
The Route of the Malignant Plasma Cell in Its Survival Niche: Exploring “Multiple Myelomas”

 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
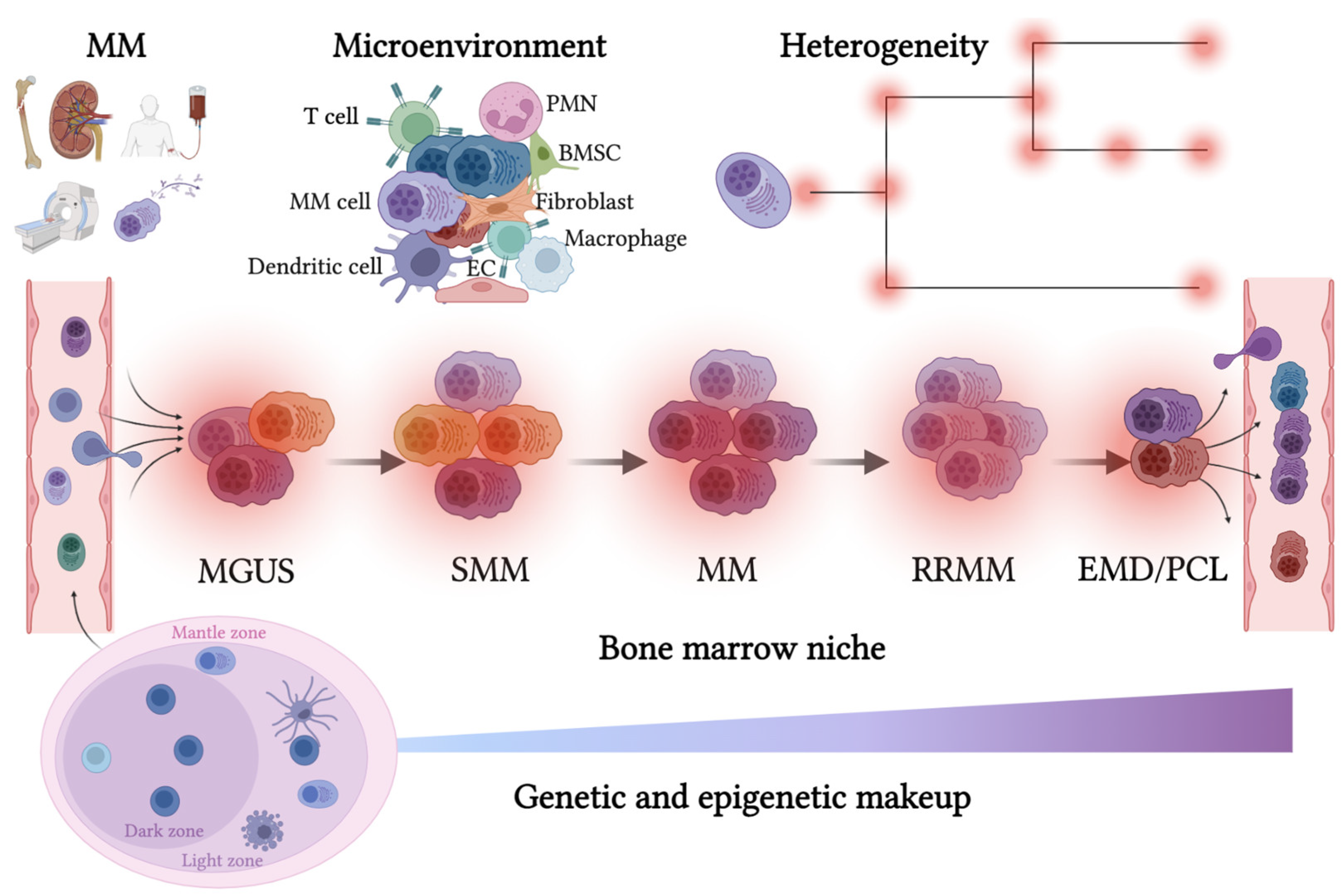
2. Cell of Origin, Homing and Soluble Cytokines in Multiple Myeloma
3. Myeloma Stemness, Genomic Evolution and Clinical Implications
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Bringhen, S.; Mateos, M.V.; Zweegman, S.; Larocca, A.; Falcone, A.P.; Oriol, A.; Rossi, D.; Cavalli, M.; Wijermans, P.; Ria, R.; et al. Age and Organ Damage Correlate with Poor Survival in Myeloma Patients: Meta-Analysis of 1435 Individual Patient Data from 4 Randomized Trials. Haematologica 2013, 98, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Melaccio, A.; Sportelli, A.; Solimando, A.G.; Dammacco, F.; Ria, R. Subcutaneous Immunoglobulins in Patients with Multiple Myeloma and Secondary Hypogammaglobulinemia: A Randomized Trial. Clin. Immunol. 2018, 191, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Nucci, M.; Anaissie, E. Infections in Patients with Multiple Myeloma in the Era of High-Dose Therapy and Novel Agents. Clin. Infect. Dis. 2009, 49, 1211–1225. [Google Scholar] [CrossRef]
- Cicco, S.; Solimando, A.G.; Leone, P.; Battaglia, S.; Ria, R.; Vacca, A.; Racanelli, V. Suspected Pericardial Tuberculosis Revealed as an Amyloid Pericardial Mass. Case Rep. Hematol. 2018, 2018, 8606430. [Google Scholar] [CrossRef]
- Kyle, R.A. “Primary” Systemic Amyloidosis and Myeloma: Discussion of Relationship and Review of 81 Cases. Arch. Intern. Med. 1961, 107, 344. [Google Scholar] [CrossRef]
- Solimando, A.G.; Sportelli, A.; Troiano, T.; Demarinis, L.; Di Serio, F.; Ostuni, A.; Dammacco, F.; Vacca, A.; Ria, R. A Multiple Myeloma That Progressed as Type I Cryoglobulinemia with Skin Ulcers and Foot Necrosis: A Case Report. Medicine 2018, 97, e12355. [Google Scholar] [CrossRef]
- Payet, J.; Livartowski, J.; Kavian, N.; Chandesris, O.; Dupin, N.; Wallet, N.; Karras, A.; Salliot, C.; Suarez, F.; Avet-Loiseau, H.; et al. Type I Cryoglobulinemia in Multiple Myeloma, a Rare Entity: Analysis of Clinical and Biological Characteristics of Seven Cases and Review of the Literature. Leuk. Lymphoma 2013, 54, 767–777. [Google Scholar] [CrossRef]
- Child, J.A.; Morgan, G.J.; Davies, F.E.; Owen, R.G.; Bell, S.E.; Hawkins, K.; Brown, J.; Drayson, M.T.; Selby, P.J. Medical Research Council Adult Leukaemia Working Party High-Dose Chemotherapy with Hematopoietic Stem-Cell Rescue for Multiple Myeloma. N. Engl. J. Med. 2003, 348, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Reale, A.; Solimando, A.G.; Mangialardi, G.; Moschetta, M.; Gelao, L.; Iodice, G.; Vacca, A. Induction Therapy and Stem Cell Mobilization in Patients with Newly Diagnosed Multiple Myeloma. Stem Cells Int. 2012, 2012, 607260. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved Survival in Multiple Myeloma and the Impact of Novel Therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma Bone Disease: From Biology Findings to Treatment Approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Vacca, A.; Ribatti, D. A Comprehensive Biological and Clinical Perspective Can Drive a Patient-Tailored Approach to Multiple Myeloma: Bridging the Gaps between the Plasma Cell and the Neoplastic Niche. J. Oncol. 2020, 2020, 6820241. