
GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mutational Hotspots and Other Less Recurrent Described Mutations in GNAQ and GNA11 Genes
2.1. G Protein-Coupled Receptors
2.2. Mitogen-Activated Protein Kinase
2.3. Protein Kinase C
2.4. Phosphatidylinositol-3 Kinase/Akt
2.5. YAP and Its Upstream Triggers
3. Prognostic Value of GNAQ and GNA11 Mutated Genes in Primary Uveal Melanoma
Possible Relation between GNAQ and GNA11 Genes in Inflammation and HLA Expression in Uveal Melanoma
4. Prevalence of GNAQ and GNA11 Mutations in Metastatic Uveal Melanoma
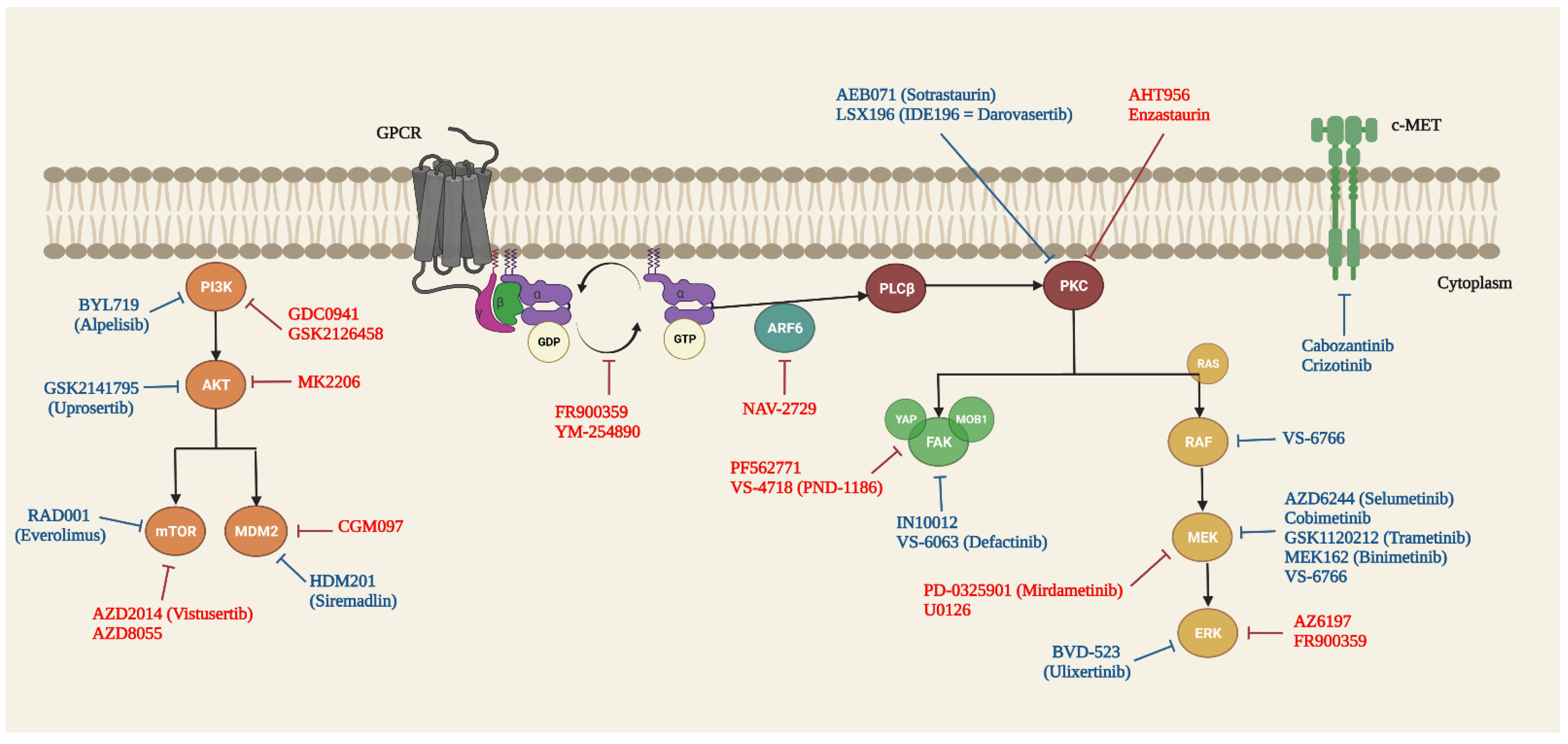
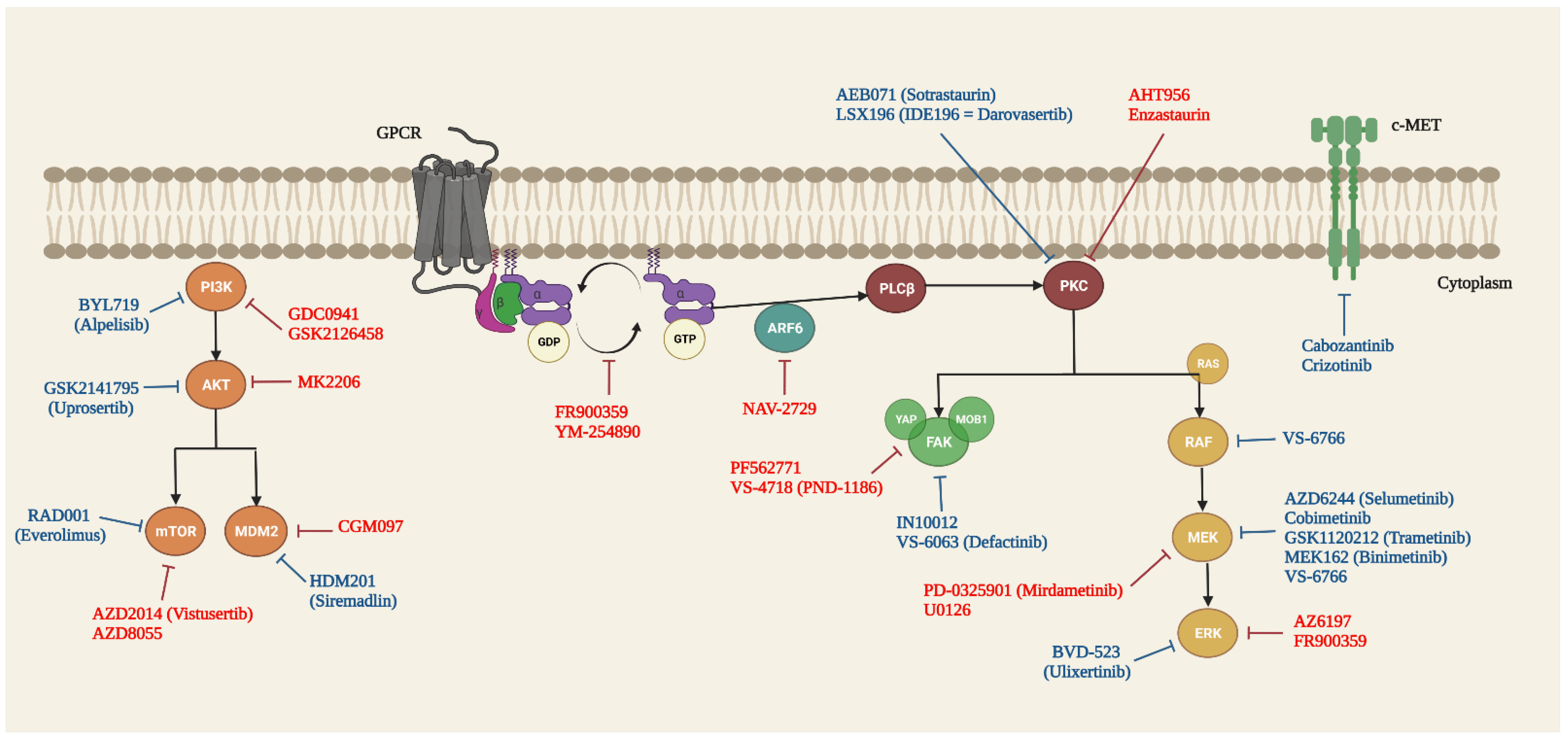
5. Targeted Therapy of GNAQ and GNA11 Mutations in UM
5.1. Guanine Nucleotide Dissociation Inhibitors (GDI)
5.2. RAS/RAF/MEK/ERK/MAPK Signalling Pathway
5.3. PLCβ/PKC Signalling Pathway
5.4. Hippo/YAP Signalling Pathway
5.5. PI3K/AKT/mTOR Signalling Pathway
5.6. Other Targets and Signalling Pathways
6. Conclusions
7. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Virgili, G.; Gatta, G.; Ciccolallo, L.; Capocaccia, R.; Biggeri, A.; Crocetti, E.; Lutz, J.M.; Paci, E.; Group, E.W. Incidence of uveal melanoma in Europe. Ophthalmology 2007, 114, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. reviews. Dis. primers 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Dogrusoz, M.; Brouwer, N.J.; de Geus, S.J.R.; Ly, L.V.; Bohringer, S.; van Duinen, S.G.; Kroes, W.G.M.; van der Velden, P.A.; Haasnoot, G.W.; Marinkovic, M.; et al. Prognostic Factors Five Years After Enucleation for Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer cell 2018, 33, 151. [Google Scholar] [CrossRef] [Green Version]
- Cruz, F., 3rd; Rubin, B.P.; Wilson, D.; Town, A.; Schroeder, A.; Haley, A.; Bainbridge, T.; Heinrich, M.C.; Corless, C.L. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003, 63, 5761–5766. [Google Scholar]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jockel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [CrossRef]
- Kilic, E.; van Gils, W.; Lodder, E.; Beverloo, H.B.; van Til, M.E.; Mooy, C.M.; Paridaens, D.; de Klein, A.; Luyten, G.P. Clinical and cytogenetic analyses in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3703–3707. [Google Scholar] [CrossRef]
- Scholes, A.G.; Damato, B.E.; Nunn, J.; Hiscott, P.; Grierson, I.; Field, J.K. Monosomy 3 in uveal melanoma: Correlation with clinical and histologic predictors of survival. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1008–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallico, M.; Raciti, G.; Longo, A.; Reibaldi, M.; Bonfiglio, V.; Russo, A.; Caltabiano, R.; Gattuso, G.; Falzone, L.; Avitabile, T. Current molecular and clinical insights into uveal melanoma (Review). Int. J. Oncol. 2021, 58. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Besaratinia, A.; Pfeifer, G.P. Uveal melanoma and GNA11 mutations: A new piece added to the puzzle. Pigment. cell melanoma Res. 2011, 24, 18–20. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Carvajal, R.D. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014, 24, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Vader, M.J.C.; Madigan, M.C.; Versluis, M.; Suleiman, H.M.; Gezgin, G.; Gruis, N.A.; Out-Luiting, J.J.; Bergman, W.; Verdijk, R.M.; Jager, M.J.; et al. GNAQ and GNA11 mutations and downstream YAP activation in choroidal nevi. Br. J. cancer 2017, 117, 884–887. