
Knockdown of 15-bp Deletion-Type v-raf Murine Sarcoma Viral Oncogene Homolog B1 mRNA in Pancreatic Ductal Adenocarcinoma Cells Repressed Cell Growth In Vitro and Tumor Volume In Vivo

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis of siRNAs
2.2. Cell Culture
2.3. Construction of Plasmids for Complete-Matched (CM) and Seed-Matched (SM) Luciferase Reporter Assays
2.4. Measurements of RNAi and Off-Target Activities by Dual Luciferase Reporter Assays
2.5. Quantitative Reverse Transcription-Polymerase Chain Reaction
2.6. Western Blotting
2.7. Microarray
2.8. Xenograft Mouse Model
2.9. Serum Degradation Assay
3. Results
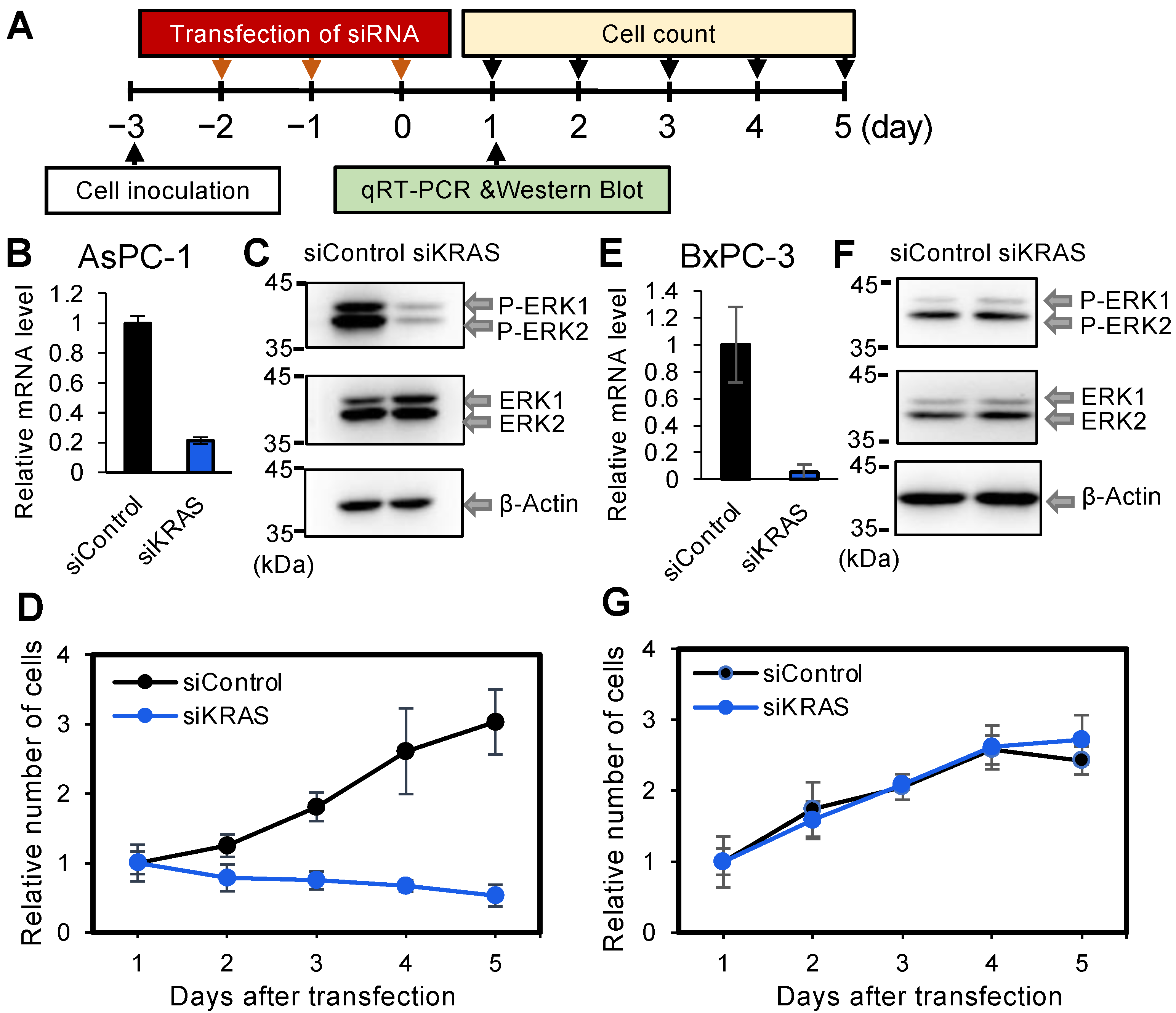
3.1. Effect of Kirsten Rat Sarcoma Viral Oncogene Homolog Knockdown in PDAC-Derived Cell Lines AsPC-1 and BxPC-3, with or without Mut Kirsten Rat Sarcoma Viral Oncogene Homolog
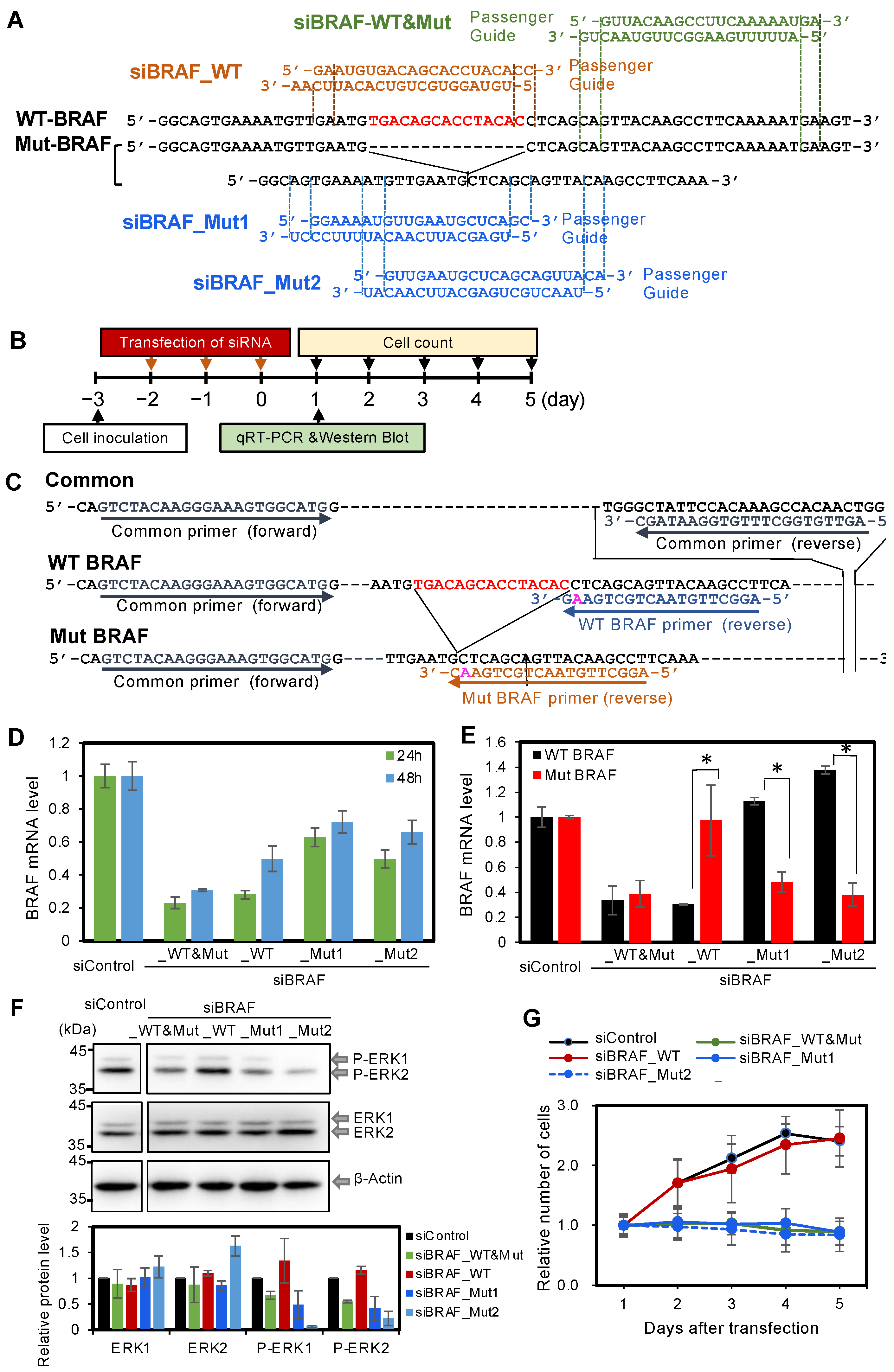
3.2. Effect of Knockdown of Mut BRAF in BxPC-3 Cells
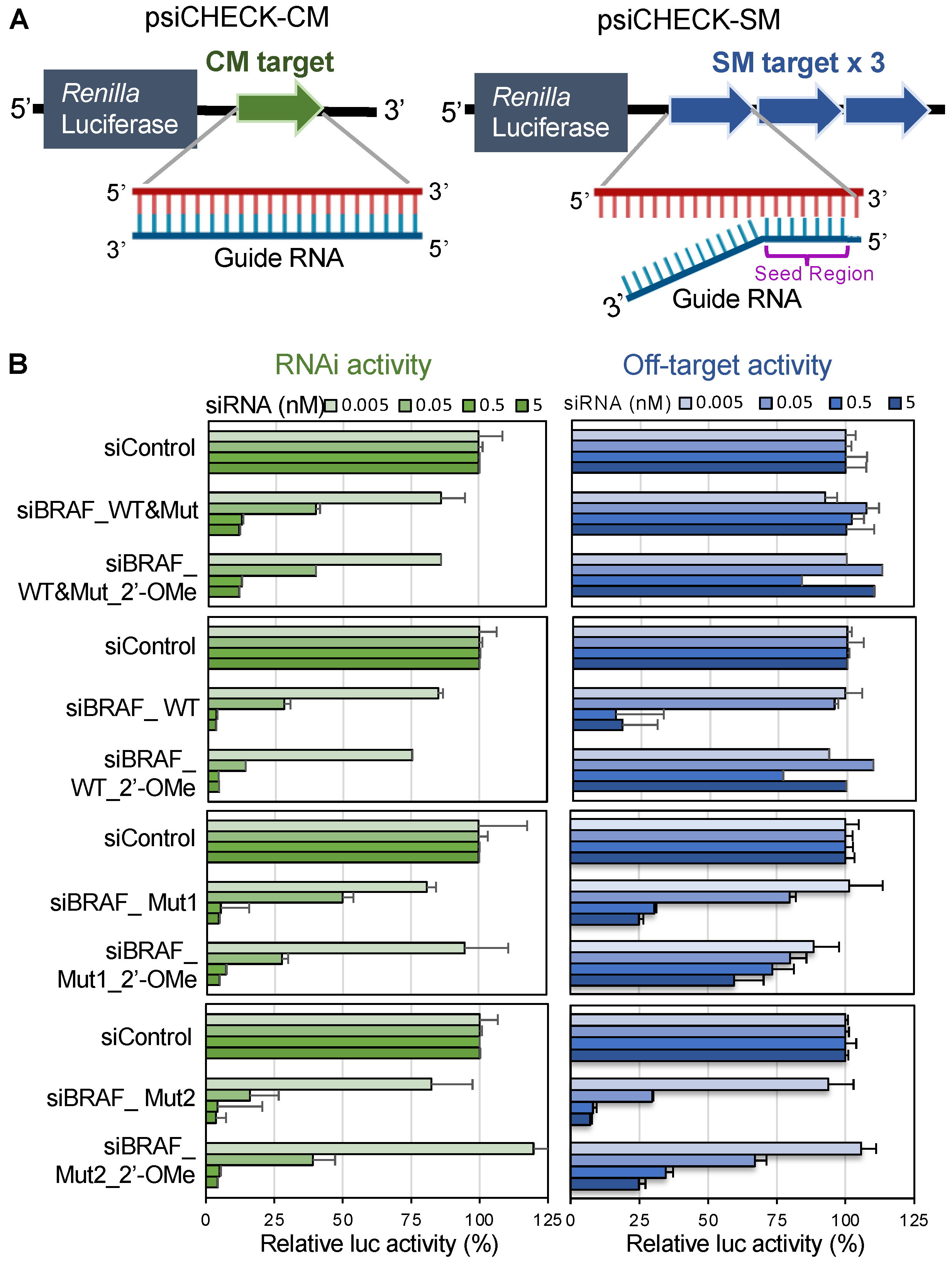
3.3. Effects of v-Raf Murine Sarcoma Viral Oncogene Homolog B1 siRNAs on RNAi and off-Target Activities
3.4. Effects of v-Raf Murine Sarcoma Viral Oncogene Homolog B1 siRNA with 2′-OMe Modification in the Seed Region on RNAi and off-Target Activities
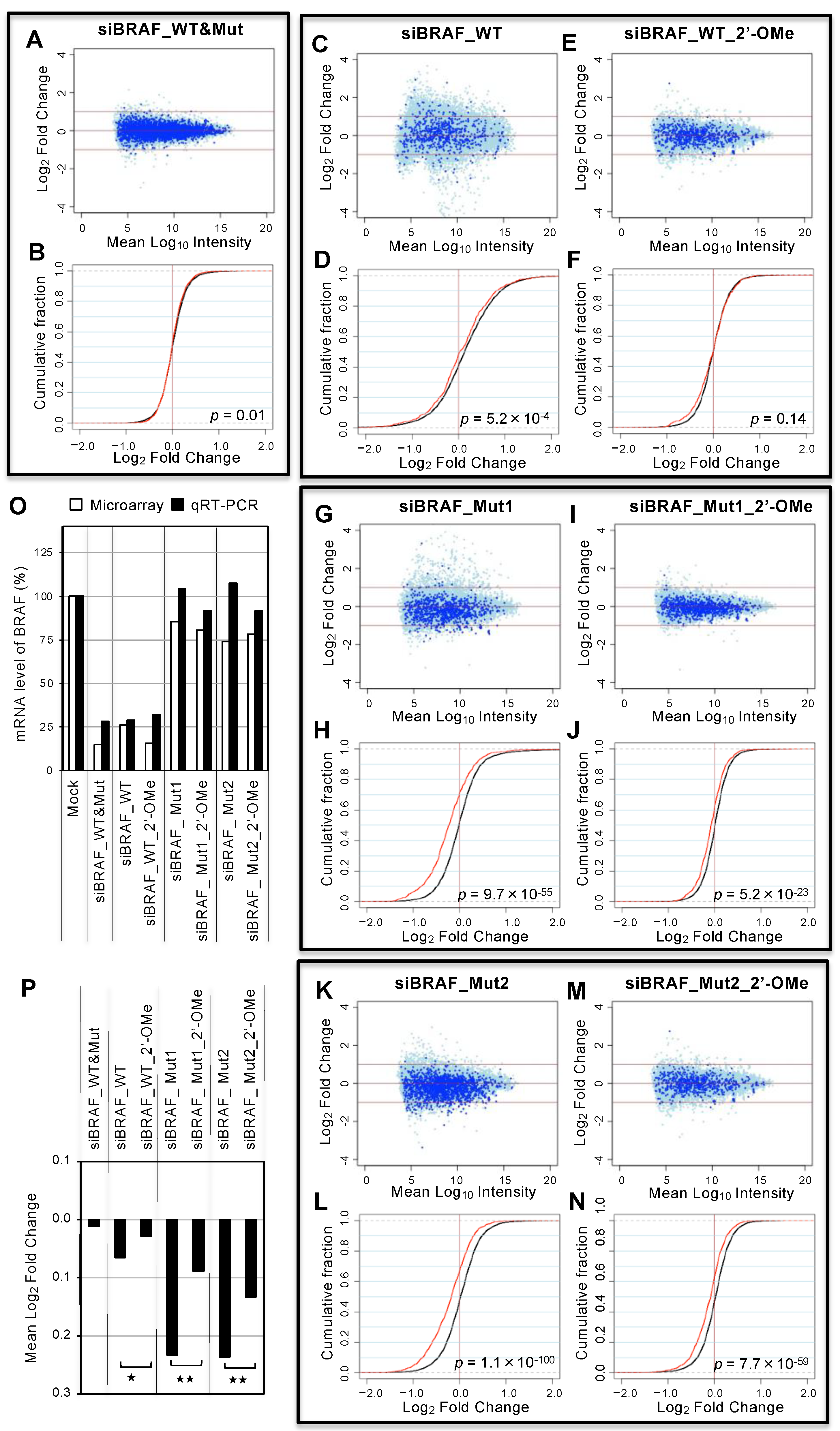
3.5. Genome-wide Analyses of siRNA Off-Target Effects by Microarray
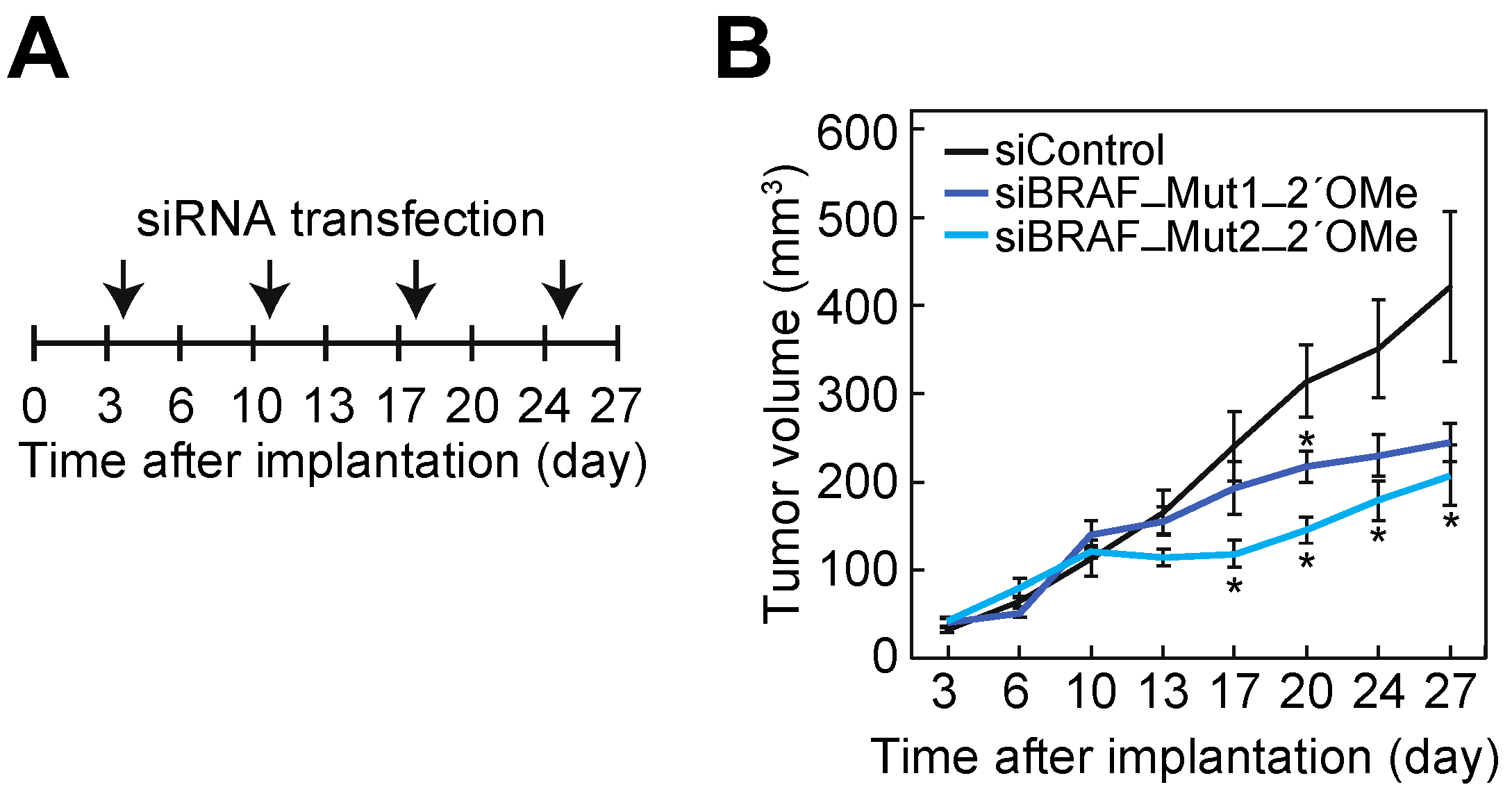
