
CDK4/CDK6 Inhibitors Synergize with Midostaurin, Avapritinib, and Nintedanib in Inducing Growth Inhibition in KIT D816V+ Neoplastic Mast Cells

 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Reagents
2.2. Isolation of Primary Neoplastic Cells
2.3. Culture of Human Cell Lines
2.4. Quantitative Polymerase Chain Reaction and Western Blot Analysis
2.5. Evaluation of Drug Effects on Cell Proliferation
2.6. Evaluation of Apoptosis in Drug-Exposed Cells
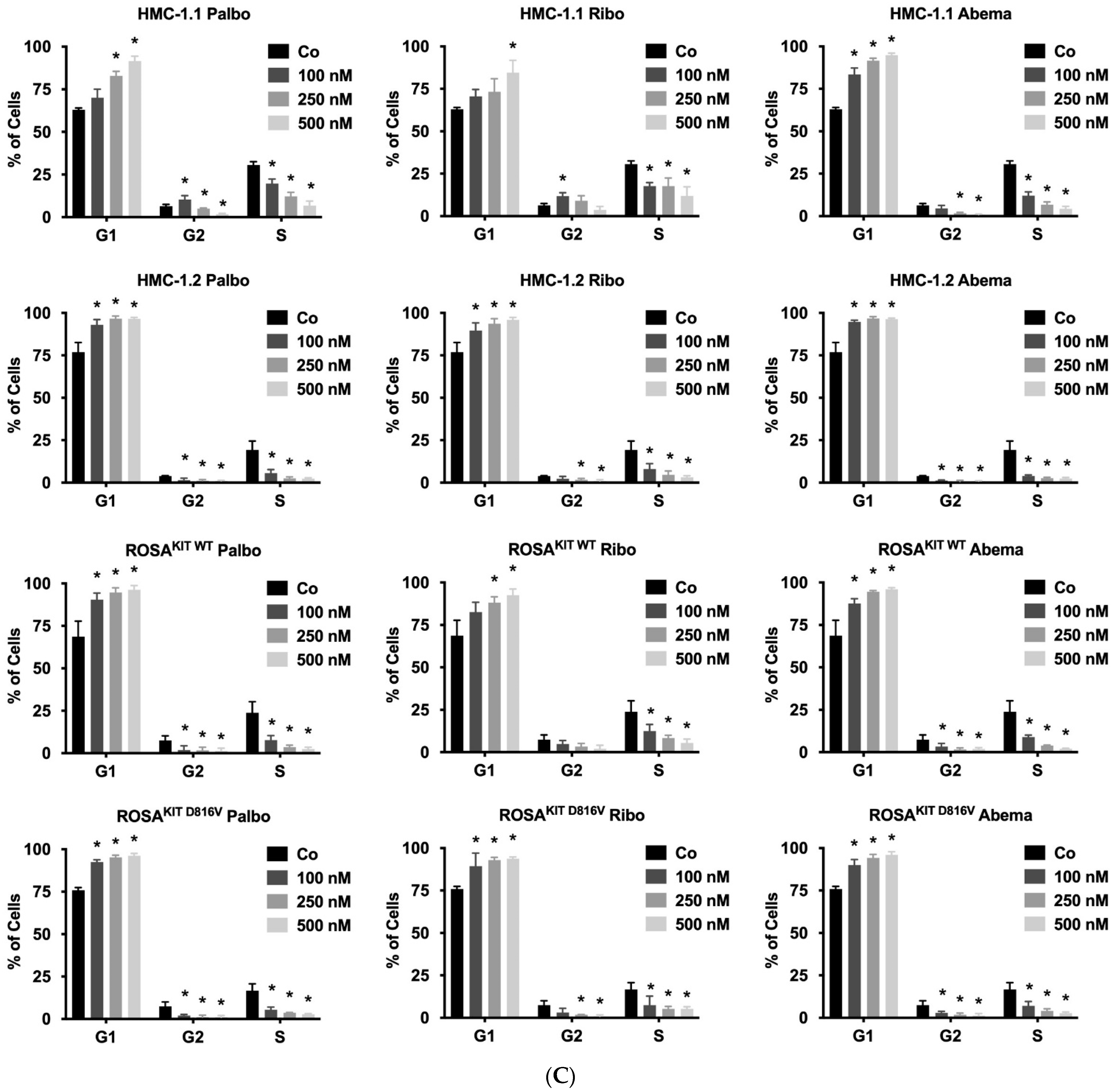
2.7. Evaluation of Drug-Induced Cell Cycle Arrest
2.8. Measurement of Histamine Release
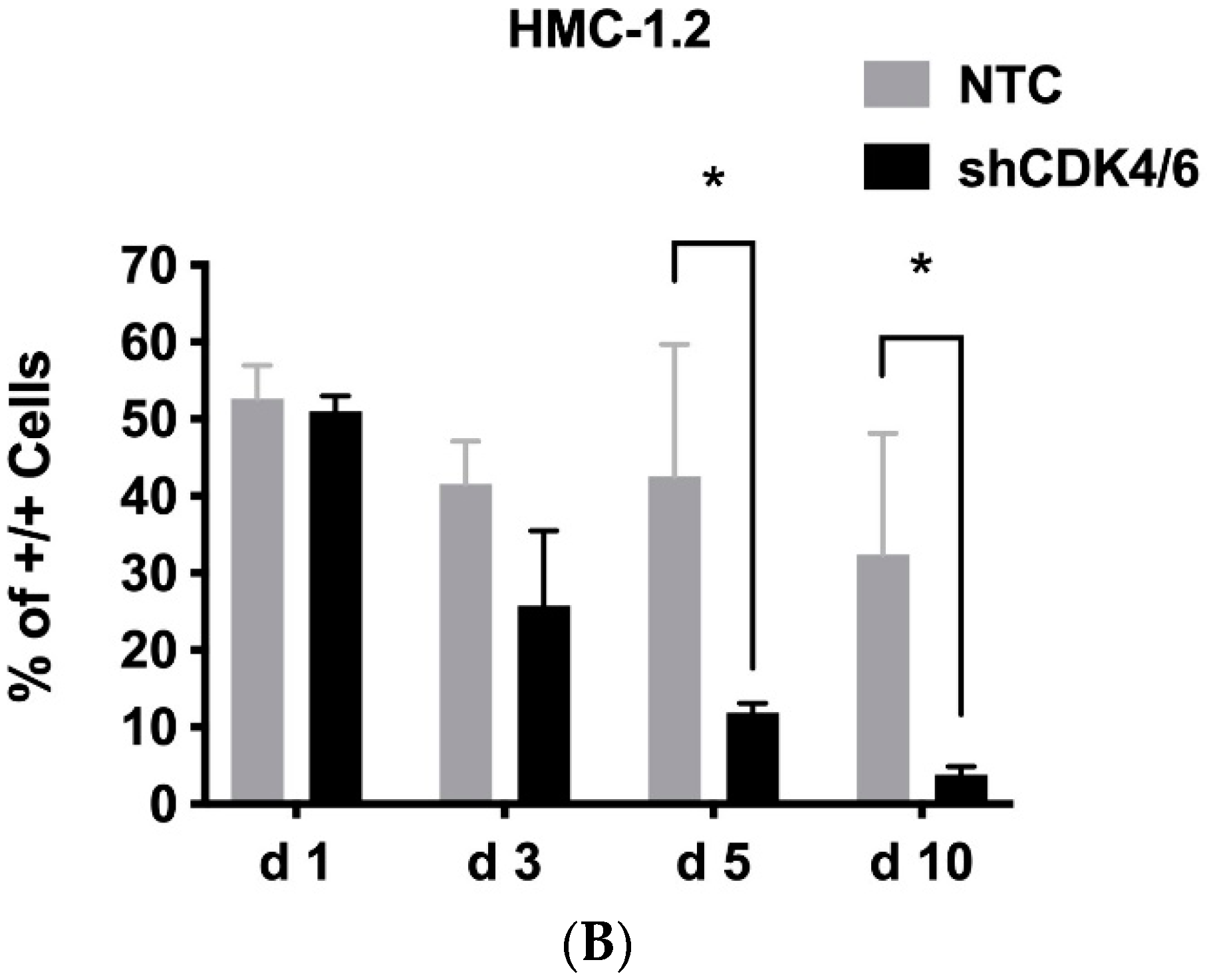
2.9. shRNA-Mediated Knockdown of CDK4 and CDK6
2.10. Statistical Analysis
3. Results
3.1. Identification of CDK4/CDK6 as Potential Therapeutic Targets in Neoplastic MC
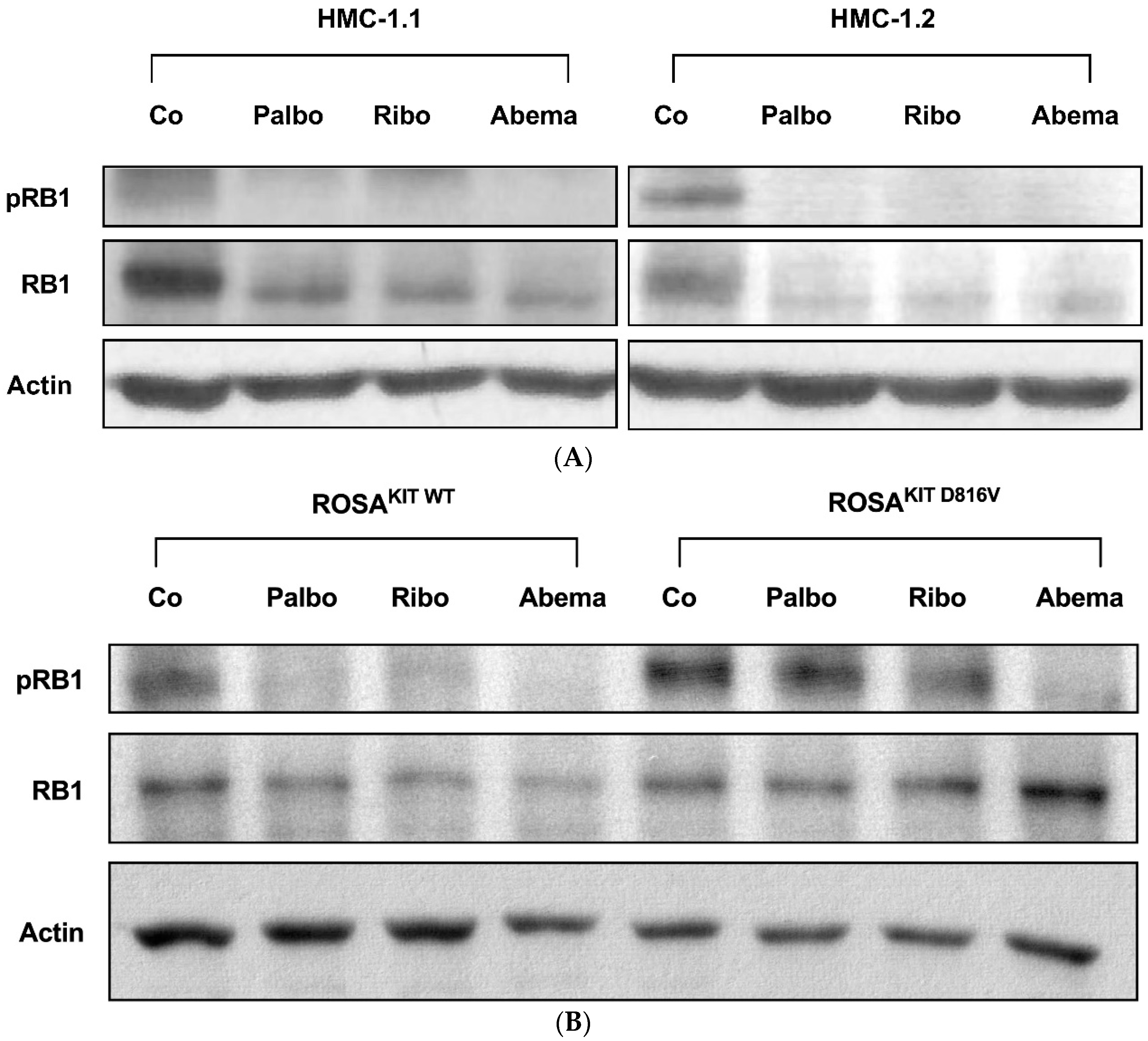
3.2. Palbociclib, Ribociclib and Abemaciclib Disrupt CDK4/CDK6 Signaling in Neoplastic MC
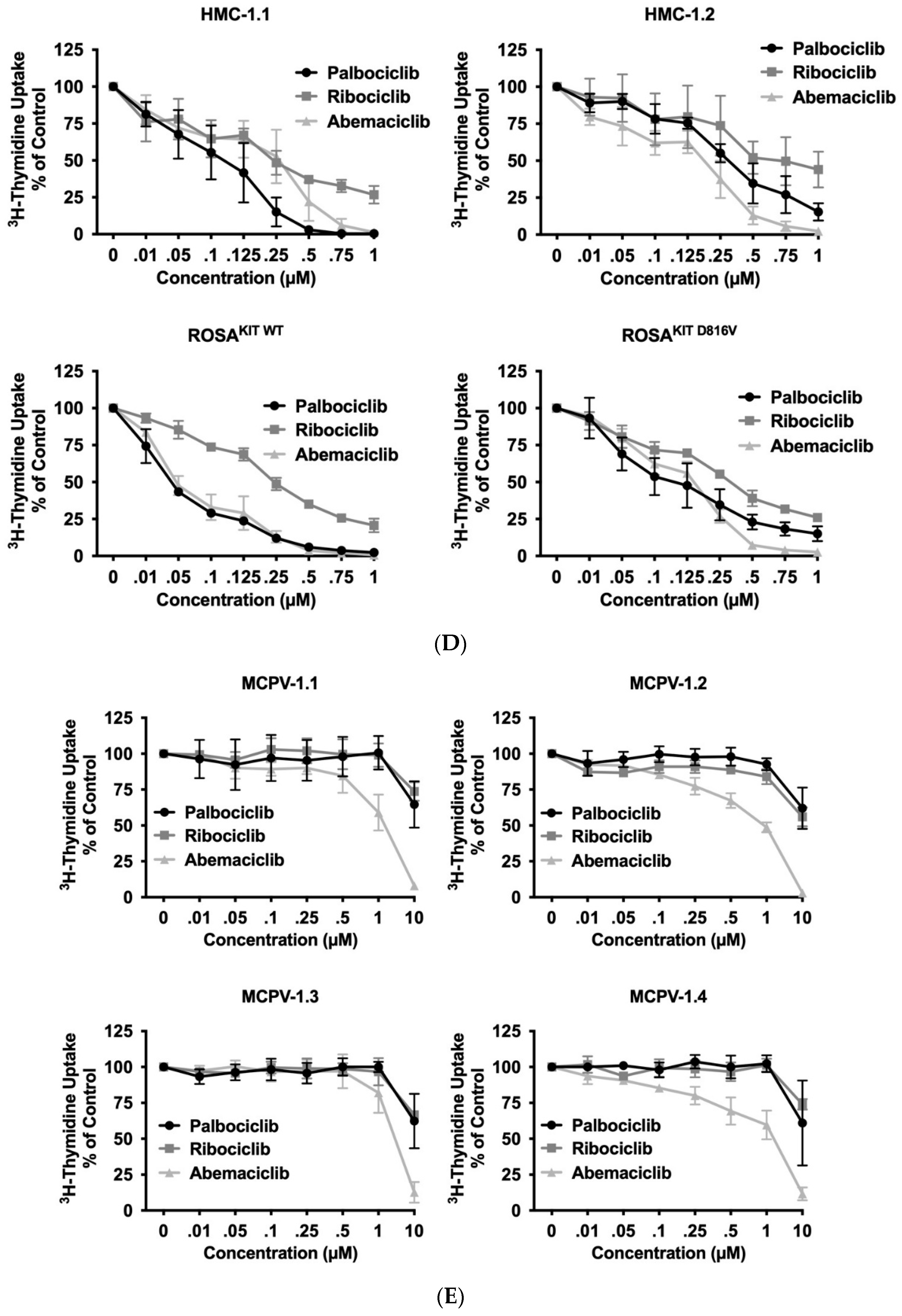
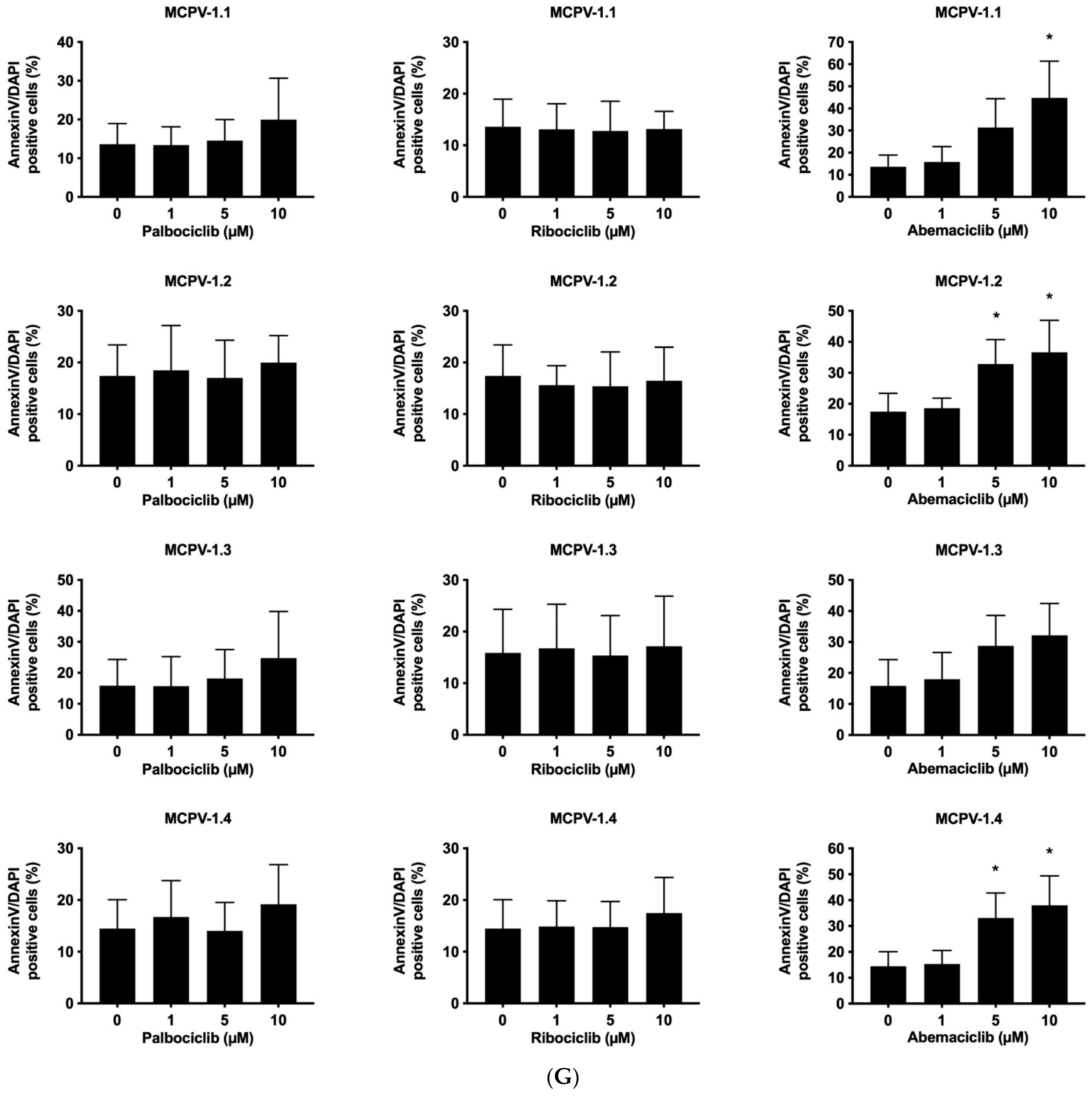
3.3. CDK4/CDK6 Inhibitors Counteract Proliferation and Survival in the Human MC Lines HMC-1 and ROSA Whereas Only Abemaciclib Is Effective in Drug-Resistant MCPV-1 Cells
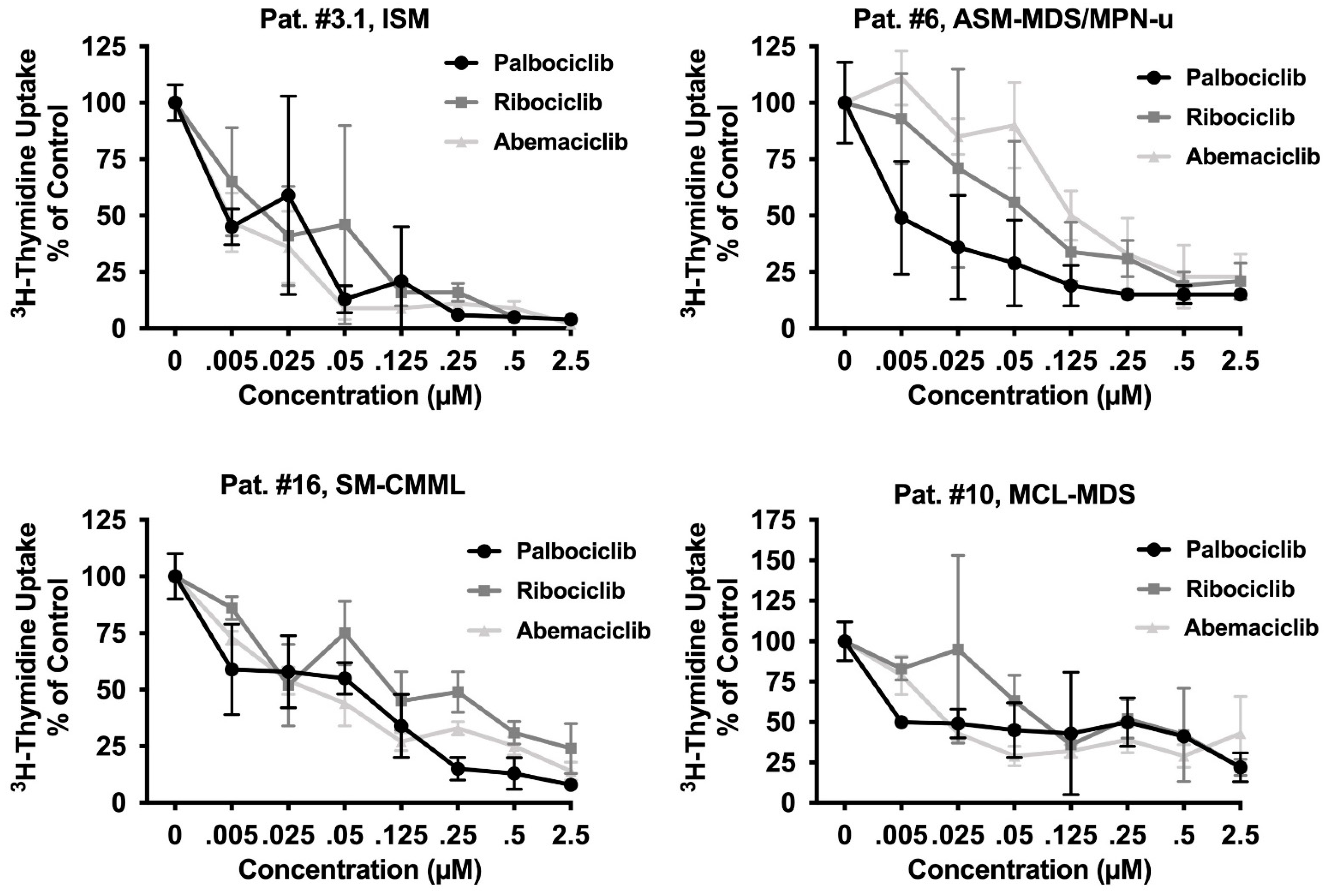
3.4. CDK4/CDK6 Inhibitors Block the Proliferation of Primary Neoplastic Cells Isolated from Patients with Various Subtypes of SM including Relapsed MCL
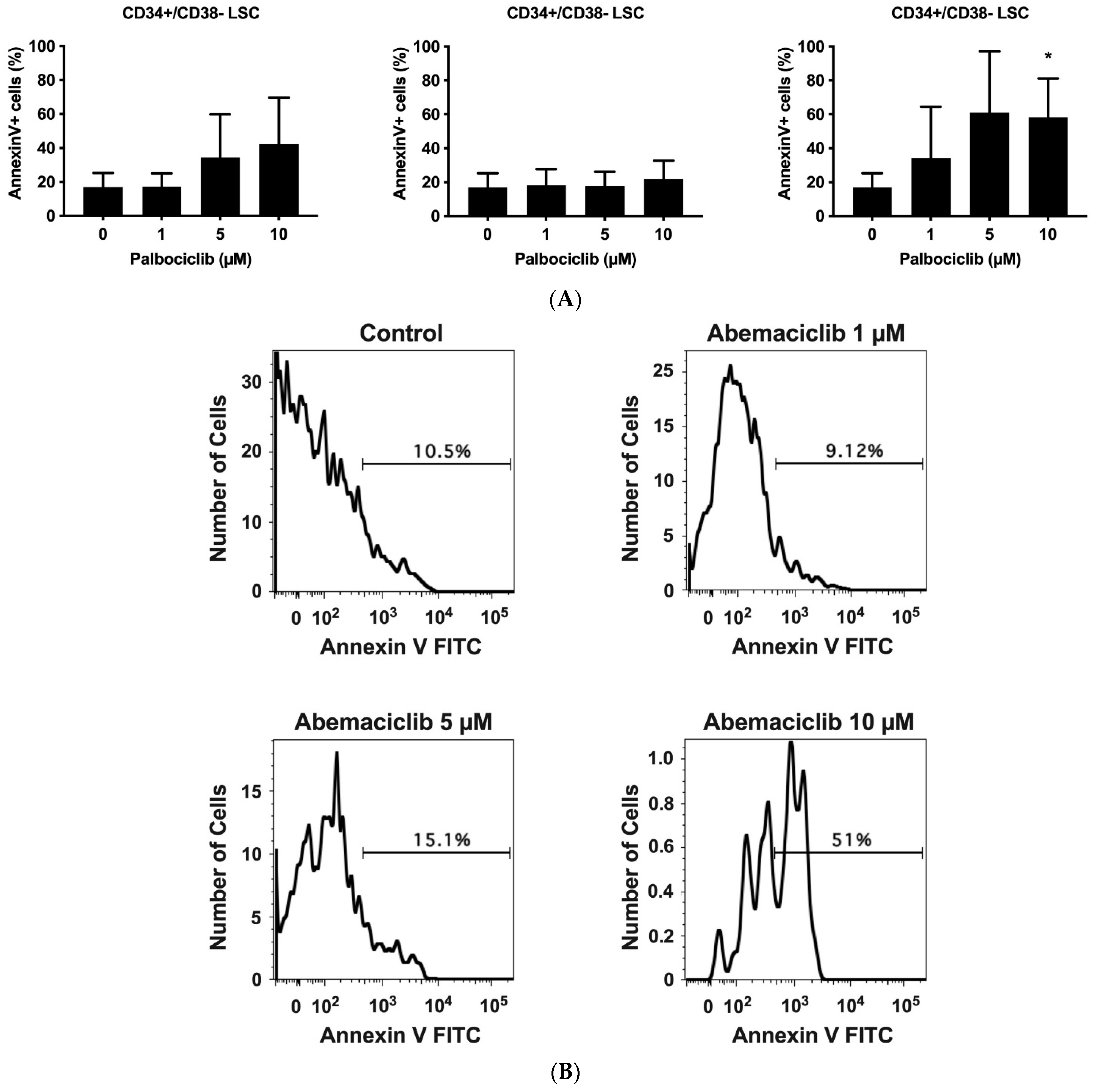
3.5. CDK4/CDK6 Inhibitors Induce Apoptosis in CD34+/CD38− Leukemic Stem Cells (LSCs) in AdvSM
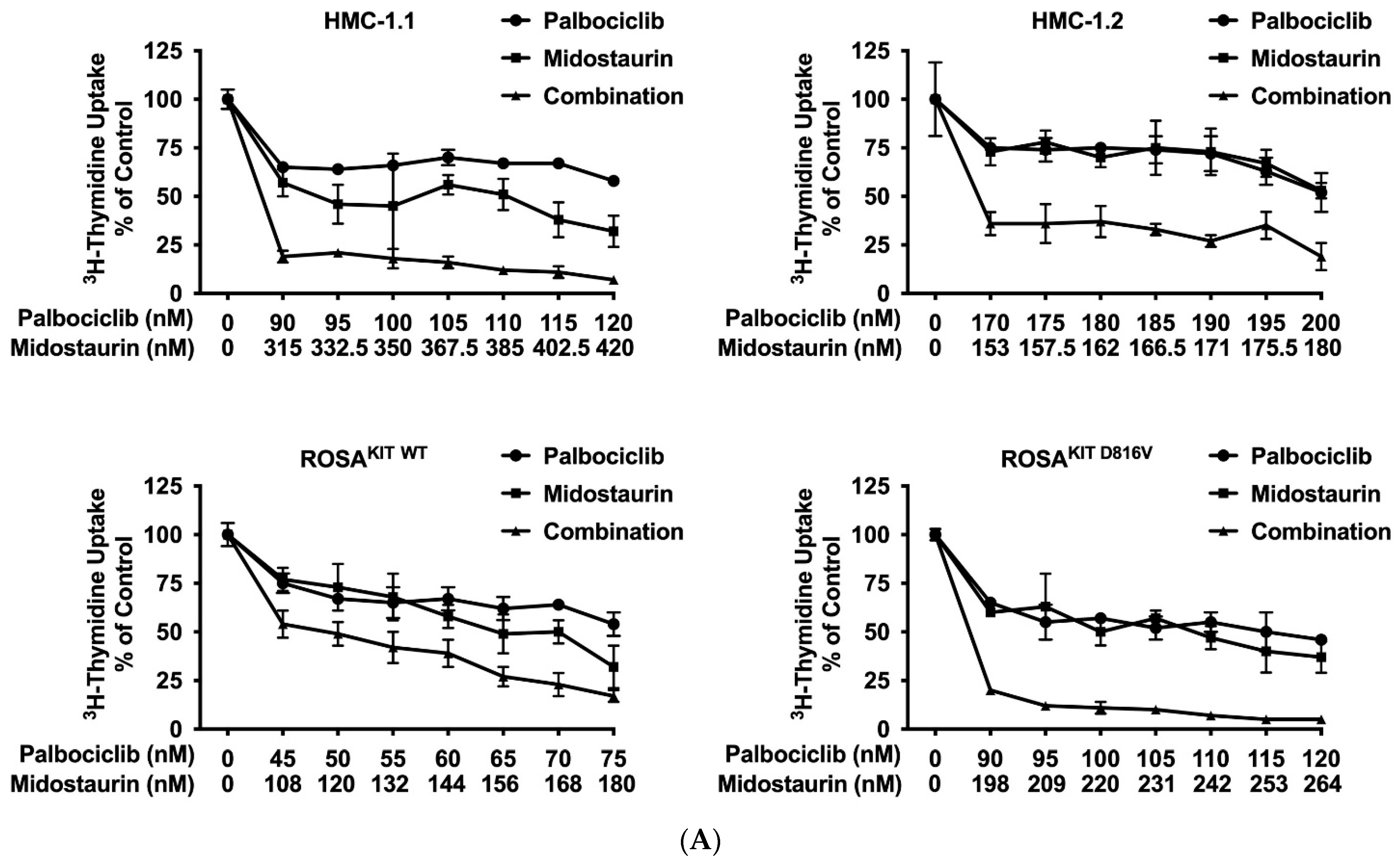
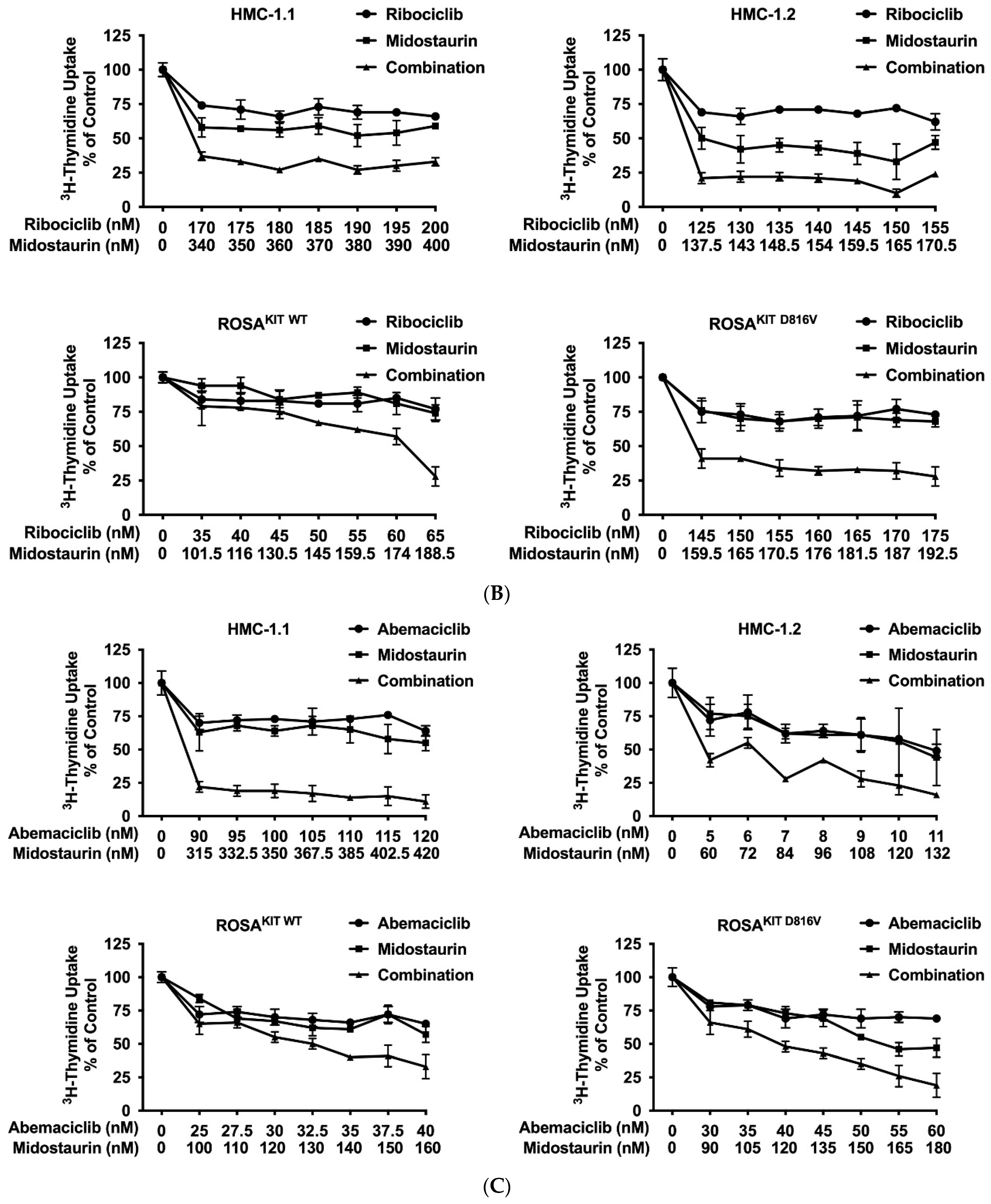
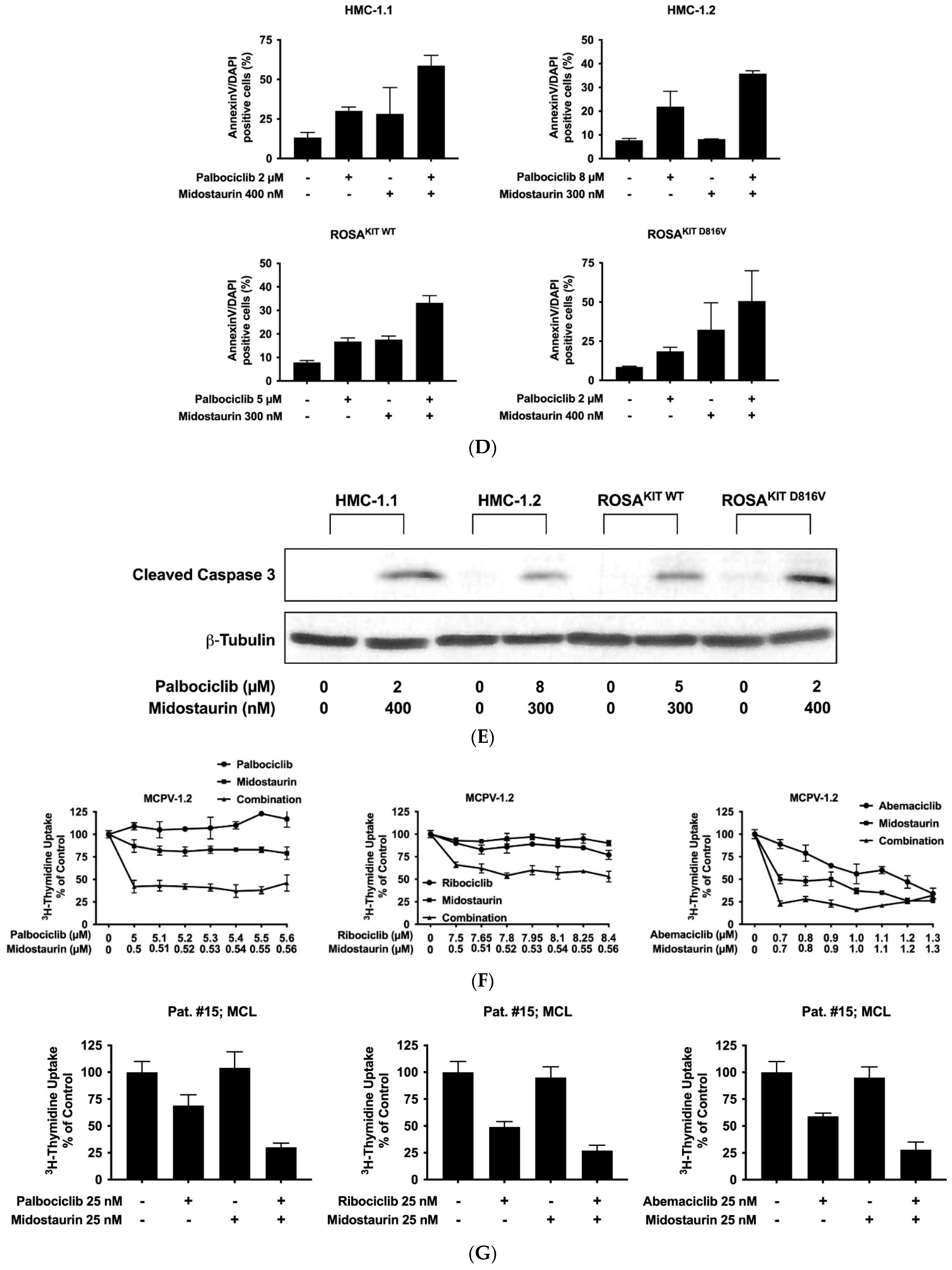
3.6. Inhibition of CDK4/CDK6 Sensitizes Neoplastic MCs against Midostaurin
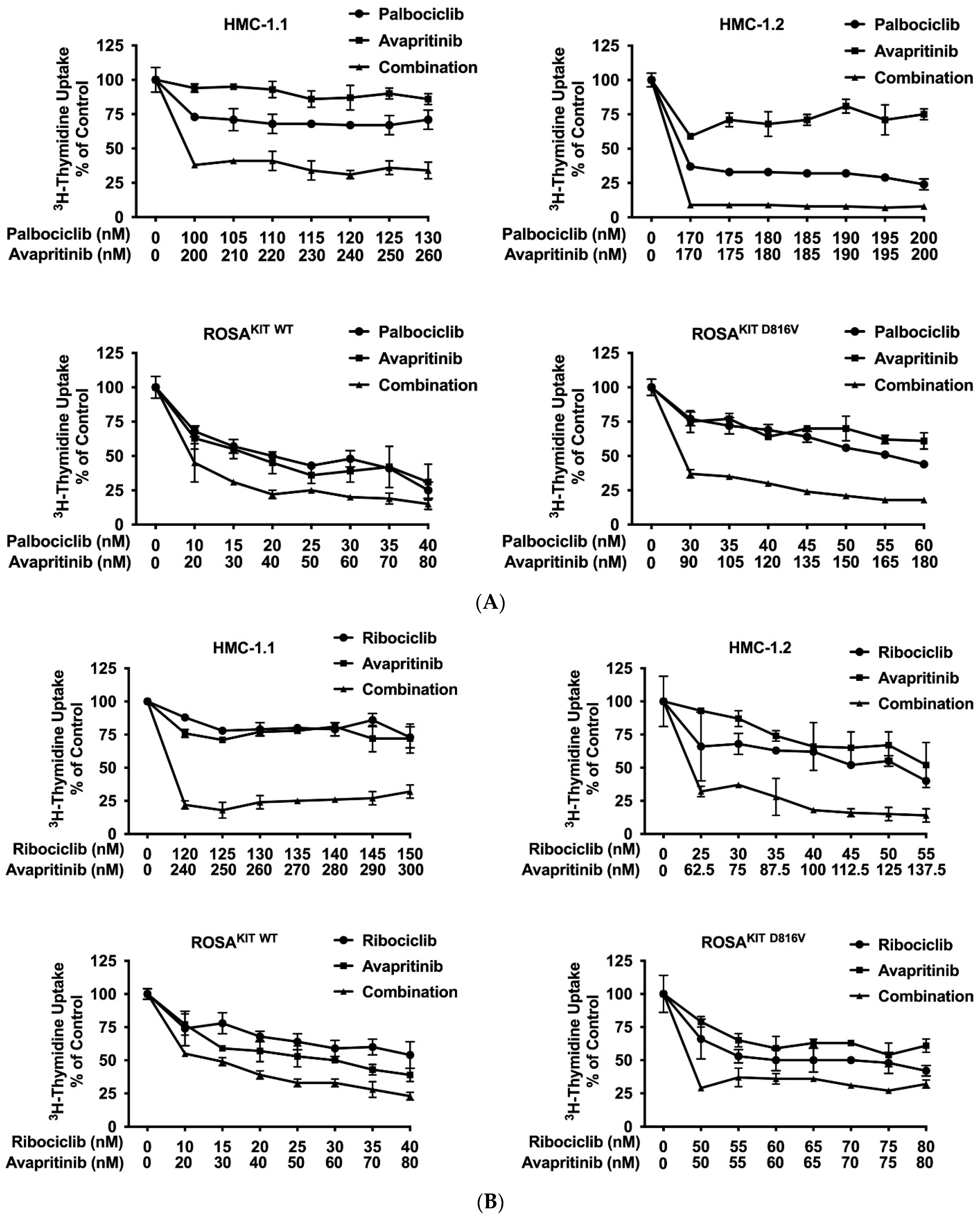
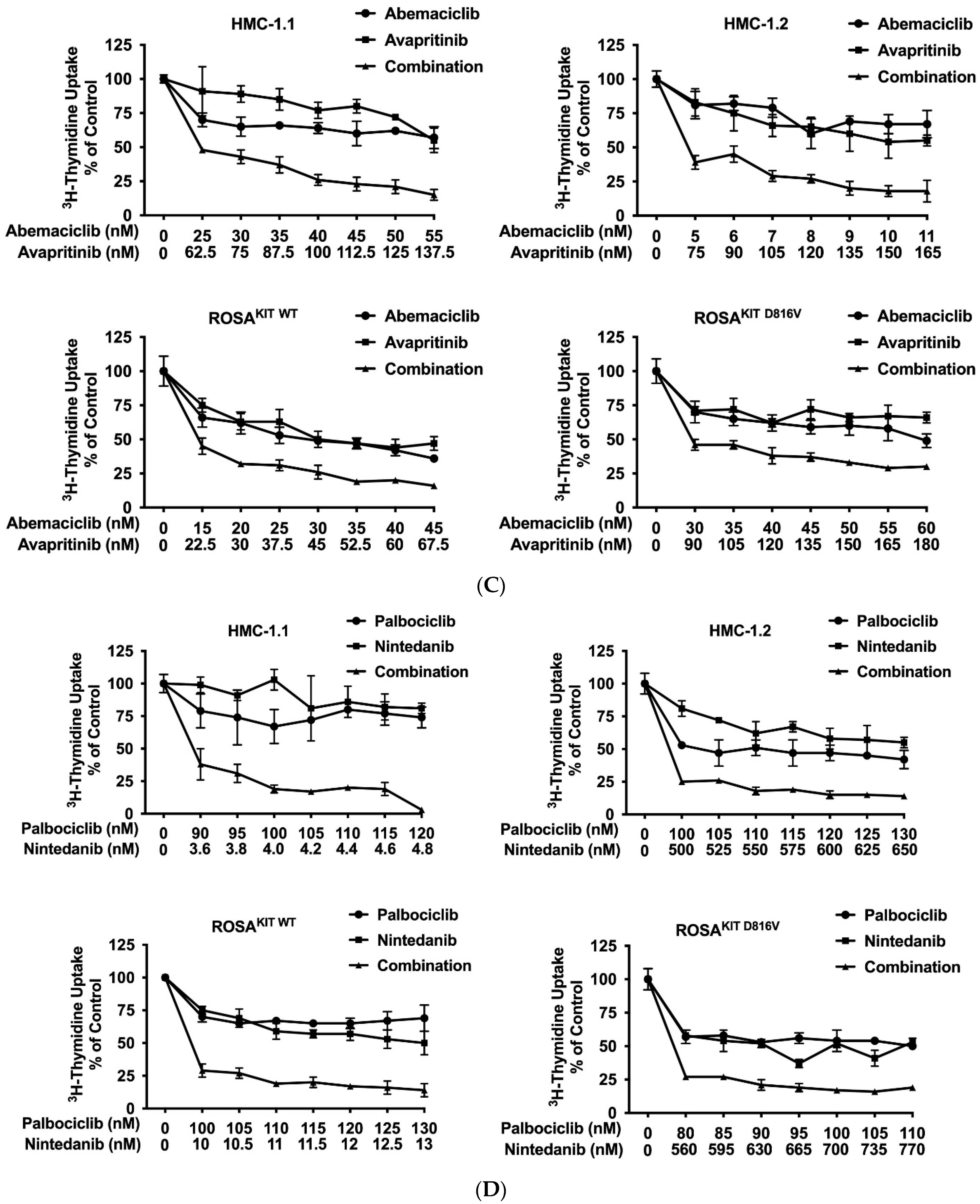
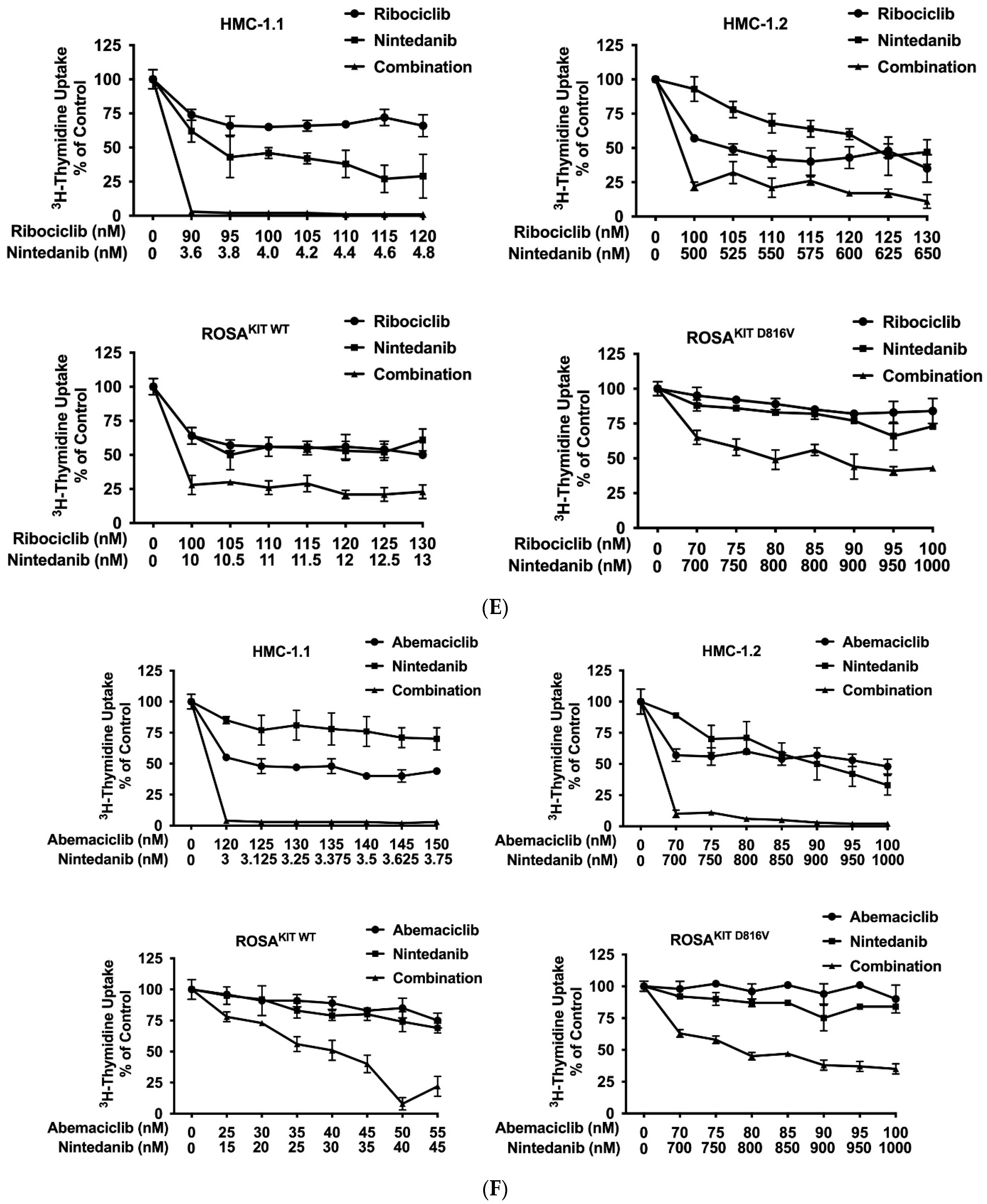
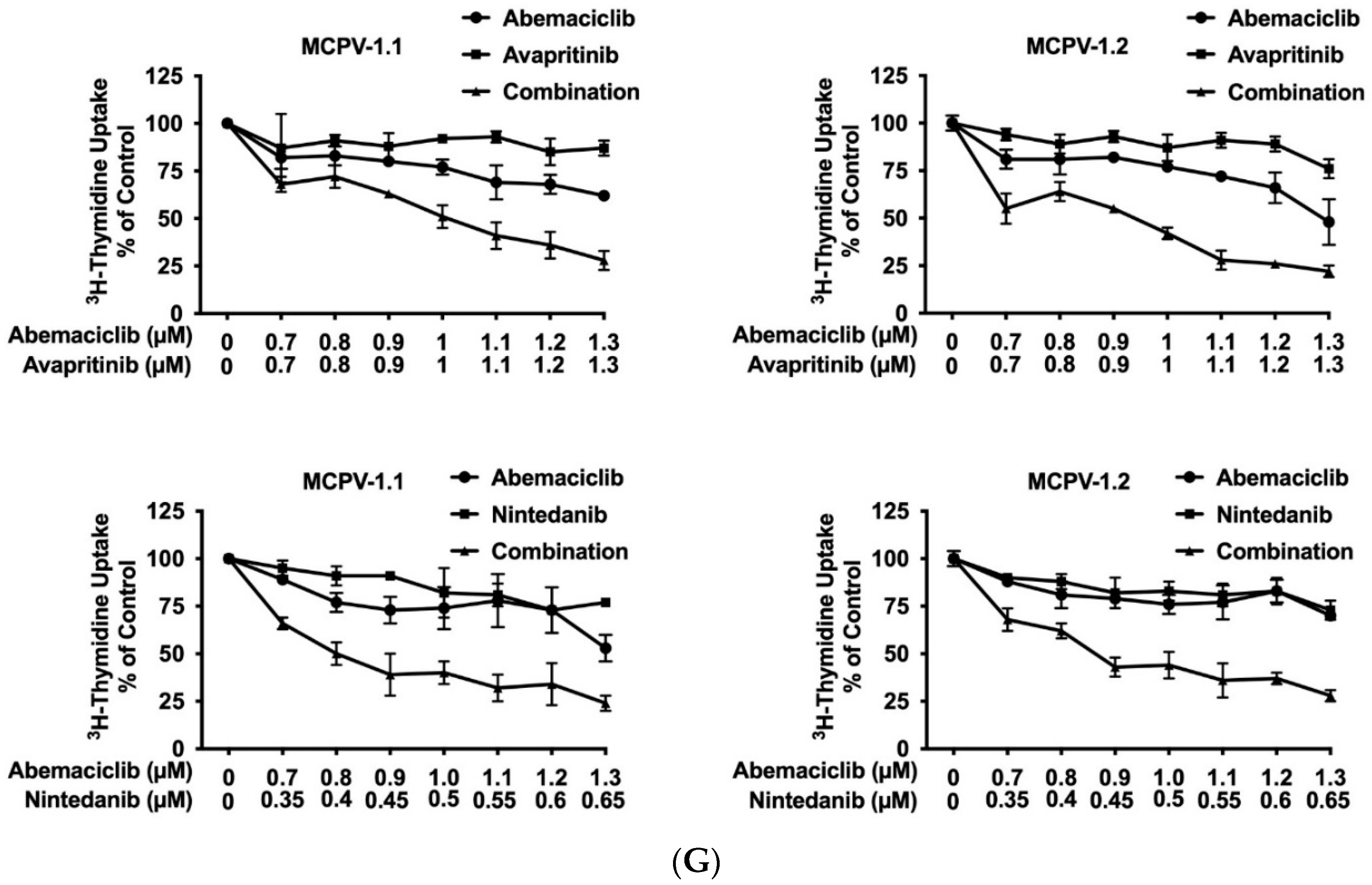
3.7. CDK4/CDK6 Inhibitors Synergize with the New KIT-Targeting Drugs Avapritinib and Nintedanib in Producing Growth Inhibition in Neoplastic MC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Metcalfe, D.D. Classification and diagnosis of mastocytosis: Current status. J. Investig. Dermatol. 