
Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material & Methods
2.1. Cell Culture
2.2. Genomic Disruption of GCLc and GPx4 Using CRISPR-Cas9
2.3. GSH Measurement
2.4. Proliferation Assay
2.5. Clonogenicity Assay
2.6. Immunoblotting
2.7. Cell Death and Lipid Hydroperoxidation
2.8. ROS Analysis
2.9. Tumor Xenograft Studies
2.10. Boyden Chamber
2.11. Statistical Analysis
3. Results
3.1. Genetic Deletion of the Catalytic Subunit of GCL Leads to the Collapse of GSH Intracellular Pool and Abolishes Growth of the PDAC Cell Lines
3.2. Genetic Deletion of GCLc Leads to Non-Ferroptotic Cell Death and Is Not Characterized by Accumulation of Lipid Hydroperoxides
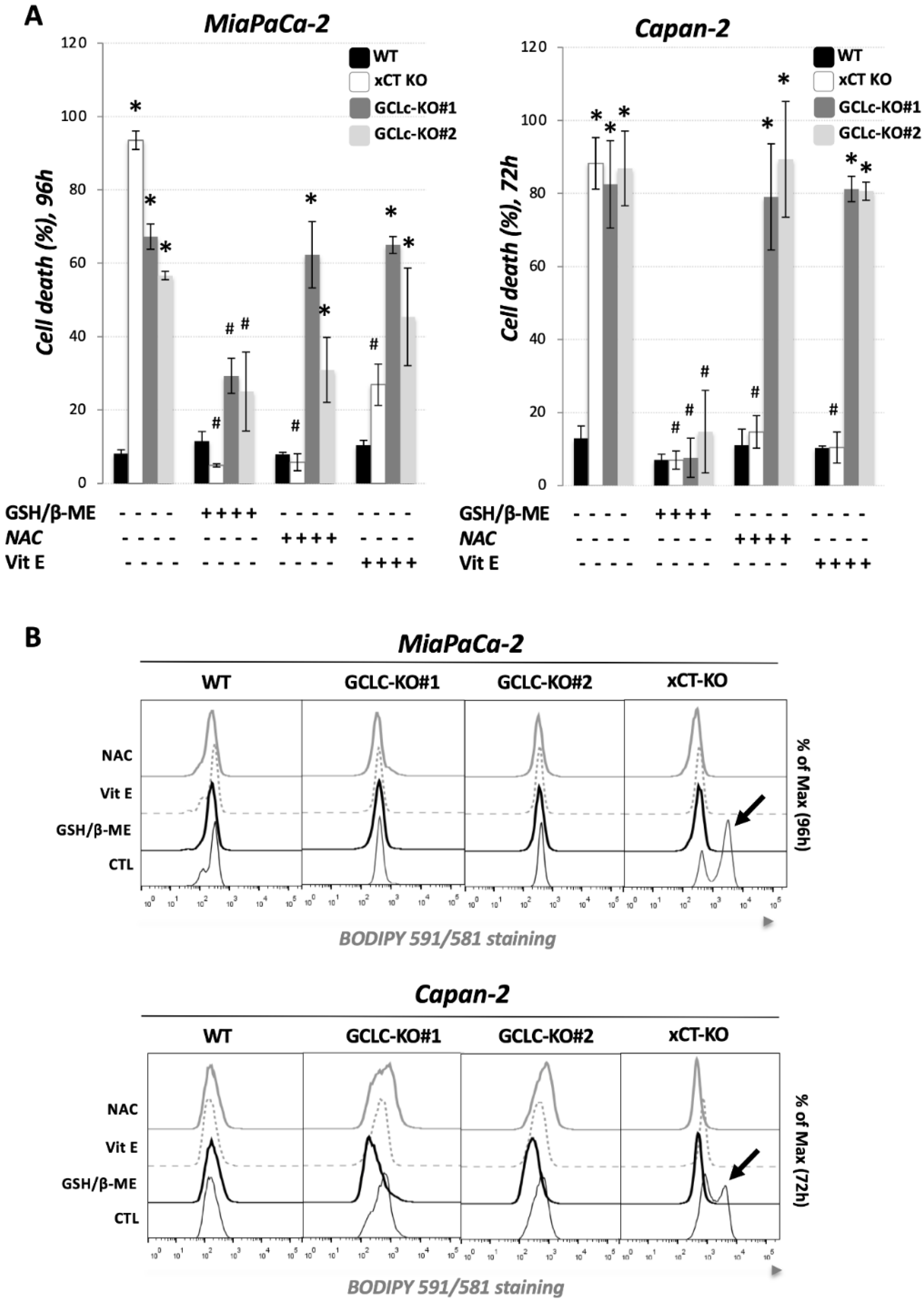
3.3. Glutathione Depletion Sensitizes Cells to ROS-Dependent Apoptosis but Not to Ferroptosis Cell Death
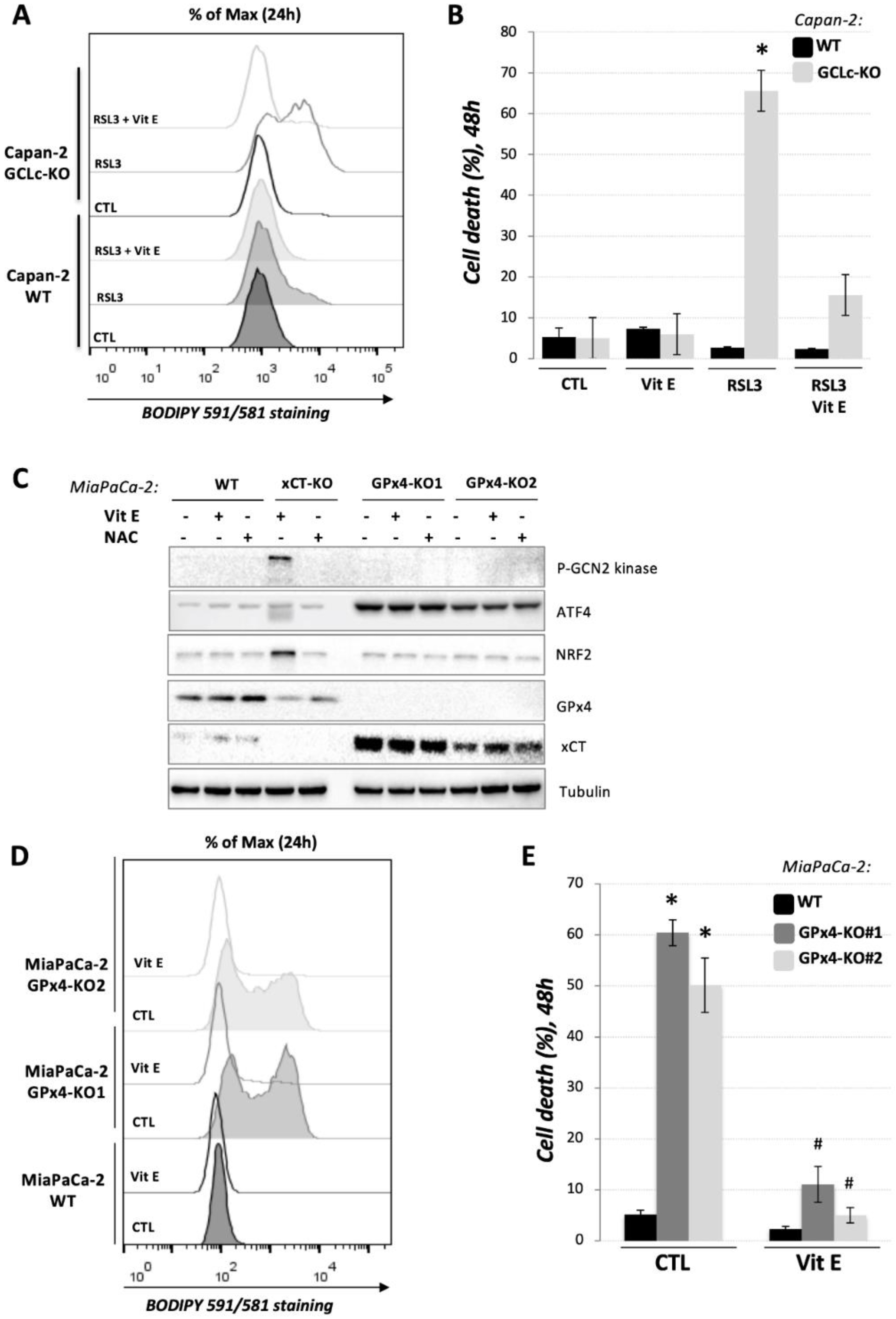
3.4. GCLc-KO Cells Remains Highly Sensitive to GPx4 Inhibition