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts Control Reactivation of Dormant Myeloma Cells by Remodelling the Endosteal Niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A Niche-Dependent Myeloid Transcriptome Signature Defines Dormant Myeloma Cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel Targeting of Phospho-CMET Overcomes Drug Resistance and Induces Antitumor Activity in Multiple Myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/CMET Autocrine Loop is Operative in Multiple Myeloma Bone Marrow Endothelial Cells and May Represent a Novel Therapeutic Target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Noll, J.E.; Williams, S.A.; Purton, L.E.; Zannettino, A.C.W. Tug of War in the Haematopoietic Stem Cell Niche: Do Myeloma Plasma Cells Compete for the HSC Niche? Blood Cancer J. 2012, 2, e91. [Google Scholar] [CrossRef]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone Marrow Endothelial Cells Sustain a Tumor-Specific CD8+ T Cell Subset with Suppressive Function in Myeloma Patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef]
- Hose, D.; Rème, T.; Hielscher, T.; Moreaux, J.; Messner, T.; Seckinger, A.; Benner, A.; Shaughnessy, J.D.; Barlogie, B.; Zhou, Y.; et al. Proliferation is a Central Independent Prognostic Factor and Target for Personalized and Risk-Adapted Treatment in Multiple Myeloma. Haematologica 2011, 96, 87–95. [Google Scholar] [CrossRef]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-Cell RNA Sequencing Reveals Compromised Immune Microenvironment in Precursor Stages of Multiple Myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Yoneda, T.; Hiraga, T. Crosstalk between Cancer Cells and Bone Microenvironment in Bone Metastasis. Biochem. Biophys. Res. Commun. 2005, 328, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Faict, S.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Schots, R.; Vanderkerken, K.; Menu, E. Extracellular Vesicle Cross-Talk in the Bone Marrow Microenvironment: Implications in Multiple Myeloma. Oncotarget 2016, 7, 38927–38945. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment Drug Resistance in Multiple Myeloma: Emerging New Players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting Angiogenesis in Multiple Myeloma by the VEGF and HGF Blocking DARPin® Protein MP0250: A Preclinical Study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the Bone Marrow Microenvironment in Multiple Myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Desantis, V.; Di Marzo, L.; Craparotta, I.; Beltrame, L.; Marchini, S.; Annese, T.; Visino, F.; Arciuli, M.; Saltarella, I.; et al. Bone Marrow Fibroblasts Overexpress MiR-27b and MiR-214 in Step with Multiple Myeloma Progression, Dependent on Tumour Cell-Derived Exosomes. J. Pathol. 2019, 247, 241–253. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple Myeloma Exosomes Establish a Favourable Bone Marrow Microenvironment with Enhanced Angiogenesis and Immunosuppression: MM Exosomes Enhance Angiogenesis and Immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef]
- Drexler, H.G.; Matsuo, Y. Malignant Hematopoietic Cell Lines: In Vitro Models for the Study of Multiple Myeloma and Plasma Cell Leukemia. Leuk. Res. 2000, 24, 681–703. [Google Scholar] [CrossRef]
- Rossi, M.; Botta, C.; Arbitrio, M.; Grembiale, R.D.; Tagliaferri, P.; Tassone, P. Mouse Models of Multiple Myeloma: Technologic Platforms and Perspectives. Oncotarget 2018, 9, 20119–20133. [Google Scholar] [CrossRef]
- Beedie, S.L.; Rore, H.M.; Barnett, S.; Chau, C.H.; Luo, W.; Greig, N.H.; Figg, W.D.; Vargesson, N. In Vivo Screening and Discovery of Novel Candidate Thalidomide Analogs in the Zebrafish Embryo and Chicken Embryo Model Systems. Oncotarget 2016, 7, 33237–33245. [Google Scholar] [CrossRef]