3.6. Suppression of Cell Growth of BxPC-3 Cells by siBRAF-Muts in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luchini, C.; Paolino, G.; Mattiolo, P.; Piredda, L.M.; Cavaliere, A.; Gaule, M.; Melisi, D.; Salvia, R.; Malleo, G.; Shin, J., II; et al. KRAS wild-type pancreatic ductal adenocarcinoma: Molecular pathology and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2020, 39, 227. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Bannaura, S.F.; Uddin, M.H.; Nagasaka, M.; Fazili, F.; AI-Hollak, M.N.; Philip, P.A.; El-Rayes, B.; Azmi, A.S. Targeting KRAS in pancreati cancer: New drugs on the horizon. Cancer Metastasis Rev. 2022, 40, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Tuveson, D.A. Modelling oncogenic Ras/Raf signalling in the mouse. Curr. Opin. Genet. Dev. 2009, 19, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Stolze, B.; Reinhart, S.; Bulllinger, L.; Fröhling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61 and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. In Cold Spring Harbor Perspectives in Medicine; Cold Spring Horbor Press: New York, NY, USA, 2018; p. a031435. [Google Scholar]
- Sevilla, A.; Morales, M.C.; Ezkurra, A.P.; Rasero, J.; Velasco, V.; Galan, G.C.; Sánchez-Diez, A.; Mujika, K.; Penas, C.; Smith, I.; et al. BRAF V600E mutational load as a prognosis biomarker in malignant melanoma. PLoS ONE 2020, 15, e0230136. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Yasothan, U.; Kirkpatrick, P. Vemurafenib. Nat. Rev. Drug Discuv. 2011, 10, 811–812. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Kirkwood, J.M. Targeted therapies: BREAKing a path for progress—Dabrafenib confirms class effect. Nat. Rev. Clin. Oncol. 2012, 9, 496–497. [Google Scholar] [CrossRef]