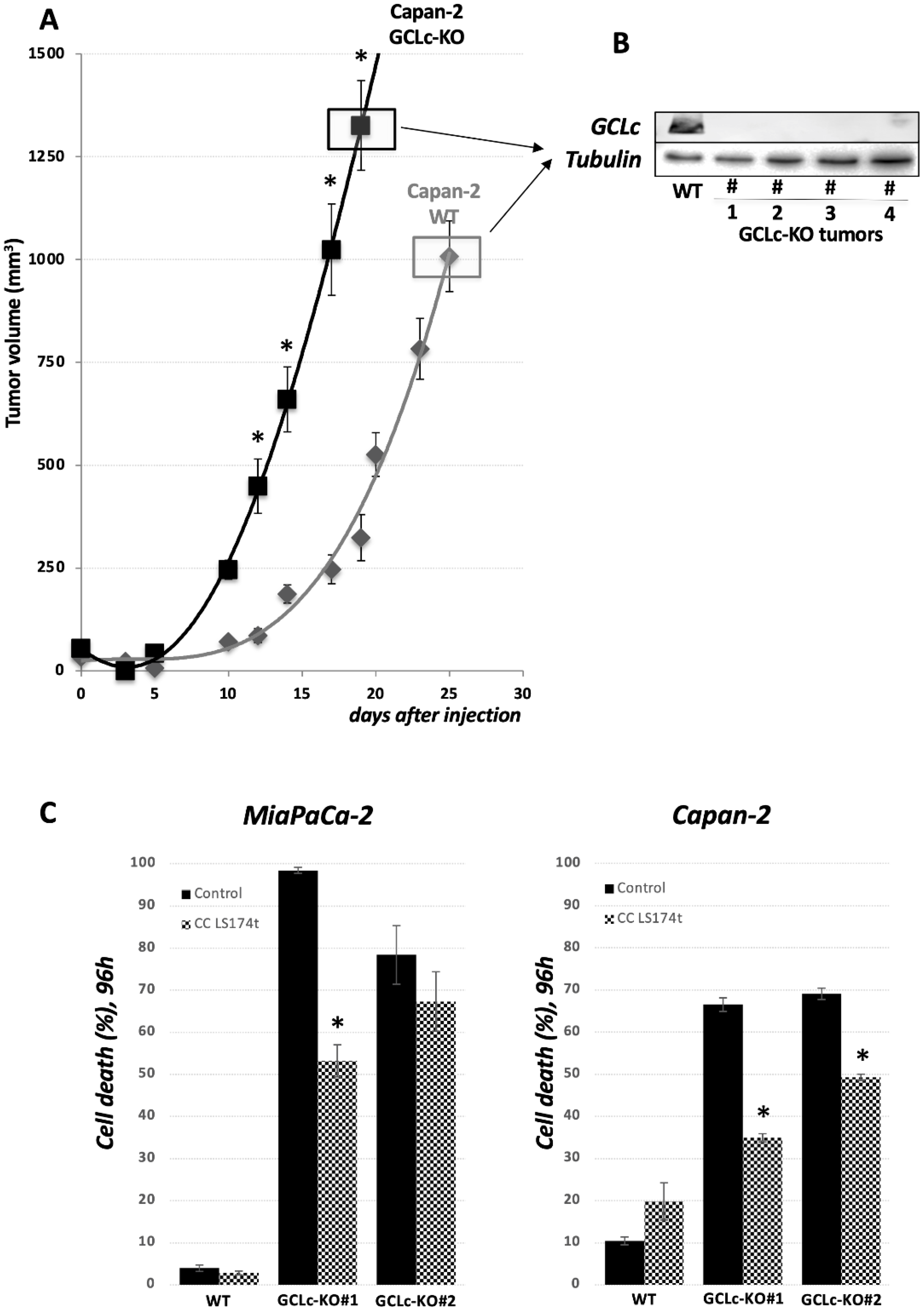
3.5. GCLc Is Dispensable for PDAC Cell Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Dalton, T.P.; Dieter, M.Z.; Yang, Y.; Shertzer, H.G.; Nebert, D.W. Knockout of the Mouse Glutamate Cysteine Ligase Catalytic Subunit (Gclc) Gene: Embryonic Lethal When Homozygous, and Proposed Model for Moderate Glutathione Deficiency When Heterozygous. Biochem. Biophys. Res. Commun. 2000, 279, 324–329. [Google Scholar] [CrossRef]
- Botta, D.; White, C.C.; Vliet-Gregg, P.; Mohar, I.; Shi, S.; McGrath, M.B.; McConnachie, L.A.; Kavanagh, T.J. Modulating GSH Synthesis Using Glutamate Cysteine Ligase Transgenic and Gene-Targeted Mice. Drug Metab. Rev. 2008, 40, 465–477. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1166–1181.e6. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [Green Version]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine–glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef] [Green Version]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Daher, B.; Vučetić, M.; Pouysségur, J. Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptotic Cell Death. Front. Oncol. 2020, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Bilton, R.; Mazure, N.; Trottier, E.; Hattab, M.; Déry, M.A.; Richard, D.E.; Pouysségur, J.; Brahimi-Horn, M.C. Arrest-defective-1 Protein, an Acetyltransferase, Does Not Alter Stability of Hypoxia-inducible Factor (HIF)-1α and Is Not Induced by Hypoxia or HIF. J. Biol. Chem. 2005, 280, 31132–31140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Vučković, A.M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.S.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [Green Version]
- Meira, W.; Daher, B.; Parks, S.; Cormerais, Y.; Durivault, J.; Tambutte, E.; Pouyssegur, J.; Vučetić, M. A Cystine-Cysteine Intercellular Shuttle Prevents Ferroptosis in xCTKO Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 1434. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Hernandez, J.I.; Almeida, A.; Esteban, M.D.; Fernandez, E.; Bolanos, J. Knockdown of Glutamate-Cysteine Ligase by Small Hairpin RNA Reveals That Both Catalytic and Modulatory Subunits Are Essential for the Survival of Primary Neurons. J. Biol. Chem. 2005, 280, 38992–39001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Cidlowski, J.A. SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: Glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J. Biol. Chem. 2006, 281, 29542–29557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanigan, M.H. Gamma-Glutamyl Transpeptidase: Redox Regulation and Drug Resistance. Adv. Cancer Res. 2014, 122, 103. [Google