- Jridi, I.; Catacchio, I.; Majdoub, H.; Shahbazzadeh, D.; El Ayeb, M.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Vacca, A.; Borchani, L. The Small Subunit of Hemilipin2, a New Heterodimeric Phospholipase A2 from Hemiscorpius Lepturus Scorpion Venom, Mediates the Antiangiogenic Effect of the Whole Protein. Toxicon 2017, 126, 38–46. [Google Scholar] [CrossRef]
- Ferrarini, M.; Mazzoleni, G.; Steimberg, N.; Belloni, D.; Ferrero, E. Innovative Models to Assess Multiple Myeloma Biology and the Impact of Drugs. In Multiple Myeloma—A Quick Reflection on the Fast Progress; Hajek, R., Ed.; InTech: London, UK, 2013; ISBN 978-953-51-1083-5. [Google Scholar]
- Loda, S.; Krebs, J.; Danhof, S.; Schreder, M.; Solimando, A.G.; Strifler, S.; Rasche, L.; Kortüm, M.; Kerscher, A.; Knop, S.; et al. Exploration of Artificial Intelligence Use with ARIES in Multiple Myeloma Research. J. Clin. Med. 2019, 8, 999. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Wei, C.; Hresko, R.C.; Bajpai, R.; Heitmeier, M.; Matulis, S.M.; Nooka, A.K.; Rosen, S.T.; Hruz, P.W.; Schiltz, G.E.; et al. In Silico Modeling-Based Identification of Glucose Transporter 4 (GLUT4)-Selective Inhibitors for Cancer Therapy. J. Biol. Chem. 2015, 290, 14441–14453. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Storti, P.; Bolzoni, M.; Palma, B.D.; Bonomini, S. Angiogenesis and Multiple Myeloma. Cancer Microenviron 2011, 4, 325–337. [Google Scholar] [CrossRef]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting Vasculogenesis to Prevent Progression in Multiple Myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of MTOR Complex 2 Restrains Tumor Angiogenesis in Multiple Myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and Immunotherapy in Multiple Myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T Cells Eliminate Myeloma and Confer Selective Fratricide of SLAMF7+ Normal Lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Mörsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for Cell Adhesion-Mediated Drug Resistance of Multiple Myeloma Cells in Vivo. Int. J. Biol. Markers 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of Clonogenic Multiple Myeloma Cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef]
- Steinbrunn, T.; Siegmund, D.; Andrulis, M.; Grella, E.; Kortüm, M.; Einsele, H.; Wajant, H.; Bargou, R.C.; Stühmer, T. Integrin-Linked Kinase is Dispensable for Multiple Myeloma Cell Survival. Leuk. Res. 2012, 36, 1165–1171. [Google Scholar] [CrossRef]
- Boise, L.H.; Kaufman, J.L.; Bahlis, N.J.; Lonial, S.; Lee, K.P. The Tao of Myeloma. Blood 2014, 124, 1873–1879. [Google Scholar] [CrossRef]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Schatz, D.G.; Oettinger, M.A.; Baltimore, D. The V(D)J Recombination Activating Gene, RAG-1. Cell 1989, 59, 1035–1048. [Google Scholar] [CrossRef]
- Oettinger, M.; Schatz, D.; Gorka, C.; Baltimore, D. RAG-1 and RAG-2, Adjacent Genes That Synergistically Activate V(D)J Recombination. Science 1990, 248, 1517–1523. [Google Scholar] [CrossRef]
- MacLennan, I.C.M. Germinal Centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Armitage, R.J.; Fanslow, W.C.; Strockbine, L.; Sato, T.A.; Clifford, K.N.; Macduff, B.M.; Anderson, D.M.; Gimpel, S.D.; Davis-Smith, T.; Maliszewski, C.R.; et al. Molecular and Biological Characterization of a Murine Ligand for CD40. Nature 1992, 357, 80–82. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; World Health Organization classification of tumours; International Agency for Research on Cancer: Lyon, France, 2017; ISBN 978-92-832-4494-3. [Google Scholar]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2018, 9, 788. [Google Scholar] [CrossRef]