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. New Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Patient survival in uveal melanoma is not affected by oncogenic mutations in GNAQ and GNA11. Br. J. cancer 2013, 109, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Kilic, E.; Vaarwater, J.; Bastian, B.C.; Garbe, C.; de Klein, A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br. J. cancer 2009, 101, 813–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griewank, K.G.; van de Nes, J.; Schilling, B.; Moll, I.; Sucker, A.; Kakavand, H.; Haydu, L.E.; Asher, M.; Zimmer, L.; Hillen, U.; et al. Genetic and clinico-pathologic analysis of metastatic uveal melanoma. Mod. Pathol. 2014, 27, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapadula, D.; Farias, E.; Randolph, C.E.; Purwin, T.J.; McGrath, D.; Charpentier, T.H.; Zhang, L.; Wu, S.; Terai, M.; Sato, T.; et al. Effects of Oncogenic Galphaq and Galpha11 Inhibition by FR900359 in Uveal Melanoma. Mol Cancer Res 2019, 17, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Makepeace, C.M.; Kaltenbronn, K.M.; Choi, J.; Hernandez-Aya, L.; Weilbaecher, K.N.; Piggott, K.D.; Rao, P.K.; Yuede, C.M.; Dixon, A.J.; et al. Targeting primary and metastatic uveal melanoma with a G protein inhibitor. J. Biol. Chem. 2021, 296, 100403. [Google Scholar] [CrossRef]
- Hitchman, T.D.; Bayshtok, G.; Ceraudo, E.; Moore, A.R.; Lee, C.; Jia, R.; Wang, N.; Pachai, M.R.; Shoushtari, A.N.; Francis, J.H.; et al. Combined Inhibition of Galphaq and MEK Enhances Therapeutic Efficacy in Uveal Melanoma. Clin. cancer Res. 2021, 27, 1476–1490. [Google Scholar] [CrossRef]
- Neelature Sriramareddy, S.; Smalley, K.S.M. MEK-ing the Most of It: Strategies to Co-target Galphaq and MAPK in Uveal Melanoma. Clin. cancer Res. 2021, 27, 1217–1219. [Google Scholar] [CrossRef]
- Sharma, A.; Stei, M.M.; Frohlich, H.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961. [Google Scholar] [CrossRef]
- Schneider, B.; Riedel, K.; Zhivov, A.; Huehns, M.; Zettl, H.; Guthoff, R.F.; Junemann, A.; Erbersdobler, A.; Zimpfer, A. Frequent and Yet Unreported GNAQ and GNA11 Mutations are Found in Uveal Melanomas. Pathol. Oncol. Res. 2019, 25, 1319–1325. [Google Scholar] [CrossRef]
- Populo, H.; Vinagre, J.; Lopes, J.M.; Soares, P. Analysis of GNAQ mutations, proliferation and MAPK pathway activation in uveal melanomas. Br. J. Ophthalmol. 2011, 95, 715–719. [Google Scholar] [CrossRef]
- Silva-Rodriguez, P.; Bande, M.; Fernandez-Diaz, D.; Lago-Baameiro, N.; Pardo, M.; Jose Blanco-Teijeiro, M.; Dominguez, F.; Loidi, L.; Pineiro, A. Role of somatic mutations and chromosomal aberrations in the prognosis of uveal melanoma in a Spanish patient cohort. Acta Ophthalmol 2021. [Google Scholar] [CrossRef]
- Psinakis, F.; Katseli, A.; Koutsandrea, C.; Frangia, K.; Florentin, L.; Apostolopoulou, D.; Dimakopoulou, K.; Papakonstantinou, D.; Georgopoulou, E.; Brouzas, D. Uveal Melanoma: GNAQ and GNA11 Mutations in a Greek Population. Anticancer Res. 2017, 37, 5719–5726. [Google Scholar] [CrossRef] [PubMed]
- Ominato, J.; Fukuchi, T.; Sato, A.; Yamaguchi, N.; Kobayashi, K.; Cho, H.; Oyama, T.; Ajioka, Y. The Role of Mutation Rates of GNAQ or GNA11 in Cases of Uveal Melanoma in Japan. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wei, W.B.; Li, B.; Gao, F.; Zhang, Z.; Jonas, J.B. Oncogenic GNAQ and GNA11 mutations in uveal melanoma in Chinese. PloS ONE 2014, 9, e109699. [Google Scholar] [CrossRef] [PubMed]
- Cerne, J.Z.; Hartig, S.M.; Hamilton, M.P.; Chew, S.A.; Mitsiades, N.; Poulaki, V.; McGuire, S.E. Protein kinase C inhibitors sensitize GNAQ mutant uveal melanoma cells to ionizing radiation. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2130–2139. [Google Scholar] [CrossRef] [Green Version]