1991, 96, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Horny, H.P.; Valent, P. Diagnosis of mastocytosis: General histopathological aspects, morphological criteria, and immunohistochemical findings. Leuk. Res. 2001, 25, 543–551. [Google Scholar] [CrossRef]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- Escribano, L.; Akin, C.; Castells, M.; Orfao, A.; Metcalfe, D.D. Mastocytosis: Current concepts in diagnosis and treatment. Ann. Hematol. 2002, 81, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Sperr, W.R.; Escribano, L.; Arock, M.; Horny, H.P.; Bennett, J.M.; Metcalfe, D.D. Aggressive systemic mastocytosis and related mast cell disorders: Current treatment options and proposed response criteria. Leuk. Res. 2003, 27, 635–641. [Google Scholar] [CrossRef]
- Arock, M.; Akin, C.; Hermine, O.; Valent, P. Current treatment options in patients with mastocytosis: Status in 2015 and future perspectives. Eur. J. Haematol. 2015, 94, 474–490. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Oude Elberink, J.N.G.; Gorska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y.; et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of the c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Kluin-Nelemans, H.C.; Oldhoff, J.M.; Van Doormaal, J.J.; Van ’t Wout, J.W.; Verhoef, G.; Gerrits, W.B.; van Dobbenburgh, O.A.; Pasmans, S.G.; Fijnheer, R. Cladribine therapy for systemic mastocytosis. Blood 2003, 102, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Brockow, K.; D’Ambrosio, C.; Kirshenbaum, A.S.; Ma, Y.; Longley, B.J.; Metcalfe, D.D. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp. Hematol. 2003, 31, 686–692. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Ustun, C.; Arock, M.; Kluin-Nelemans, H.C.; Reiter, A.; Sperr, W.R.; George, T.; Horny, H.P.; Hartmann, K.; Sotlar, K.; Damaj, G.; et al. Advanced systemic mastocytosis: From molecular and genetic progress to clinical practice. Haematologica 2016, 101, 1133–1143. [Google Scholar] [CrossRef]
- Gotlib, J. Tyrosine Kinase Inhibitors in the Treatment of Eosinophilic Neoplasms and Systemic Mastocytosis. Hematol. Oncol. Clin. N. Am. 2017, 31, 643–661. [Google Scholar] [CrossRef]
- Shomali, W.; Gotlib, J. The new tool “KIT” in advanced systemic mastocytosis. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 127–136. [Google Scholar] [CrossRef]