- Ria, R.; Solimando, A.; Melaccio, A.; Sportelli, A.; Vacca, A. Angiogenesis and Antiangiogenesis in Multiple Myeloma. In Update on Multiple Myeloma; Ahmed Al-Anazi, K., Ed.; IntechOpen: London, UK, 2019; ISBN 978-1-78985-217-2. [Google Scholar]
- Gadó, K.; Domján, G.; Hegyesi, H.; Falus, A. Role of INTERLEUKIN-6 in the Pathogenesis of Multiple Myeloma. Cell Biol. Int. 2000, 24, 195–209. [Google Scholar] [CrossRef]
- Chng, W.J.; Kumar, S.; Vanwier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.-H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular Dissection of Hyperdiploid Multiple Myeloma by Gene Expression Profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef]
- Hallek, M.; Bergsagel, P.L.; Anderson, K.C. Multiple Myeloma: Increasing Evidence for a Multistep Transformation Process. Blood 1998, 91, 3–21. [Google Scholar] [CrossRef]
- Klein, B.; Zhang, X.G.; Lu, Z.Y.; Bataille, R. Interleukin-6 in Human Multiple Myeloma. Blood 1995, 85, 863–872. [Google Scholar] [CrossRef]
- Bakkus, M.H.; Heirman, C.; Van Riet, I.; Van Camp, B.; Thielemans, K. Evidence That Multiple Myeloma Ig Heavy Chain VDJ Genes Contain Somatic Mutations but Show No Intraclonal Variation. Blood 1992, 80, 2326–2335. [Google Scholar] [CrossRef]
- Urbańska-Ryś, H.; Wiersbowska, A.; Stepień, H.; Robak, T. Relationship between Circulating Interleukin-10 (IL-10) with Interleukin-6 (IL-6) Type Cytokines (IL-6, Interleukin-11 (IL-11), Oncostatin M (OSM)) and Soluble Interleukin-6 (IL-6) Receptor (SIL-6R) in Patients with Multiple Myeloma. Eur. Cytokine Netw. 2000, 11, 443–451. [Google Scholar]
- Zhang, X.G.; Gu, J.J.; Lu, Z.Y.; Yasukawa, K.; Yancopoulos, G.D.; Turner, K.; Shoyab, M.; Taga, T.; Kishimoto, T.; Bataille, R. Ciliary Neurotropic Factor, Interleukin 11, Leukemia Inhibitory Factor, and Oncostatin M Are Growth Factors for Human Myeloma Cell Lines Using the Interleukin 6 Signal Transducer Gp130. J. Exp. Med. 1994, 179, 1337–1342. [Google Scholar] [CrossRef]
- Burger, R.; Günther, A.; Klausz, K.; Staudinger, M.; Peipp, M.; Penas, E.M.M.; Rose-John, S.; Wijdenes, J.; Gramatzki, M. Due to Interleukin-6 Type Cytokine Redundancy Only Glycoprotein 130 Receptor Blockade Efficiently Inhibits Myeloma Growth. Haematologica 2017, 102, 381–390. [Google Scholar] [CrossRef]
- Chatterjee, M.; Hönemann, D.; Lentzsch, S.; Bommert, K.; Sers, C.; Herrmann, P.; Mathas, S.; Dörken, B.; Bargou, R.C. In the Presence of Bone Marrow Stromal Cells Human Multiple Myeloma Cells Become Independent of the IL-6/Gp130/STAT3 Pathway. Blood 2002, 100, 3311–3318. [Google Scholar] [CrossRef]
- Cao, Y.; Luetkens, T.; Kobold, S.; Hildebrandt, Y.; Gordic, M.; Lajmi, N.; Meyer, S.; Bartels, K.; Zander, A.R.; Bokemeyer, C.; et al. The Cytokine/Chemokine Pattern in the Bone Marrow Environment of Multiple Myeloma Patients. Exp. Hematol. 2010, 38, 860–867. [Google Scholar] [CrossRef]
- Jiang, G.-M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The Relationship between Autophagy and the Immune System and its Applications for Tumor Immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef]
- Hideshima, T. Novel Therapies Targeting the Myeloma Cell and Its Bone Marrow Microenvironment. Semin. Oncol. 2001, 28, 607–612. [Google Scholar] [CrossRef]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting Autophagy in Multiple Myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone Marrow Niches in Haematological Malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- de Jong, M.M.E.; Kellermayer, Z.; Papazian, N.; Tahri, S.; Hofste op Bruinink, D.; Hoogenboezem, R.; Sanders, M.A.; van de Woestijne, P.C.; Bos, P.K.; Khandanpour, C.; et al. The Multiple Myeloma Microenvironment Is Defined by an Inflammatory Stromal Cell Landscape. Nat. Immunol. 2021, 22, 769–780. [Google Scholar] [CrossRef]