- Terai, M.; Shimada, A.; Chervoneva, I.; Hulse, L.; Danielson, M.; Swensen, J.; Orloff, M.; Wedegaertner, P.B.; Benovic, J.L.; Aplin, A.E.; et al. Prognostic Values of G-Protein Mutations in Metastatic Uveal Melanoma. Cancers 2021, 13, 5794. [Google Scholar] [CrossRef]
- Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.M.; Mascarelli, F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: Interaction with B-Raf/ERK. Investig. Ophthalmol. Vis. Sci. 2010, 51, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Moleirinho, S.; Hoxha, S.; Mandati, V.; Curtale, G.; Troutman, S.; Ehmer, U.; Kissil, J.L. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. eLife 2017, 6. [Google Scholar] [CrossRef]
- Rimoldi, D.; Salvi, S.; Lienard, D.; Lejeune, F.J.; Speiser, D.; Zografos, L.; Cerottini, J.C. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003, 63, 5712–5715. [Google Scholar]
- Lee, C.H.; Park, D.; Wu, D.; Rhee, S.G.; Simon, M.I. Members of the Gq alpha subunit gene family activate phospholipase C beta isozymes. J. Biol. Chem. 1992, 267, 16044–16047. [Google Scholar] [CrossRef]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef]
- Mouti, M.A.; Dee, C.; Coupland, S.E.; Hurlstone, A.F. Minimal contribution of ERK1/2-MAPK signalling towards the maintenance of oncogenic GNAQQ209P-driven uveal melanomas in zebrafish. Oncotarget 2016, 7, 39654–39670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boru, G.; Cebulla, C.M.; Sample, K.M.; Massengill, J.B.; Davidorf, F.H.; Abdel-Rahman, M.H. Heterogeneity in Mitogen-Activated Protein Kinase (MAPK) Pathway Activation in Uveal Melanoma With Somatic GNAQ and GNA11 Mutations. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2474–2480. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhu, M.; Fletcher, J.A.; Giobbie-Hurder, A.; Hodi, F.S. The protein kinase C inhibitor enzastaurin exhibits antitumor activity against uveal melanoma. PloS ONE 2012, 7, e29622. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, J.; Zhu, M.; Fletcher, J.A.; Hodi, F.S. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol. cancer Ther. 2012, 11, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, Q.; Dang, K.; Ma, S.; Cotton, J.L.; Yang, S.; Zhu, L.J.; Deng, A.C.; Ip, Y.T.; Johnson, R.L.; et al. YAP/TAZ Activation Drives Uveal Melanoma Initiation and Progression. Cell Rep. 2019, 29, 3200–3211 e3204. [Google Scholar] [CrossRef] [Green Version]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Zembowicz, A.; Mihm, M.C. Dermal dendritic melanocytic proliferations: An update. Histopathol. 2004, 45, 433–451. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Fitch, K.R.; Fuchs, H.; de Angelis, M.H.; Barsh, G.S. Effects of G-protein mutations on skin color. Nat. Genet. 2004, 36, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Daniels, A.B.; Lee, J.E.; MacConaill, L.E.; Palescandolo, E.; Van Hummelen, P.; Adams, S.M.; DeAngelis, M.M.; Hahn, W.C.; Gragoudas, E.S.; Harbour, J.W.; et al. High throughput mass spectrometry-based mutation profiling of primary uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6991–6996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Dono, M.; Angelini, G.; Cecconi, M.; Amaro, A.; Esposito, A.I.; Mirisola, V.; Maric, I.; Lanza, F.; Nasciuti, F.; Viaggi, S.; et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: Detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br. J. cancer 2014, 110, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Ewens, K.G.; Kanetsky, P.A.; Richards-Yutz, J.; Purrazzella, J.; Shields, C.L.; Ganguly, T.; Ganguly, A. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5160–5167. [Google Scholar] [CrossRef] [Green Version]