- Wu, C.P.; Lusvarghi, S.; Wang, J.C.; Hsiao, S.H.; Huang, Y.H.; Hung, T.H.; Ambudkar, S.V. Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines. Mol. Pharm. 2019, 16, 3040–3052. [Google Scholar] [CrossRef]
- Lübke, J.; Naumann, N.; Kluger, S.; Schwaab, J.; Metzgeroth, G.; Evans, E.; Gardino, A.K.; Lengauer, C.; Hofmann, W.K.; Fabarius, A.; et al. Inhibitory effects of midostaurin and avapritinib on myeloid progenitors derived from patients with KIT D816V positive advanced systemic mastocytosis. Leukemia 2019, 33, 1195–1205. [Google Scholar] [CrossRef]
- Peter, B.; Winter, G.E.; Blatt, K.; Bennett, K.L.; Stefanzl, G.; Rix, U.; Eisenwort, G.; Hadzijusufovic, E.; Gridling, M.; Dutreix, C.; et al. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia 2016, 30, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Hartmann, K.; George, T.I.; Sotlar, K.; Peter, B.; Gleixner, K.V.; Blatt, K.; Sperr, W.R.; Manley, P.W.; et al. Midostaurin: A magic bullet that blocks mast cell expansion and activation. Ann. Oncol. 2017, 28, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Gotlib, J.; Akin, C.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Menssen, H.D.; Redhu, S.; Knoll, S.; et al. Midostaurin improves quality of life and mediator-related symptoms in advanced systemic mastocytosis. J. Allergy Clin. Immunol. 2020, 146, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Winger, B.A.; Cortopassi, W.A.; Ruiz, D.G.; Ding, L.; Jang, K.; Leyte-Vidal, A.; Zhang, N.; Esteve-Puig, R.; Jacobson, M.P.; Shah, N.P. ATP-Competitive Inhibitors Midostaurin and Avapritinib Have Distinct Resistance Profiles in Exon 17-Mutant KIT. Cancer Res. 2019, 79, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; George, T.I.; Gotlib, J. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 2020, 135, 1365–1376. [Google Scholar] [CrossRef]
- Dhillon, S. Avapritinib: First Approval. Drugs 2020, 80, 433–439. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Radia, D.H.; George, T.I.; Robinson, W.A.; Quiery, A.T.; Drummond, M.W.; Bose, P.; Hexner, E.O.; Winton, E.F.; Horny, H.P.; et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: The phase 1 EXPLORER trial. Nat. Med. 2021, 27, 2183–2191. [Google Scholar] [CrossRef]