- Desantis, V.; Frassanito, M.A.; Tamma, R.; Saltarella, I.; Di Marzo, L.; Lamanuzzi, A.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; et al. Rhu-Epo down-Regulates pro-Tumorigenic Activity of Cancer-Associated Fibroblasts in Multiple Myeloma. Ann. Hematol. 2018, 97, 1251–1258. [Google Scholar] [CrossRef]
- Nefedova, Y.; Landowski, T.H.; Dalton, W.S. Bone Marrow Stromal-Derived Soluble Factors and Direct Cell Contact Contribute to de Novo Drug Resistance of Myeloma Cells by Distinct Mechanisms. Leukemia 2003, 17, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Giannico, D.; Leone, P.; Solimando, A.G.; Maiorano, E.; Caporusso, C.; Duda, L.; Tamma, R.; Mallamaci, R.; Susca, N.; et al. HB-EGF-EGFR Signaling in Bone Marrow Endothelial Cells Mediates Angiogenesis Associated with Multiple Myeloma. Cancers 2020, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Kong, Y.; Yang, G.; Gao, L.; Shi, J. Multiple Myeloma Cancer Stem Cells. Oncotarget 2016, 7, 35466–35477. [Google Scholar] [CrossRef] [PubMed]
- Mahtouk, K.; Jourdan, M.; De Vos, J.; Hertogh, C.; Fiol, G.; Jourdan, E.; Rossi, J.-F.; Klein, B. An Inhibitor of the EGF Receptor Family Blocks Myeloma Cell Growth Factor Activity of HB-EGF and Potentiates Dexamethasone or Anti-IL-6 Antibody-Induced Apoptosis. Blood 2004, 103, 1829–1837. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic Cells Accumulate in the Bone Marrow of Myeloma Patients Where They Protect Tumor Plasma Cells from CD8+ T-Cell Killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The Dormant Cancer Cell Life Cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The Immune System and Cancer Evasion Strategies: Therapeutic Concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the Initiation of Multiple Myeloma. Nat. Commun. 2020, 11, 1917. [Google Scholar] [CrossRef]
- Lathia, J.D.; Li, M.; Sinyuk, M.; Alvarado, A.G.; Flavahan, W.A.; Stoltz, K.; Rosager, A.M.; Hale, J.; Hitomi, M.; Gallagher, J.; et al. High-Throughput Flow Cytometry Screening Reveals a Role for Junctional Adhesion Molecule a as a Cancer Stem Cell Maintenance Factor. Cell Rep. 2014, 6, 117–129. [Google Scholar] [CrossRef]
- van Driel, M.; Günthert, U.; Stauder, R.; Joling, P.; Lokhorst, H.M.; Bloem, A.C. CD44 Isoforms Distinguish between Bone Marrow Plasma Cells from Normal Individuals and Patients with Multiple Myeloma at Different Stages of Disease. Leukemia 1998, 12, 1821–1828. [Google Scholar] [CrossRef]
- Wang, L.; Zuo, X.; Xie, K.; Wei, D. The Role of CD44 and Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Brandl, A.; Mattenheimer, K.; Graf, C.; Ritz, M.; Ruckdeschel, A.; Stühmer, T.; Mokhtari, Z.; Rudelius, M.; Dotterweich, J.; et al. JAM-A as a Prognostic Factor and New Therapeutic Target in Multiple Myeloma. Leukemia 2018, 32, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Da Vià, M.C.; Borrelli, P.; Leone, P.; Di Lernia, G.; Tabares Gaviria, P.; Brandl, A.; Pedone, G.L.; Rauert-Wunderlich, H.; Lapa, C.; et al. Central Function for JAM-a in Multiple Myeloma Patients with Extramedullary Disease. Blood 2018, 132, 4455. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Croci, G.; Borrelli, P.; Tabares Gaviria, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Adhesion-Mediated Multiple Myeloma (MM) Disease Progression: Junctional Adhesion Molecule a Enhances Angiogenesis and Multiple Myeloma Dissemination and Predicts Poor Survival. Blood 2019, 134, 855. [Google Scholar] [CrossRef]