- Decatur, C.L.; Ong, E.; Garg, N.; Anbunathan, H.; Bowcock, A.M.; Field, M.G.; Harbour, J.W. Driver Mutations in Uveal Melanoma: Associations With Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016, 134, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer cell 2017, 32, 204–220 e215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staby, K.M.; Gravdal, K.; Mork, S.J.; Heegaard, S.; Vintermyr, O.K.; Krohn, J. Prognostic impact of chromosomal aberrations and GNAQ, GNA11 and BAP1 mutations in uveal melanoma. Acta Ophthalmol 2018, 96, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.; Rice, M.; Toomey, S.; Horgan, N.; Hennessey, B.T.; Larkin, A. An insight into the molecular genetics of a uveal melanoma patient cohort. J. cancer Res. Clin. Oncol. 2018, 144, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Smit, K.N.; van Poppelen, N.M.; Vaarwater, J.; Verdijk, R.; van Marion, R.; Kalirai, H.; Coupland, S.E.; Thornton, S.; Farquhar, N.; Dubbink, H.J.; et al. Combined mutation and copy-number variation detection by targeted next-generation sequencing in uveal melanoma. Mod. Pathol. 2018, 31, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Afshar, A.R.; Damato, B.E.; Stewart, J.M.; Zablotska, L.B.; Roy, R.; Olshen, A.B.; Joseph, N.M.; Bastian, B.C. Next-Generation Sequencing of Uveal Melanoma for Detection of Genetic Alterations Predicting Metastasis. Transl. Vis. Sci. Technol. 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Piaggio, F.; Tozzo, V.; Bernardi, C.; Croce, M.; Puzone, R.; Viaggi, S.; Patrone, S.; Barla, A.; Coviello, D.; Jager, M.J.; et al. Secondary Somatic Mutations in G-Protein-Related Pathways and Mutation Signatures in Uveal Melanoma. Cancers 2019, 11, 1688. [Google Scholar] [CrossRef] [Green Version]
- Thornton, S.; Coupland, S.E.; Olohan, L.; Sibbring, J.S.; Kenny, J.G.; Hertz-Fowler, C.; Liu, X.; Haldenby, S.; Heimann, H.; Hussain, R.; et al. Targeted Next-Generation Sequencing of 117 Routine Clinical Samples Provides Further Insights into the Molecular Landscape of Uveal Melanoma. Cancers 2020, 12, 1039. [Google Scholar] [CrossRef] [Green Version]
- Isaacson, A.L.; Sompallae, R.R.; Guseva, N.V.; Bellizzi, A.M.; Bossler, A.D.; Ma, D. Genomic Profiling of Metastatic Uveal Melanoma Shows Frequent Coexisting BAP1 or SF3B1 and GNAQ/GNA11 Mutations and Correlation With Prognosis. Am. J. Clin. Pathol. 2022. [Google Scholar] [CrossRef]
- Piaggio, F.; Croce, M.; Reggiani, F.; Monti, P.; Bernardi, C.; Ambrosio, M.; Banelli, B.; Dogrusoz, M.; Jockers, R.; Bordo, D.; et al. In uveal melanoma Galpha-protein GNA11 mutations convey a shorter disease-specific survival and are more strongly associated with loss of BAP1 and chromosomal alterations than Galpha-protein GNAQ mutations. Eur. J. cancer 2022, 170, 27–41. [Google Scholar] [CrossRef]
- van Weeghel, C.; Wierenga, A.P.A.; Versluis, M.; van Hall, T.; van der Velden, P.A.; Kroes, W.G.M.; Pfeffer, U.; Luyten, G.P.M.; Jager, M.J. Do GNAQ and GNA11 Differentially Affect Inflammation and HLA Expression in Uveal Melanoma? Cancers 2019, 11, 1127. [Google Scholar] [CrossRef] [Green Version]