- Gotlib, J.; Reiter, A.; Radia, D.H.; Deininger, M.W.; George, T.I.; Panse, J.; Vannucchi, A.M.; Platzbecker, U.; Alvarez-Twose, I.; Mital, A.; et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: Interim analysis of the phase 2 PATHFINDER trial. Nat. Med. 2021, 27, 2192–2199. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Mayerhofer, M.; Sonneck, K.; Gruze, A.; Samorapoompichit, P.; Baumgartner, C.; Lee, F.Y.; Aichberger, K.J.; Manley, P.W.; Fabbro, D.; et al. Synergistic growth-inhibitory effects of two tyrosine kinase inhibitors, dasatinib and PKC412, on neoplastic mast cells expressing the D816V-mutated oncogenic variant of KIT. Haematologica 2007, 92, 1451–1459. [Google Scholar] [CrossRef][Green Version]
- Peter, B.; Gleixner, K.V.; Cerny-Reiterer, S.; Herrmann, H.; Winter, V.; Hadzijusufovic, E.; Ferenc, V.; Schuch, K.; Mirkina, I.; Horny, H.P.; et al. Polo-like kinase-1 as a novel target in neoplastic mast cells: Demonstration of growth-inhibitory effects of small interfering RNA and the Polo-like kinase-1 targeting drug BI 2536. Haematologica 2011, 96, 672–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gleixner, K.V.; Peter, B.; Blatt, K.; Suppan, V.; Reiter, A.; Radia, D.; Hadzijusufovic, E.; Valent, P. Synergistic growth-inhibitory effects of ponatinib and midostaurin (PKC412) on neoplastic mast cells carrying KIT D816V. Haematologica 2013, 98, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS ONE 2012, 7, e43090. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.M.; Maric, I.; Simakova, O.; Bai, Y.; Chan, E.C.; Olivares, N.; Carter, M.; Maric, D.; Robyn, J.; Metcalfe, D.D. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica 2011, 96, 459–463. [Google Scholar] [CrossRef]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Damaj, G.; Joris, M.; Chandesris, O.; Hanssens, K.; Soucie, E.; Canioni, D.; Kolb, B.; Durieu, I.; Gyan, E.; Livideanu, C.; et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suffering systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS ONE 2014, 9, e85362. [Google Scholar] [CrossRef] [PubMed]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lübke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef]