- Canella, A.; Cordero Nieves, H.; Sborov, D.W.; Cascione, L.; Radomska, H.S.; Smith, E.; Stiff, A.; Consiglio, J.; Caserta, E.; Rizzotto, L.; et al. HDAC Inhibitor AR-42 Decreases CD44 Expression and Sensitizes Myeloma Cells to Lenalidomide. Oncotarget 2015, 6, 31134–31150. [Google Scholar] [CrossRef]
- Saltarella, I.; Lamanuzzi, A.; Reale, A.; Vacca, A.; Ria, R. Identify Multiple Myeloma Stem Cells: Utopia? World J. Stem Cells 2015, 7, 84–95. [Google Scholar] [CrossRef]
- Brioli, A.; Giles, H.; Pawlyn, C.; Campbell, J.P.; Kaiser, M.F.; Melchor, L.; Jackson, G.H.; Gregory, W.M.; Owen, R.G.; Child, J.A.; et al. Serum Free Immunoglobulin Light Chain Evaluation as a Marker of Impact from Intraclonal Heterogeneity on Myeloma Outcome. Blood 2014, 123, 3414–3419. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome Translocations in Multiple Myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-Cell Genetic Analysis Reveals the Composition of Initiating Clones and Phylogenetic Patterns of Branching and Parallel Evolution in Myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef]
- Solimando, A.G.; Ribatti, D.; Vacca, A.; Einsele, H. Targeting B-Cell Non Hodgkin Lymphoma: New and Old Tricks. Leuk. Res. 2016, 42, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Ocqueteau, M.; San Miguel, J.F.; González, M.; Almeida, J.; Orfao, A. Do Myelomatous Plasma Cells Really Express Surface Immunoglobulins? Haematologica 1996, 81, 460–463. [Google Scholar] [PubMed]
- Johnsen, H.E.; Bøgsted, M.; Schmitz, A.; Bødker, J.S.; El-Galaly, T.C.; Johansen, P.; Valent, P.; Zojer, N.; Van Valckenborgh, E.; Vanderkerken, K.; et al. The Myeloma Stem Cell Concept, Revisited: From Phenomenology to Operational Terms. Haematologica 2016, 101, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Kopen, G.C.; Prockop, D.J.; Phinney, D.G. Marrow Stromal Cells Migrate throughout Forebrain and Cerebellum, and They Differentiate into Astrocytes after Injection into Neonatal Mouse Brains. Proc. Natl. Acad. Sci. USA 1999, 96, 10711–10716. [Google Scholar] [CrossRef]
- Wallace, S.R.; Oken, M.M.; Lunetta, K.L.; Panoskaltsis-Mortari, A.; Masellis, A.M. Abnormalities of Bone Marrow Mesenchymal Cells in Multiple Myeloma Patients. Cancer 2001, 91, 1219–1230. [Google Scholar] [CrossRef]
- Ghosh, N.; Matsui, W. Cancer Stem Cells in Multiple Myeloma. Cancer Lett. 2009, 277, 1–7. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial Genome Sequencing and Analysis of Multiple Myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Morgan, G.J.; Child, J.A.; Gregory, W.M.; Szubert, A.J.; Cocks, K.; Bell, S.E.; Navarro-Coy, N.; Drayson, M.T.; Owen, R.G.; Feyler, S.; et al. Effects of Zoledronic Acid versus Clodronic Acid on Skeletal Morbidity in Patients with Newly Diagnosed Multiple Myeloma (MRC Myeloma IX): Secondary Outcomes from a Randomised Controlled Trial. Lancet Oncol. 2011, 12, 743–752. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Steinbrunn, T.; Stühmer, T.; Gattenlöhner, S.; Rosenwald, A.; Mottok, A.; Unzicker, C.; Einsele, H.; Chatterjee, M.; Bargou, R.C. Mutated RAS and Constitutively Activated Akt Delineate Distinct Oncogenic Pathways, Which Independently Contribute to Multiple Myeloma Cell Survival. Blood 2011, 117, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. Int. J. Mol. Sci. 2020, 21, 3084. [Google Scholar] [CrossRef]