- Triozzi, P.L.; Eng, C.; Singh, A.D. Targeted therapy for uveal melanoma. Cancer Treat. Rev. 2008, 34, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallone, F.; Sacchetti, M.; Lambiase, A.; Moramarco, A. Molecular Insights and Emerging Strategies for Treatment of Metastatic Uveal Melanoma. Cancers 2020, 12, 2761. [Google Scholar] [CrossRef] [PubMed]
- Seedor, R.S.; Orloff, M.; Sato, T. Genetic Landscape and Emerging Therapies in Uveal Melanoma. Cancers 2021, 13, 5503. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Lin, V.; Toumi, E.; Wang, K.; Zhu, H.; Conway, R.M.; Madigan, M.C.; Murray, M.; Cherepanoff, S.; Zhou, F.; et al. Development of new therapeutic options for the treatment of uveal melanoma. FEBS J. 2021, 288, 6226–6249. [Google Scholar] [CrossRef]
- Zhang, H.; Nielsen, A.L.; Boesgaard, M.W.; Harpsoe, K.; Daly, N.L.; Xiong, X.F.; Underwood, C.R.; Haugaard-Kedstrom, L.M.; Brauner-Osborne, H.; Gloriam, D.E.; et al. Structure-activity relationship and conformational studies of the natural product cyclic depsipeptides YM-254890 and FR900359. Eur J. Med. Chem 2018, 156, 847–860. [Google Scholar] [CrossRef]
- Schrage, R.; Schmitz, A.L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Bullesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. [Google Scholar] [CrossRef] [Green Version]
- Onken, M.D.; Makepeace, C.M.; Kaltenbronn, K.M.; Kanai, S.M.; Todd, T.D.; Wang, S.; Broekelmann, T.J.; Rao, P.K.; Cooper, J.A.; Blumer, K.J. Targeting nucleotide exchange to inhibit constitutively active G protein alpha subunits in cancer cells. Sci Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Nagai, K.; Arao, N.; Kawasaki, T.; Saito, T.; Moritani, Y.; Takasaki, J.; Hayashi, K.; Fujita, S.; Suzuki, K.; et al. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J. Antibiot (Tokyo) 2003, 56, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.F.; Zhang, H.; Boesgaard, M.W.; Underwood, C.R.; Brauner-Osborne, H.; Stromgaard, K. Structure-Activity Relationship Studies of the Natural Product Gq/11 Protein Inhibitor YM-254890. ChemMedChem. 2019, 14, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Weng, L.; Bastian, B.C.; Chen, X. Functional characterization of uveal melanoma oncogenes. Oncogene 2021, 40, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Larribere, L.; Utikal, J. Update on GNA Alterations in Cancer: Implications for Uveal Melanoma Treatment. Cancers 2020, 12, 1524. [Google Scholar] [CrossRef] [PubMed]
- Zuidervaart, W.; van Nieuwpoort, F.; Stark, M.; Dijkman, R.; Packer, L.; Borgstein, A.M.; Pavey, S.; van der Velden, P.; Out, C.; Jager, M.J.; et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br. J. cancer 2005, 92, 2032–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, G.; Pratilas, C.A.; Qin, L.X.; Tadi, M.; Surriga, O.; Carvajal, R.D.; Schwartz, G.K. Identification of unique MEK-dependent genes in GNAQ mutant uveal melanoma involved in cell growth, tumor cell invasion, and MEK resistance. Clin. cancer Res. 2012, 18, 3552–3561. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Bastholt, L.; Robert, C.; Sosman, J.; Larkin, J.; Hersey, P.; Middleton, M.; Cantarini, M.; Zazulina, V.; Kemsley, K.; et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. cancer Res. 2012, 18, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. Jama 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Schwartz, G.K.; Mann, H.; Smith, I.; Nathan, P.D. Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC cancer 2015, 15, 467. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Decaudin, D.; El Botty, R.; Diallo, B.; Massonnet, G.; Fleury, J.; Naguez, A.; Raymondie, C.; Davies, E.; Smith, A.; Wilson, J.; et al. Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget 2018, 9, 21674–21686. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet. Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.; Stang, S.L.; Zheng, Y.; Beswick, N.S.; Stone, J.C. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 2003, 102, 1414–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer cell 2017, 31, 685–696 e686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperno-Neumann, S.; Kapiteijn, E.; Larkin, J.M.G.; Carvajal, R.D.; Luke, J.J.; Seifert, H.; Roozen, I.; Zoubir, M.; Yang, L.; Choudhury, S.; et al. Phase I dose-escalation study of the protein kinase C (PKC) inhibitor AEB071 in patients with metastatic uveal melanoma. J. Clin. Oncol. 2014, 32, 9030. [Google Scholar] [CrossRef]
- Sagoo, M.S.; Harbour, J.W.; Stebbing, J.; Bowcock, A.M. Combined PKC and MEK inhibition for treating metastatic uveal melanoma. Oncogene 2014, 33, 4722–4723. [Google Scholar] [CrossRef] [Green Version]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Nemati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget 2016, 7, 33542–33556. [Google Scholar] [CrossRef] [Green Version]
- Kapiteijn, E.; Carlino, M.; Boni, V.; Loirat, D.; Speetjens, F.; Park, J.; Calvo, E.; Carvajal, R.; Nyakas, M.; Gonzalez-Maffe, J.; et al. Abstract CT068: A Phase I trial of LXS196, a novel PKC inhibitor for metastatic uveal melanoma. Cancer Res. 2019, 79 (Suppl. 13), CT068. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; Konig, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer cell 2019, 35, 457–472 e455. [Google Scholar] [CrossRef] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. reviews. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagi, C.M.; Christensen, J.; Cohen, D.P.; Roberts, W.G.; Wilkie, D.; Swanson, T.; Tuthill, T.; Andresen, C.J. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol. Ther. 2009, 8, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, J.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Honda, T.; Kamat, A.A.; Han, L.Y.; Kim, T.J.; Lu, C.; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007, 67, 10976–10983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, J.S.; Acosta, M.; Saddawi-Konefka, R.; Kishore, A.; Gomes, F.; Arang, N.; Tiago, M.; Coma, S.; Lubrano, S.; Wu, X.; et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clin. cancer Res. 2021, 27, 3190–3200. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Nemati, F.; Gentien, D.; Nicolas, A.; Dumont, A.; Carita, G.; Camonis, J.; Desjardins, L.; Cassoux, N.; Piperno-Neumann, S.; et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol. Oncol. 2014, 8, 1508–1520. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Ong, L.T.; Schoder, H.; Singh-Kandah, S.; Abbate, K.T.; Postow, M.A.; Callahan, M.K.; Wolchok, J.; Chapman, P.B.; Panageas, K.S.; et al. A phase 2 trial of everolimus and pasireotide long-acting release in patients with metastatic uveal melanoma. Melanoma Res. 2016, 26, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Musi, E.; Ambrosini, G.; de Stanchina, E.; Schwartz, G.K. The phosphoinositide 3-kinase alpha selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol. cancer Ther. 2014, 13, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Khan, S.; Komatsubara, K.; Feun, L.; Acquavella, N.; Singh-Kandah, S.; Negri, T.; Nesson, A.; Abbate, K.; Cremers, S.; et al. A Phase Ib Study of Sotrastaurin, a PKC Inhibitor, and Alpelisib, a PI3Kalpha Inhibitor, in Patients with Metastatic Uveal Melanoma. Cancers 2021, 13, 5504. [Google Scholar] [CrossRef]