- Chen, S.-H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.S.; Whalen, M.W.; Özen, A.; Wongchenko, J.M.; Yin, J.; Yen, I.; Schaefer, G.; Mayfield, D.J.; Chmielecki, J.; Stephens, J.P.; et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 2016, 29, 477–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojnowski, L.; Zimmer, A.M.; Bech, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endotherial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, A.; Montgomery, K.M.; Kostas, A.S.; Driver, E.S.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Linsley, S.P. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-Stranded RNA Directs the ATP-Dependent Cleavage of mRNA at 21 to 23 Nucleotide Intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21-and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Ketting, R.F.; Fischer, S.E.J.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H.A. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Lührmann, R.; Tuschl, T. Single-Stranded Antisense siRNAs Guide Target RNA Cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a Link Between Genetic and Biochemical Analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [Green Version]
- Doi, N.; Zenno, S.; Ueda, R.; Ohki-Hamazaki, H.; Ui-tei, K.; Saigo, K. Short-Interfering-RNA-Mediated Gene Silencing in Mammalian Cells Requires Dicer and eIF2C Translation Initiation Factors. Curr. Biol. 2003, 13, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 Is the Catalytic Engine of Mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Rivas, F.V.; Tolia, N.H.; Song, J.J.; Aragon, J.P.; Liu, J.; Hannon, G.J.; Joshua-Tor, L. Purified Argonaute2 and an siRNA form recombinant human RISC. Nat. Struct. Mol. Biol. 2005, 12, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Izaurraide, E. RNAi: Finding the elusive endonuclease. RNA 2004, 10, 1675–1679. [Google Scholar] [CrossRef] [Green Version]
- Hutnagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.J.; Martins, S.; Sarmento, B. siRNA as a tool to improve the treatment of brain diseases: Mechanism, targets and delivery. Ageing Res. Rev. 2015, 21, 43–54. [Google Scholar] [CrossRef]
- Lin, X.; Ruan, X.; Anderson, G.M.; McDowell, A.J.; Kroeger, E.P.; Fesik, W.S.; Shen, Y. siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 2005, 33, 4527–4535. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.L.; Linsley, P.S. Noise amidst the silence: Off-target effect os siRNA? Trends Genet. 2004, 20, 521–524. [Google Scholar] [CrossRef]
- Ui-Tei, K.; Naito, Y.; Zenno, S.; Nishi, K.; Yamato, K.; Takahashi, F.; Juni, A.; Saigo, K. Functional dissection of siRNA seequence by systematic DNA substitution: Modified siRNA with a DNA seed arm is a powerful tool for mammalian gene silencing with significantly reduced off-target effect. Nucleic Acids Res. 2008, 36, 2136–2151. [Google Scholar] [CrossRef]