- Arock, M.; Sotlar, K.; Gotlib, J.; Sperr, W.R.; Hartmann, K.; Schwartz, L.B.; Akin, C.; Horny, H.P.; Valent, P. New developments in the field of mastocytosis and mast cell activation syndromes: A summary of the Annual Meeting of the European Competence Network on Mastocytosis (ECNM) 2019. Leuk. Lymphoma 2020, 61, 1075–1083. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Kumarasamy, V.; Witkiewicz, A.K.; Knudsen, E.S. Chemotherapy and CDK4/6 Inhibitors: Unexpected Bedfellows. Mol. Cancer Ther. 2020, 19, 1575–1588. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular pathways: Targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Laderian, B.; Fojo, T. CDK4/6 Inhibition as a therapeutic strategy in breast cancer: Palbociclib, ribociclib, and abemaciclib. Semin. Oncol. 2017, 44, 395–403. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.C.; Cai, S.; Ajamie, R.T.; Burke, T.; Beckmann, R.P.; Chan, E.M.; De Dios, A.; Wishart, G.N.; Gelbert, L.M.; Cronier, D.M. Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin. Cancer Res. 2014, 20, 3763–3774. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women with HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef]
- Scheicher, R.; Hoelbl-Kovacic, A.; Bellutti, F.; Tigan, A.S.; Prchal-Murphy, M.; Heller, G.; Schneckenleithner, C.; Salazar-Roa, M.; Zöchbauer-Müller, S.; Zuber, J.; et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 2015, 125, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Aleem, E.; Arceci, R.J. Targeting cell cycle regulators in hematologic malignancies. Front. Cell Dev. Biol. 2015, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Zeidner, J.F. Cyclin-dependent kinase (CDK) 9 and 4/6 inhibitors in acute myeloid leukemia (AML): A promising therapeutic approach. Expert Opin. Investig. Drugs 2019, 28, 989–1001. [Google Scholar] [CrossRef]
- Schneeweiss-Gleixner, M.; Byrgazov, K.; Stefanzl, G.; Berger, D.; Eisenwort, G.; Lucini, C.B.; Herndlhofer, S.; Preuner, S.; Obrova, K.; Pusic, P.; et al. CDK4/CDK6 inhibition as a novel strategy to suppress the growth and survival of BCR-ABL1T315I+ clones in TKI-resistant CML. EBioMedicine 2019, 50, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Schneeweiss, M.; Peter, B.; Bibi, S.; Eisenwort, G.; Smiljkovic, D.; Blatt, K.; Jawhar, M.; Berger, D.; Stefanzl, G.; Herndlhofer, S.; et al. The KIT and PDGFRA switch-control inhibitor DCC-2618 blocks growth and survival of multiple neoplastic cell types in advanced mastocytosis. Haematologica 2018, 103, 799–809. [Google Scholar] [CrossRef]