Scholar] [PubMed] [Green Version]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorino, M.; Roveri, A.; Benazzi, L.; Bosello, V.; Mauri, P.; Toppo, S.; Tosatto, S.C.; Ursini, F. Functional interaction of phospholipid hydroperoxide glutathione peroxidase with sperm mitochondrion-associated cysteine-rich protein discloses the adjacent cysteine motif as a new substrate of the selenoperoxidase. J. Biol. Chem. 2005, 280, 38395–38402. [Google Scholar] [CrossRef] [Green Version]
- Godeas, C.; Tramer, F.; Micali, F.; Roveri, A.; Maiorino, M.; Nisii, C.; Sandri, G.; Panfili, E. Phospholipid Hydroperoxide Glutathione Peroxidase (PHGPx) in Rat Testis Nuclei Is Bound to Chromatin. Biochem. Mol. Med. 1996, 59, 118–124. [Google Scholar] [CrossRef]
- Vucetic, M.; Daher, B.; Cassim, S.; Meira, W.; Pouyssegur, J. Together we stand, apart we fall: How cell-to-cell contact/interplay provides resistance to ferroptosis. Cell Death Dis. 2020, 11, 789. [Google Scholar] [CrossRef]
- Batsios, G.; Najac, C.; Cao, P.; Viswanath, P.; Subramani, E.; Saito, Y.; Gillespie, A.M.; Yoshihara, H.; Larson, P.; Sando, S.; et al. In vivo detection of γ-glutamyl-transferase up-regulation in glioma using hyperpolarized γ-glutamyl-[1-13C]glycine. Sci. Rep. 2020, 10, 6244. [Google Scholar] [CrossRef] [Green Version]
- Bachhawat, A.K.; Kaur, A. Glutathione Degradation. Antioxid. Redox Signal. 2017, 27, 1200–1216. [Google Scholar] [CrossRef]
- Haddad, M.; Hervé, V.; Ben Khedher, M.R.; Rabanel, J.-M.; Ramassamy, C. Glutathione: An Old and Small Molecule with Great Functions and New Applications in the Brain and in Alzheimer’s Disease. Antioxid. Redox Signal. 2021, 35, 270–292. [Google Scholar] [CrossRef] [PubMed]
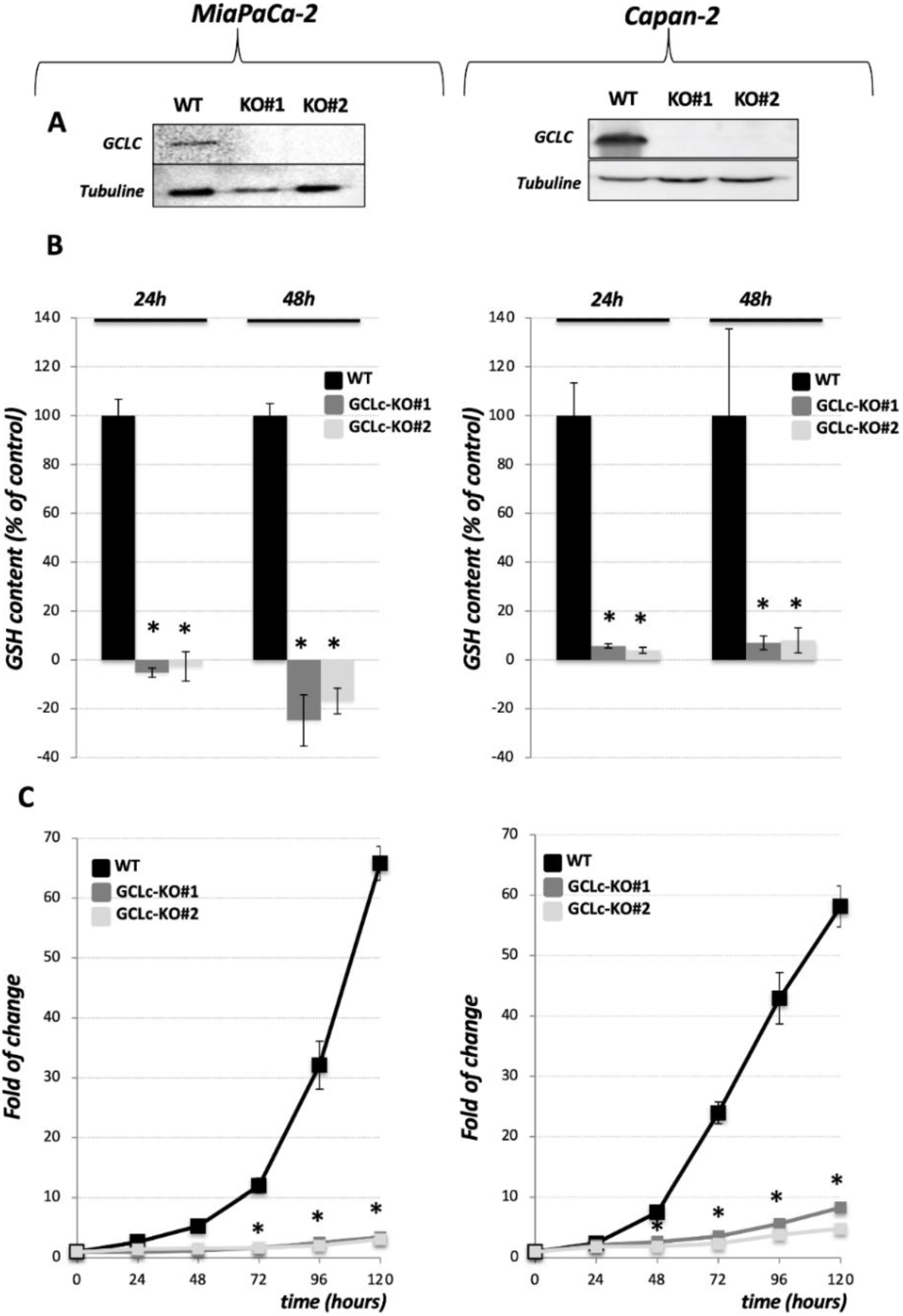
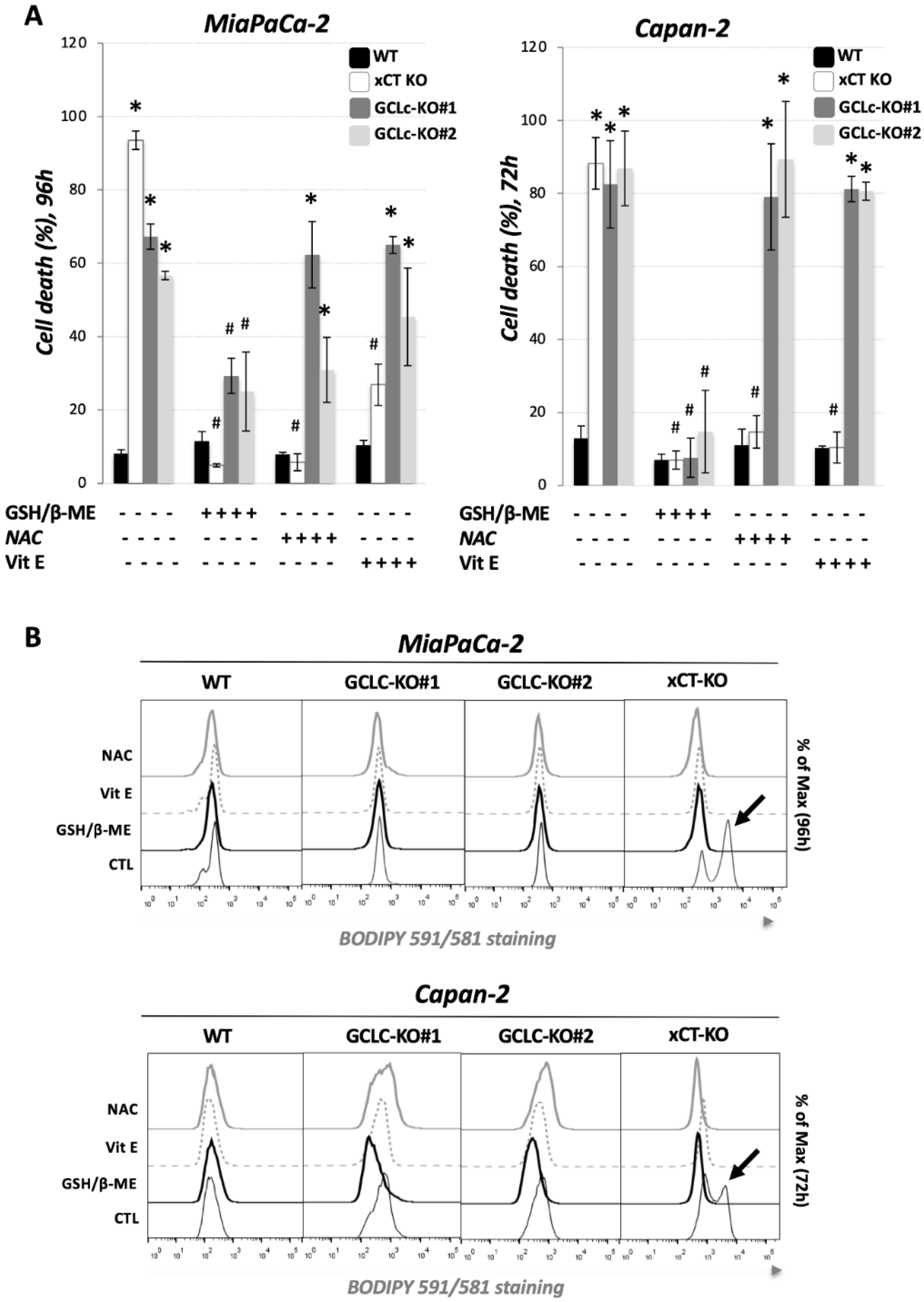