- Ho, A.L.; Musi, E.; Ambrosini, G.; Nair, J.S.; Deraje Vasudeva, S.; de Stanchina, E.; Schwartz, G.K. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PloS ONE 2012, 7, e40439. [Google Scholar] [CrossRef] [Green Version]
- Khalili, J.S.; Yu, X.; Wang, J.; Hayes, B.C.; Davies, M.A.; Lizee, G.; Esmaeli, B.; Woodman, S.E. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin. cancer Res. 2012, 18, 4345–4355. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.H.; Shi, D.S.; Grossmann, A.H.; Sorensen, L.K.; Tong, Z.; Mleynek, T.M.; Rogers, A.; Zhu, W.; Richards, J.R.; Winter, J.M.; et al. ARF6 Is an Actionable Node that Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer cell 2016, 29, 889–904. [Google Scholar] [CrossRef] [Green Version]
- Daud, A.; Kluger, H.M.; Edelman, G.; Gordon, M.S.; Schimmoller, F.; Weitzman, A.; Samuel, T.A.; Moussa, A.H.; Flaherty, K.; Shapiro, G. Activity of cabozantinib in metastatic uveal melanoma: Updated results from a phase II randomized discontinuation trial (RDT). J. Clin. Oncol. 2013, 31, 9094. [Google Scholar] [CrossRef]
- Luke, J.J.; Olson, D.J.; Allred, J.B.; Strand, C.A.; Bao, R.; Zha, Y.; Carll, T.; Labadie, B.W.; Bastos, B.R.; Butler, M.O.; et al. Randomized Phase II Trial and Tumor Mutational Spectrum Analysis from Cabozantinib versus Chemotherapy in Metastatic Uveal Melanoma (Alliance A091201). Clin. cancer Res. 2020, 26, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surriga, O.; Rajasekhar, V.K.; Ambrosini, G.; Dogan, Y.; Huang, R.; Schwartz, G.K. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol. cancer Ther. 2013, 12, 2817–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Lutzky, J.; Shoushtari, A.N.; Jeter, J.M.; Chiuzan, C.; Sender, N.; Blumberg, L.E.; Nesson, A.; Singh-Kandah, S.V.; Hernandez, S.; et al. Adjuvant crizotinib in high-risk uveal melanoma following definitive therapy. J. Clin. Oncol. 2020, 38, 10075. [Google Scholar] [CrossRef]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Green, D.L.; Hawkins, B.S.; Hayman, J.; Jaiyesimi, I.; et al. Screening for metastasis from choroidal melanoma: The Collaborative Ocular Melanoma Study Group Report 23. J. Clin. Oncol. 2004, 22, 2438–2444. [Google Scholar] [CrossRef]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vidal, C.; Fernandez-Diaz, D.; Fernandez-Marta, B.; Lago-Baameiro, N.; Pardo, M.; Silva, P.; Paniagua, L.; Blanco-Teijeiro, M.J.; Pineiro, A.; Bande, M. Treatment of Metastatic Uveal Melanoma: Systematic Review. Cancers 2020, 12, 2557. [Google Scholar] [CrossRef]
- Mergener, S.; Siveke, J.T.; Pena-Llopis, S. Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma. Int. J. Mol. Sci. 2021, 22, 6727. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Chr | Mutations | Exon Involved | Percentage of UM with Mutations |
|---|---|---|---|---|
| GNAQ | 9q21.2 | p.(Thr96Ser) p.(Pro170Ser) p.(Gln176Arg) p.(Arg183Cys) p.(Arg183His) p.(Ile189Thr) p.(Pro193Leu) | 4 | 24.2–53.3% [17,28,30,31,32,33] |
| p.(Met203Val) p.(Gln209Leu) p.(Gln209Pro) p.(Gln209Met) p.(Gln209His) p.(Gln209Ile) p.(Phe228Leu) p.(Val344Met) | 5 | |||
| GNA11 | Chr | Mutations | Exon involved | |
| 19p13.3 | p.(Gly48Leu) | 2 | 24.2–60% [19,28,30,31,32,33,35] | |
| p.(Arg166His) p.(Arg183Cys) p.(Arg183His) | 4 | |||
| p.(Gln209Leu) p.(Gln209Pro) p.(Gln209Tyr) p.(Glu221Asp) p.(Glu234Lys) | 5 | |||
| p.(Arg338His) | 7 |
| Mutation Rate | Relation with Metastasis | ||
|---|---|---|---|