- Ui-Tei, K.; Naito, Y.; Nishi, K.; Juni, A.; Saigo, K. Thermodynamic stability and Watson-Crick base pairing in the seed duplex are major determinants of the efficiency of the siRNA- based off-target effect. Nucleic Acids Res. 2008, 36, 2136–2151. [Google Scholar] [CrossRef] [PubMed]
- Kamola, J.P.; Nakano, Y.; Takahashi, T.; Wilson, A.P.; Ui-Tei, K. The siRNA Non-seed Region and Its Target Sequences Are Auxiliary Determinants of Off-Target Effects. PLoS Comput. Biol. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, D.A.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Ayadi, L.; Galvanin, A.; Pichot, F.; Marchand, V.; Motorin, Y. RNA ribose methylation (2′-O-methylation): Occurrence, biosynthesis and biological functions. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Iribe, H.; Miyamoto, K.; Takahashi, T.; Kobayashi, Y.; Leo, J.; Aida, M.; Ui-Tei, K. Chemical Modification of the siRNA Seed Region Suppresses Off-Target Effects by Steric Hindrance to Base-Pairing with Targets. ACS Omega 2017, 2, 2055–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, O.H.; Azizian, N.; Guo, M.; Capello, M.; Deng, D.; Zang, F.; Fry, J.; Katz, M.H.; Fleming, J.B.; Lee, J.E.; et al. Prognostic and Functional Significance of MAP4K5 in Pancreatic Cancer. PLoS ONE 2016, 11, e0152300. [Google Scholar] [CrossRef]