- Nefedova, Y.; Cheng, P.; Alsina, M.; Dalton, W.S.; Gabrilovich, D.I. Involvement of Notch-1 Signaling in Bone Marrow Stroma-Mediated de Novo Drug Resistance of Myeloma and Other Malignant Lymphoid Cell Lines. Blood 2004, 103, 3503–3510. [Google Scholar] [CrossRef]
- Ely, S.A.; Knowles, D.M. Expression of CD56/Neural Cell Adhesion Molecule Correlates with the Presence of Lytic Bone Lesions in Multiple Myeloma and Distinguishes Myeloma from Monoclonal Gammopathy of Undetermined Significance and Lymphomas with Plasmacytoid Differentiation. Am. J. Pathol. 2002, 160, 1293–1299. [Google Scholar] [CrossRef]
- Kaiser, U.; Auerbach, B.; Oldenburg, M. The Neural Cell Adhesion Molecule NCAM in Multiple Myeloma. Leuk. Lymphoma 1996, 20, 389–395. [Google Scholar] [CrossRef]
- Da Vià, M.C.; Solimando, A.G.; Garitano-Trojaola, A.; Barrio, S.; Munawar, U.; Strifler, S.; Haertle, L.; Rhodes, N.; Teufel, E.; Vogt, C.; et al. CIC Mutation as a Molecular Mechanism of Acquired Resistance to Combined BRAF-MEK Inhibition in Extramedullary Multiple Myeloma with Central Nervous System Involvement. Oncologist 2019, 25, 112–118. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J. Targeting of Adhesion Molecules as a Therapeutic Strategy in Multiple Myeloma. Curr. Cancer Drug Targets 2012, 12, 776–796. [Google Scholar] [CrossRef]
- Seibold, M.; Stühmer, T.; Kremer, N.; Mottok, A.; Scholz, C.-J.; Schlosser, A.; Leich, E.; Holzgrabe, U.; Brünnert, D.; Barrio, S.; et al. RAL GTPases Mediate Multiple Myeloma Cell Survival and Are Activated Independently of Oncogenic RAS. Haematologica 2019, 105, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a Role for CD44 and Cell Adhesion in Mediating Resistance to Lenalidomide in Multiple Myeloma: Therapeutic Implications. Leukemia 2014, 28, 373–383. [Google Scholar] [CrossRef]
- Hood, J.D.; Frausto, R.; Kiosses, W.B.; Schwartz, M.A.; Cheresh, D.A. Differential Alphav Integrin-Mediated Ras-ERK Signaling during Two Pathways of Angiogenesis. J. Cell Biol. 2003, 162, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic Patterns of Progression in Smoldering Multiple Myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and Non-Canonical WNT Signaling in Cancer Stem Cells and Their Niches: Cellular Heterogeneity, Omics Reprogramming, Targeted Therapy and Tumor Plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Spaan, I.; Raymakers, R.A.; van de Stolpe, A.; Peperzak, V. Wnt Signaling in Multiple Myeloma: A Central Player in Disease with Therapeutic Potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef]
- Tai, D.; Wells, K.; Arcaroli, J.; Vanderbilt, C.; Aisner, D.L.; Messersmith, W.A.; Lieu, C.H. Targeting the WNT Signaling Pathway in Cancer Therapeutics. Oncologist 2015, 20, 1189–1198. [Google Scholar] [CrossRef]
- Ge, X.; Wang, X. Role of Wnt Canonical Pathway in Hematological Malignancies. J. Hematol. Oncol. 2010, 3, 33. [Google Scholar] [CrossRef]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt Pathways in Cancer Stem Cells: Clinical Update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT Signalling Pathways as Therapeutic Targets in Cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of Venetoclax as Targeted Therapy for Relapsed/Refractory t(11;14) Multiple Myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising Efficacy and Acceptable Safety of Venetoclax plus Bortezomib and Dexamethasone in Relapsed/Refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef]