- Butterfield, J.H.; Weiler, D.; Dewald, G.; Gleich, G.J. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk. Res. 1988, 12, 345–355. [Google Scholar] [CrossRef]
- Saleh, R.; Wedeh, G.; Herrmann, H.; Bibi, S.; Cerny-Reiterer, S.; Sadovnik, I.; Blatt, K.; Hadzijusufovic, E.; Jeanningros, S.; Blanc, C.; et al. A new human mast cell line expressing a functional IgE receptor converts to factor-independence and tumorigenicity by KIT D816V-transfection. Blood 2014, 124, 111–120. [Google Scholar] [CrossRef]
- Hoermann, G.; Blatt, K.; Greiner, G.; Putz, E.M.; Berger, A.; Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Walz, C.; Hoetzenecker, K.; et al. CD52 is a molecular target in advanced systemic mastocytosis. FASEB J. 2014, 28, 3540–3551. [Google Scholar] [CrossRef]
- Greiner, G.; Witzeneder, N.; Berger, A.; Schmetterer, K.; Eisenwort, G.; Schiefer, A.I.; Roos, S.; Popow-Kraupp, T.; Müllauer, L.; Zuber, J.; et al. CCL2 is a KIT D816V-dependent modulator of the bone marrow microenvironment in systemic mastocytosis. Blood 2017, 129, 371–382. [Google Scholar] [CrossRef]
- Eisenwort, G.; Sadovnik, I.; Schwaab, J.; Jawhar, M.; Keller, A.; Stefanzl, G.; Berger, D.; Blatt, K.; Hoermann, G.; Bilban, M.; et al. Identification of a leukemia-initiating stem cell in human mast cell leukemia. Leukemia 2019, 33, 2673–2684. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinform. 2022, 24, S1672-0229. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997–3011. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneira, D.B.; Mayhew, C.N.; Thangavel, C.; Sotillo, E.; Reed, C.A.; Graña, X.; Knudsen, E.S. Proliferative suppression by CDK4/6 inhibition: Complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 2010, 138, 1920–1930. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1437. [Google Scholar] [CrossRef]
- Overed-Sayer, C.; Miranda, E.; Dunmore, R.; Liarte Marin, E.; Beloki, L.; Rassl, D.; Parfrey, H.; Carruthers, A.; Chahboub, A.; Koch, S.; et al. Inhibition of mast cells: A novel mechanism by which nintedanib may elicit anti-fibrotic effects. Thorax 2020, 75, 754–763. [Google Scholar] [CrossRef]