- Uhr, K.; Smissen, W.J.C.; Heine, A.A.J.; Ozturk, B.; Jaarsveld, M.T.M.; Boersma, A.W.M.; Jager, A.; Wiemer, E.A.C.; Smid, M.; Foekens, J.A.; et al. MicroRNAs as possible indicators of drug sensitivity in breast cancer cell lines. PLoS ONE 2019, 14, e0216400. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Yoshida, T.; Sugimura, T.; Terada, M. Liposome-mediated in vivo gene transfer of antisense K-ras construct inhibits pancreatic tumor dissemination in the murine peritoneal cavity. Cancer Res. 1995, 55, 3806–3810. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Gautier, L.; Wu, Z. Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V., Huber, W., Irizarry, R., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 13–32. [Google Scholar]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athylmic (nude) mide. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Sapi, J.; Kovacs, L.; Drexler, D.A.; Kocsis, P.; Gajari, D.; Sapi, Z. Tumor volume estimation and quasi-continuous administration for most effective bevacizumab therapy. PLoS ONE 2015, 10, e1043190. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woddendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Fukuhara, D.; Akase, D.; Aida, M.; Ui-Tei, K. siRNA seed region is divided into two functionally different domains in RNA interference is response to 2′-OMe modifications. ACS Omega 2022, 7, 2398–2410. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O′Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Anderson, E.M.; Reynolds, A.; Ilsley-Tyree, D.; Leak, D.; Fedorov, Y.; Baskerville, S.; Maksimova, E.; Robinson, K.; Karpilow, J.; et al. 3′UTR see matches, but not overall idenity,, are associated with RNAi off-targets. Nat. Methods 2007, 27, 91–105. [Google Scholar]
- Sledz, C.A.; Hlko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R.G. Activation of the interferon system by short-intefering RNAs. Nat. Cell Biol. 2003, 5, 834–939. [Google Scholar] [CrossRef]
- Villnueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF inhibitors: Unraveling mechanism and future treatment options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Sau, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therayp of BRAF-mutant metastatic menlanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [Green Version]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Cho, K.Y.; Tiwari, R.K. Overcoming barriers for siRNA therapeutics: From Bench to bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Muijsen, R.A.M.; Collin, R.W.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarria, L.; et al. Celivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mo. Med. 2021, 13, 313243. [Google Scholar]
- Chernikov, V.I.; Vlassov, V.V.; Chernolovskaya, L.E. Current development of siRNA bioconjugates: From research to the clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y. RNA Therapy: Current Status and Future Potential. Chonnam Med. J. 2020, 56, 87–93. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Kobayashi, Y.; Asano, Y.; Sato, A.; Taniguchi, H.; Ui-Tei, K. Knockdown of 15-bp Deletion-Type v-raf Murine Sarcoma Viral Oncogene Homolog B1 mRNA in Pancreatic Ductal Adenocarcinoma Cells Repressed Cell Growth In Vitro and Tumor Volume In Vivo. Cancers 2022, 14, 3162. https://doi.org/10.3390/cancers14133162
Song J, Kobayashi Y, Asano Y, Sato A, Taniguchi H, Ui-Tei K. Knockdown of 15-bp Deletion-Type v-raf Murine Sarcoma Viral Oncogene Homolog B1 mRNA in Pancreatic Ductal Adenocarcinoma Cells Repressed Cell Growth In Vitro and Tumor Volume In Vivo. Cancers. 2022; 14(13):3162. https://doi.org/10.3390/cancers14133162
Chicago/Turabian StyleSong, Jiaxuan, Yoshiaki Kobayashi, Yoshimasa Asano, Atsushi Sato, Hiroaki Taniguchi, and Kumiko Ui-Tei. 2022. "Knockdown of 15-bp Deletion-Type v-raf Murine Sarcoma Viral Oncogene Homolog B1 mRNA in Pancreatic Ductal Adenocarcinoma Cells Repressed Cell Growth In Vitro and Tumor Volume In Vivo" Cancers 14, no. 13: 3162. https://doi.org/10.3390/cancers14133162
APA StyleSong, J., Kobayashi, Y., Asano, Y., Sato, A., Taniguchi, H., & Ui-Tei, K. (2022). Knockdown of 15-bp Deletion-Type v-raf Murine Sarcoma Viral Oncogene Homolog B1 mRNA in Pancreatic Ductal Adenocarcinoma Cells Repressed Cell Growth In Vitro and Tumor Volume In Vivo. Cancers, 14(13), 3162. https://doi.org/10.3390/cancers14133162