1 marker hamper T cell activation. | [56,57,58] |
| Extracellular matrix (ECM): collagens, proteoglycans, fibronectin, elastins, laminins, hyaluronans | Proportion of ECM in tumours, ECM composition, architecture and posttranslational modifications are highly variable from a tumour to another. | Increased amounts of collagens in ECM of pancreatic cancers is associated with poor prognosis and chemoresistance. The expression levels of certain collagen isoforms (notably increased Col I levels and decreased Col IV levels) are correlated with stage of cancers and poor prognosis. Enhanced laminin expression and its anarchic distribution as well as high hyaluronic acid levels are correlated with poor clinical outcomes. Abundant and rigid ECM in tumours can act as a barrier and protect tumour cells from therapeutic agents. The stiffness of ECM and its enrichment in hyaluronic acid and Col I isoform drive epithelial-to-mesenchymal transition and promote metastasis and drug resistance. | [59] |
| Cancer-associated adipocytes (CAAs) | Less is known about the heterogeneity in the adipocyte part of the ECM. CAAs are characterised by irregular morphologies with decreased lipid content and reduced differentiation marker expression compared to normal mature adipocytes. | Growing evidence highlight the role of CAAs in the development of certain tumour types. CAAs interact with cancers cells and induce the reprogramming of their energy metabolism, the development of chemoresistance and the secretion of adipokines that modify the behaviour of tumour cells. | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. https://doi.org/10.3390/cancers14061384
Gilson P, Merlin J-L, Harlé A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers. 2022; 14(6):1384. https://doi.org/10.3390/cancers14061384
Chicago/Turabian StyleGilson, Pauline, Jean-Louis Merlin, and Alexandre Harlé. 2022. "Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy" Cancers 14, no. 6: 1384. https://doi.org/10.3390/cancers14061384
APA StyleGilson, P., Merlin, J.-L., & Harlé, A. (2022). Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers, 14(6), 1384. https://doi.org/10.3390/cancers14061384