- Bohl, S.R.; Schmalbrock, L.K.; Bauhuf, I.; Meyer, T.; Dolnik, A.; Szyska, M.; Blätte, T.J.; Knödler, S.; Röhner, L.; Miller, D.; et al. Comprehensive CRISPR-Cas9 Screens Identify Genetic Determinants of Drug Responsiveness in Multiple Myeloma. Blood Adv. 2021, 5, 2391–2402. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Gnoni, A.; Brunetti, O.; Longo, V.; Calabrese, A.; Argentiero, A.; Calbi, R.; Solimando Antonio, G.; Licchetta, A. Immune System and Bone Microenvironment: Rationale for Targeted Cancer Therapies. Oncotarget 2020, 11, 480–487. [Google Scholar] [CrossRef][Green Version]
- Desantis, V.; Solimando, A.G.; Saltarella, I.; Sacco, A.; Giustini, V.; Bento, M.; Lamanuzzi, A.; Melaccio, A.; Frassanito, M.A.; Paradiso, A.; et al. MicroRNAs as a Potential New Preventive Approach in the Transition from Asymptomatic to Symptomatic Multiple Myeloma Disease. Cancers 2021, 13, 3650. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the Vicious Cycle within the Multiple Myeloma Ecosystem: Blocking JAM-A on Bone Marrow Endothelial Cells Restores the Angiogenic Homeostasis and Suppresses Tumor Progression. Haematologica 2020, 106, 1943–1956. [Google Scholar] [CrossRef]
- Brandl, A.; Solimando, A.; Mokhtari, Z.; Tabares, P.; Medler, J.; Manz, H.; Da Vià, M.C.; Croci, G.A.; Kurzwart, M.; Thusek, S.; et al. Junctional Adhesion Molecule-C Expression Specifies a CD138low/Neg Multiple Myeloma Cell Population in Mice and Humans. Blood Adv. 2021, 6, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Multiple Myeloma: New Insights and Therapeutic Approaches. Hematology 2000, 2000, 147–165. [Google Scholar] [CrossRef]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-Angiogenesis and Immunotherapy: Novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Solimando, A.G.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Annese, T.; Tamma, R.; Ingravallo, G.; Maiorano, E.; Vacca, A.; Specchia, G.; Ribatti, D. New Insights into Diffuse Large B-Cell Lymphoma Pathobiology. Cancers 2020, 12, 1869. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V. The Multiple Myelomas—Current Concepts in Cytogenetic Classification and Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef]


{kind=link}
| Cell Type | Cell Marker |
|---|---|
| Premalignant plasma cell | CD38+; CD19−; CD27+/−; CD20−; CD56+/− |
| Malignant plasma cell | CD38+; CD19−; CD27+/−; CD20−; CD56+/− |
| Clonotypic centroblast/centrocytes | CD38+; CD20+; CXCR4+/− |
| Clonotypic memory B-cell | CD38+; CD20+; CD27+; mIgG/A |
| Clonotypic plasmablast | CD38+; CD20+; CD27+ |
| Human myeloma cell lines | CD38+; CD19−; CD27+/−; CD20-; CD56+/− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solimando, A.G.; Da Vià, M.C.; Bolli, N.; Steinbrunn, T. The Route of the Malignant Plasma Cell in Its Survival Niche: Exploring “Multiple Myelomas”. Cancers 2022, 14, 3271. https://doi.org/10.3390/cancers14133271
Solimando AG, Da Vià MC, Bolli N, Steinbrunn T. The Route of the Malignant Plasma Cell in Its Survival Niche: Exploring “Multiple Myelomas”. Cancers. 2022; 14(13):3271. https://doi.org/10.3390/cancers14133271
Chicago/Turabian StyleSolimando, Antonio Giovanni, Matteo Claudio Da Vià, Niccolò Bolli, and Torsten Steinbrunn. 2022. "The Route of the Malignant Plasma Cell in Its Survival Niche: Exploring “Multiple Myelomas”" Cancers 14, no. 13: 3271. https://doi.org/10.3390/cancers14133271
APA StyleSolimando, A. G., Da Vià, M. C., Bolli, N., & Steinbrunn, T. (2022). The Route of the Malignant Plasma Cell in Its Survival Niche: Exploring “Multiple Myelomas”. Cancers, 14(13), 3271. https://doi.org/10.3390/cancers14133271