- Toledo, M.A.S.; Gatz, M.; Sontag, S.; Gleixner, K.V.; Eisenwort, G.; Feldberg, K.; Hamouda, A.E.I.; Kluge, F.; Guareschi, R.; Rossetti, G.; et al. Nintedanib Targets KIT D816V Neoplastic Cells Derived from Induced Pluripotent Stem cells of Systemic Mastocytosis. Blood 2021, 137, 2070–2084. [Google Scholar] [CrossRef]
- Hayes, T.K.; Luo, F.; Cohen, O.; Goodale, A.B.; Lee, Y.; Pantel, S.; Bagul, M.; Piccioni, F.; Root, D.E.; Garraway, L.A.; et al. A Functional Landscape of Resistance to MEK1/2 and CDK4/6 Inhibition in NRAS-Mutant Melanoma. Cancer Res. 2019, 79, 2352–2366. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK4K/6 inhibitor palbociclib (PD0D332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Q.; Pan, X.H.; Wang, T.T.; Wang, J.; Yang, B.; He, Q.J.; Ding, L. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol. Sin. 2021, 42, 171–178. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Age | m/f | Diagnosis, SM Variant | KIT D816V | Serum Tryptase ng/mL | BM MC Infiltration % | Palbociclib IC50 (nM) | Ribociclib IC50 (nM) | Abemaciclib IC50 (nM) |
|---|---|---|---|---|---|---|---|---|---|
| #1 | 61 | f | ISM | + | 25.4 | 10 | 12.8 | n.t. | n.t. |
| #2 | 42 | f | ISM | + | 22.7 | 2 | 201.2 | 148.3 | 109.2 |
| #3.1 | 61 | m | ISM | + | 17.5 | 3 | 7.2 | 16.7 | 4.5 |
| #3.2 | 62 | m | ISM-AML | + | 26.2 | 5 | 40.3 | 200.2 | 55.2 |
| #4.1 | 70 | m | ISM | + | 83.3 | 20 | 5.7 | n.t. | n.t. |
| #4.2 * | 70 | m | ASM | + | 119 | 5 | 94.4 | 362.1 | n.t. |
| #4.3 | 70 | m | MCL | + | 94.2 | 20 | 12.0 | 252.6 | 13.8 |
| #5 * | 78 | m | ASM-MPN-eo | + | 45.9 | 10 | 67.5 | n.t. | n.t. |
| #6 | 78 | m | ASM-MDS/MPN-u | + | 101.0 | 50 | 4.2 | 75.4 | 159.4 |
| #7 | 75 | m | ASM-MDS/MPN-u | + | 377.0 | 30 | 163.4 | n.t. | n.t. |
| #8 * | 63 | m | ASM-AML | + | 16.5 | 10 | 181.8 | n.t. | n.t. |
| #9 | 91 | m | MCL | + | 49.6 | 10 | 10.4 | n.t. | n.t. |
| #10 | 86 | m | MCL-MDS | + | 180.0 | 30 | 16.8 | 193.9 | 23.7 |
| #11 | 80 | f | ISM | + | 82.9 | 30 | 6.5 | 38.0 | 19.9 |
| #12 | 70 | m | ASM | + | 650.0 | 50 | 81.6 | 119.9 | 135.4 |
| #13 | 57 | m | ISM | + | 85.4 | 5 | 37.0 | 261.7 | 16.9 |
| #14.1 | 70 | m | SM-CMML | + | 374.0 | 20 | 2.6 | 65.8 | 11.1 |
| #14.2 | 70 | m | SM-CMML | + | 66.2 | 2 | 0.25 | 408.0 | 60.4 |
| #15 | 59 | m | MCL | + | 250.0 | 60 | 43.9 | 65.5 | 98.4 |
| #16 | 48 | m | SM-CMML | + | 670.0 | 60 | 28.2 | 134.4 | 33.3 |