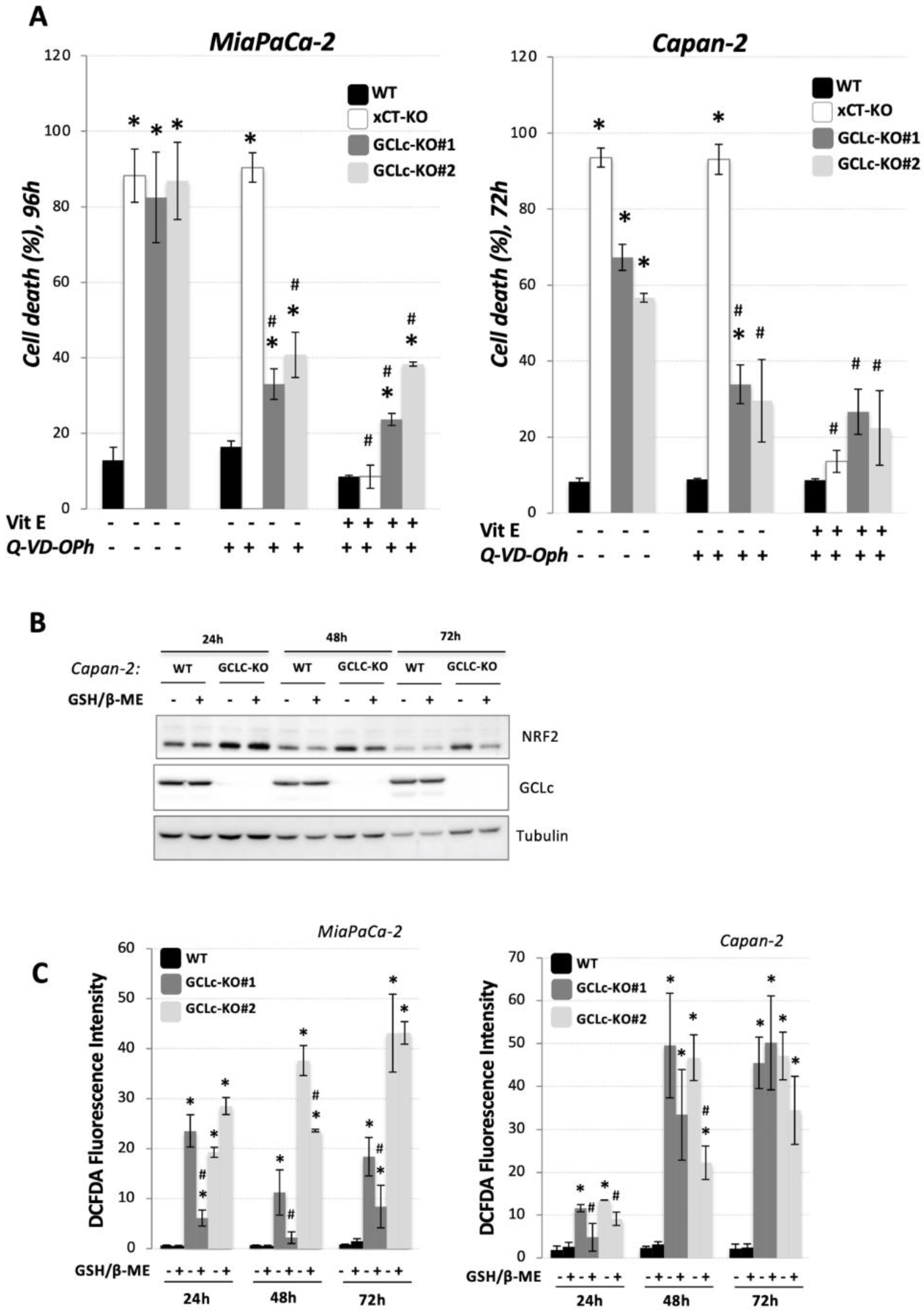
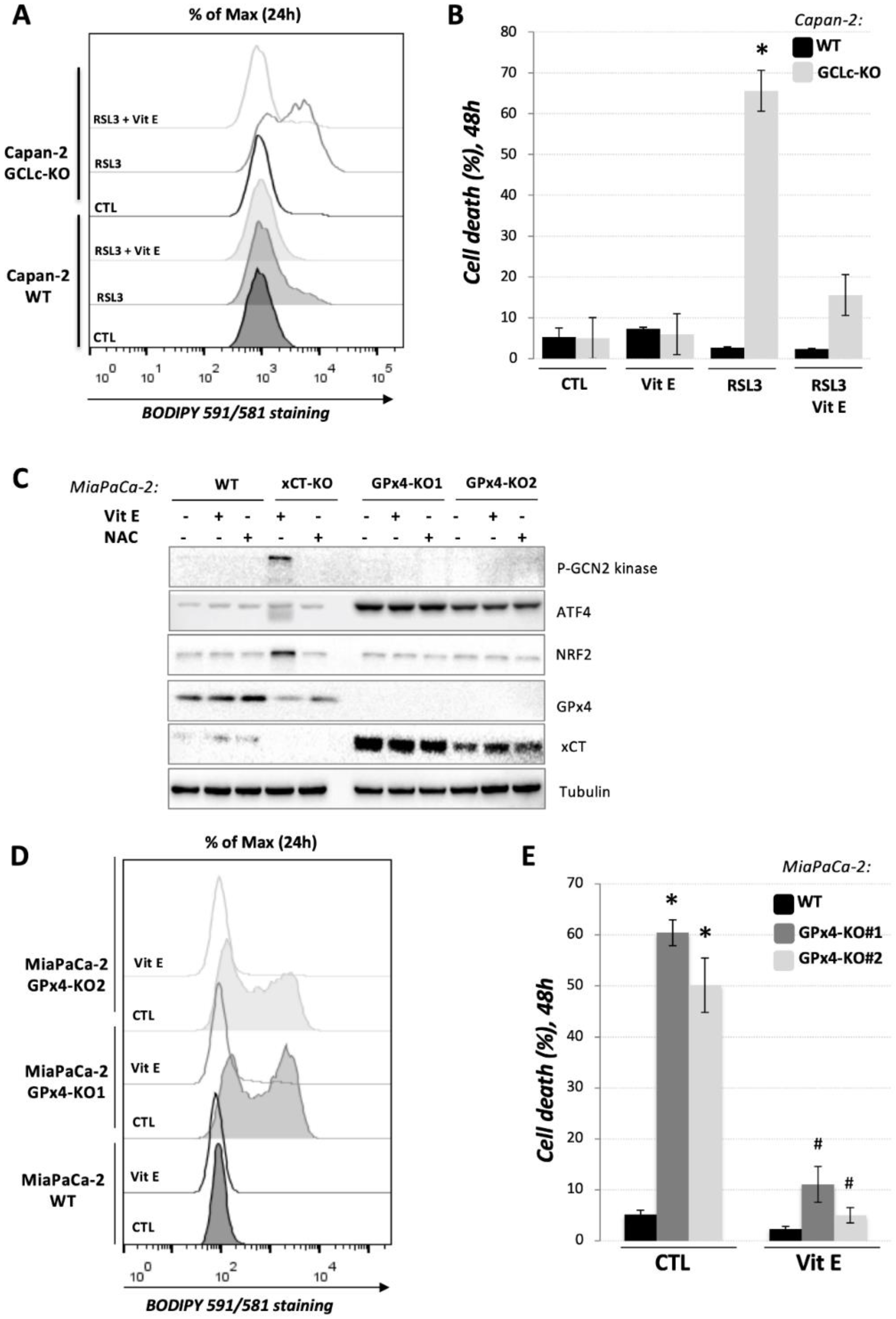
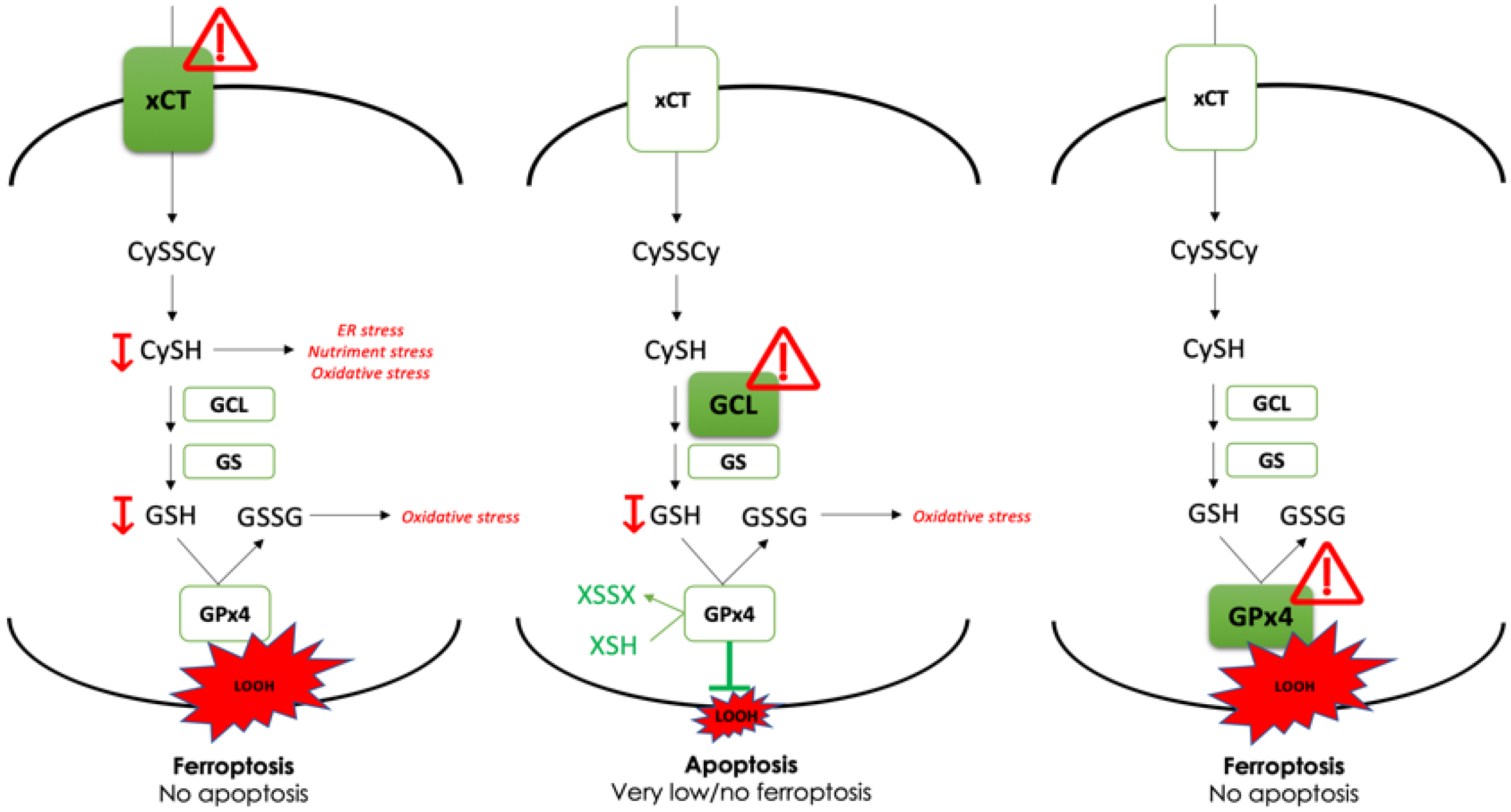
 ), revealed indispensable role of cystine transporter (xCT) and antioxidant enzyme GPx4 for ferroptosis prevention. On the contrary, genetic deletion of the main enzyme in GSH biosynthesis (GCL) proved to be substitutable in this context (see Discussion).
), revealed indispensable role of cystine transporter (xCT) and antioxidant enzyme GPx4 for ferroptosis prevention. On the contrary, genetic deletion of the main enzyme in GSH biosynthesis (GCL) proved to be substitutable in this context (see Discussion).
), revealed indispensable role of cystine transporter (xCT) and antioxidant enzyme GPx4 for ferroptosis prevention. On the contrary, genetic deletion of the main enzyme in GSH biosynthesis (GCL) proved to be substitutable in this context (see Discussion).
), revealed indispensable role of cystine transporter (xCT) and antioxidant enzyme GPx4 for ferroptosis prevention. On the contrary, genetic deletion of the main enzyme in GSH biosynthesis (GCL) proved to be substitutable in this context (see Discussion).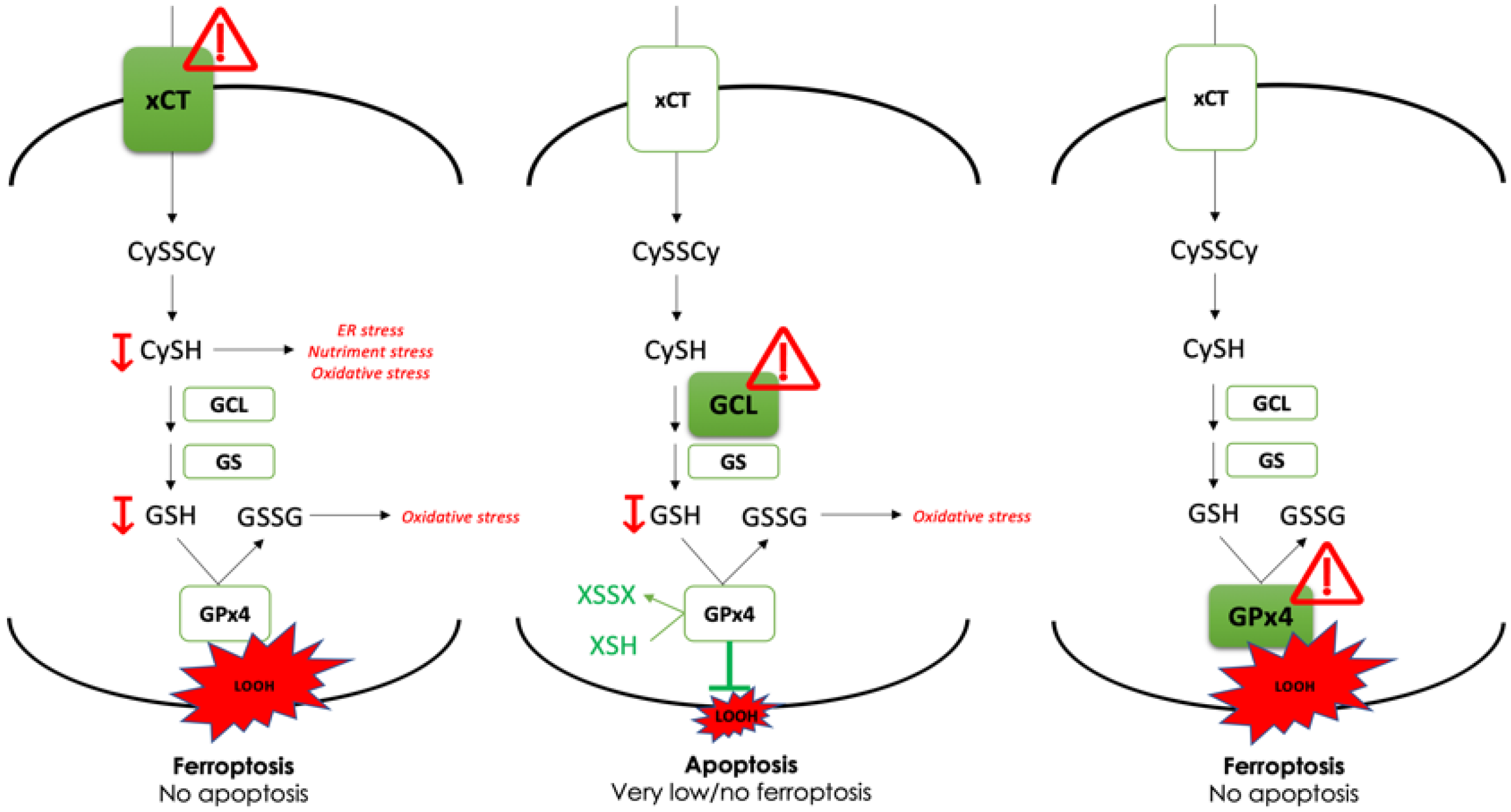

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CrGCLc_ex1 | GC CAT GCC GAC CAC GTG CGG |
| CrGCLc_ex3 | GC AAC ATG CGA AAA CGC CGG A |
| CrGPx4_ex3 | GC CCG AAC TGG TTA CAC GGG A |
| CrGPx4_ex4 | GA GAG ATC AAA GAG TTC GCC G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daher, B.; Meira, W.; Durivault, J.; Gotorbe, C.; Pouyssegur, J.; Vucetic, M. Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth. Cancers 2022, 14, 3154. https://doi.org/10.3390/cancers14133154
Daher B, Meira W, Durivault J, Gotorbe C, Pouyssegur J, Vucetic M. Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth. Cancers. 2022; 14(13):3154. https://doi.org/10.3390/cancers14133154
Chicago/Turabian StyleDaher, Boutaina, Willian Meira, Jerome Durivault, Celia Gotorbe, Jacques Pouyssegur, and Milica Vucetic. 2022. "Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth" Cancers 14, no. 13: 3154. https://doi.org/10.3390/cancers14133154
APA StyleDaher, B., Meira, W., Durivault, J., Gotorbe, C., Pouyssegur, J., & Vucetic, M. (2022). Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth. Cancers, 14(13), 3154. https://doi.org/10.3390/cancers14133154