| #17 * | 77 | m | SM-AML | + | 30.0 | 10 | n.t. | n.t. | n.t. |
| Cell Line/Cell Type | Palbociclib, IC50 | Ribociclib, IC50 | Abemaciclib, IC50 |
|---|---|---|---|
| HMC-1.1 | 92.6 ± 10.3 nM | 245.6 ± 33.4 nM | 174.5 ± 20.9 nM |
| HMC-1.2 | 296.1 ± 22.3 nM | 703.8 ± 155.0 nM | 143.8 ± 14.0 nM |
| ROSAKIT WT | 35.1 ± 2.2 nM | 262.0 ± 9.5 nM | 48.4 ± 3.5 nM |
| ROSAKIT D816V | 123.0 ± 13.1 nM | 303.3 ± 14.9 nM | 134.1 ± 5.1 nM |
| MCPV-1.1 | >10 µM | >10 µM | 1.48 ± 0.2 µM |
| MCPV-1.2 | >10 µM | >10 µM | 0.9 ± 0.07 µM |
| MCPV-1.3 | >10 µM | >10 µM | 2.83 ± 0.43 µM |
| MCPV-1.4 | >10 µM | >10 µM | 1.32 ± 0.16 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneeweiss-Gleixner, M.; Filik, Y.; Stefanzl, G.; Berger, D.; Sadovnik, I.; Bauer, K.; Smiljkovic, D.; Eisenwort, G.; Witzeneder, N.; Greiner, G.; et al. CDK4/CDK6 Inhibitors Synergize with Midostaurin, Avapritinib, and Nintedanib in Inducing Growth Inhibition in KIT D816V+ Neoplastic Mast Cells. Cancers 2022, 14, 3070. https://doi.org/10.3390/cancers14133070
Schneeweiss-Gleixner M, Filik Y, Stefanzl G, Berger D, Sadovnik I, Bauer K, Smiljkovic D, Eisenwort G, Witzeneder N, Greiner G, et al. CDK4/CDK6 Inhibitors Synergize with Midostaurin, Avapritinib, and Nintedanib in Inducing Growth Inhibition in KIT D816V+ Neoplastic Mast Cells. Cancers. 2022; 14(13):3070. https://doi.org/10.3390/cancers14133070
Chicago/Turabian StyleSchneeweiss-Gleixner, Mathias, Yüksel Filik, Gabriele Stefanzl, Daniela Berger, Irina Sadovnik, Karin Bauer, Dubravka Smiljkovic, Gregor Eisenwort, Nadine Witzeneder, Georg Greiner, and et al. 2022. "CDK4/CDK6 Inhibitors Synergize with Midostaurin, Avapritinib, and Nintedanib in Inducing Growth Inhibition in KIT D816V+ Neoplastic Mast Cells" Cancers 14, no. 13: 3070. https://doi.org/10.3390/cancers14133070
APA StyleSchneeweiss-Gleixner, M., Filik, Y., Stefanzl, G., Berger, D., Sadovnik, I., Bauer, K., Smiljkovic, D., Eisenwort, G., Witzeneder, N., Greiner, G., Hoermann, G., Schiefer, A.-I., Schwaab, J., Jawhar, M., Reiter, A., Sperr, W. R., Arock, M., Valent, P., & Gleixner, K. V. (2022). CDK4/CDK6 Inhibitors Synergize with Midostaurin, Avapritinib, and Nintedanib in Inducing Growth Inhibition in KIT D816V+ Neoplastic Mast Cells. Cancers, 14(13), 3070. https://doi.org/10.3390/cancers14133070