| Published Study | GNAQ (%) | GNA11 (%) | |
| Van Raamsdonk et al. (2009) [17] | 46 | - | - |
| Bauer et al. (2009) [21] | No relation between GNAQ-exon 5 mutations and DFS in UM (log-rank; p-value = 0.273). | ||
| Van Raamsdonk et al. (2010) [19] | 48 | 34 | Inverse relationship for GNA11 p.Q209 mutations with metastatic lesions (no statistical data). |
| Pópulo et al. (2011) [29] | 36 | - | No associations between the GNAQ mutations and prognostic parameters. |
| Daniels et al. (2012) [52] | 47 | 44 | - |
| Furney et al. (2013) [53] | 25 | 58 | - |
| Harbour et al. (2013) [54] | 42 | 52 | - |
| Koopmans et al. (2013) [20] | 50 | 43 | No relation between patient survival in UM and mutations in GNAQ and GNA11 (log-rank p-value = 0.466). |
| Martin et al. (2013) [55] | 45 | 40 | - |
| Dono et al. (2014) [56] | 42 | 33 | GNAQ is inversely associated with M3 monosomy and metastasis. Mutations in GNA11 are related with a more aggressive tumour phenotype (no statistical data). |
| Ewens et al. (2014) [57] | 46 | 35 | GNA11 mutations are positively associated with metastatic status after UM treatment (odds ratio 2.5, 95% confidence interval 1.1–5.5). |
| Xiaolin Xu et al. (2014) [33] | 18 | 20 | Metastasis-free survival is not significantly associated with GNAQ/11 mutations (p-value = 0.94). |
| Johansson et al. (2015) [5] | 29 | 50 | - |
| Decatur et al. (2016) [58] | 44 | 44 | GNAQ and GNA11 are not associated with prognosis. |
| Moore et al. (2016) [59] | 43 | 49 | - |
| Royer-Bertrand et al. (2016) [60] | 58 | 42 | - |
| Yavuzyigitoglu et al. (2016) [61] | 49 | 45 | - |
| Robertson et al. (2017) [62] | 50 | 45 | - |
| Kajersti et al. (2017) [63] | 40 | 36 | Mutations in GNAQ are inversely associated with progression to metastasis (log-rank test; p-value = 0.09). |
| Psinakis et al. (2017) [31] | 18 | 24 | No correlation between mutation status and metastasis or OS time of patients. |
| Staby et al. (2018) [63] | 41 | 35 | GNA11 mutations are more frequent in the metastatic group (not statistically significative). |
| Kennedy et al. (2018) [64] | 32 | 53 | Suggestion of a bias towards GNA11 p.Q209L mutations in metastasis. |
| Smit et al. (2018) [65] | 42 | 44 | - |
| Ominato et al. (2018) [32] | 26 | 31 | - |
| Afshar et al. (2019) [66] | 58 | 42 | No statistically significant association between M3 and mutations in GNAQ (p-value = 0.200) and GNA11 (p-value = 0.200). |
| Piaggio et al. (2019) [67] | 48 | 46 | - |
| Schneider et al. (2019) [28] | 20 | 44 | Significant prolonged OS in UM with GNAQ exon 5 wildtype vs. mutated GNAQ exon 5-UM (p-value = 0.018) (not confirmed by multivariate analysis). |
| Thornton et al. (2020) [68] | 53 | 39 | |
| Silva et al. (2021) [30] | 52 | 35 | No correlation between mutations and metastasis or OS time (GNAQ log-rank p-value = 0.88; GNA11 Log-rank p-value = 0.51). |
| Isaacson et al. (2022) [69] | 44 | 52 | Time to first metastasis (GNA11 vs. GNAQ; 77.8 vs. 43.1 months). OS (GNA11 vs. GNAQ; 79.8 vs. 33.7 months). |
| Piaggio et al. (2022) [70] | 51.14 | 48.86 | GNA11 mutated UM has worse prognosis (HR = 1.97 (95%CI 1.12–3.46), p = 0.02). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Rodríguez, P.; Fernández-Díaz, D.; Bande, M.; Pardo, M.; Loidi, L.; Blanco-Teijeiro, M.J. GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers 2022, 14, 3066. https://doi.org/10.3390/cancers14133066
Silva-Rodríguez P, Fernández-Díaz D, Bande M, Pardo M, Loidi L, Blanco-Teijeiro MJ. GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers. 2022; 14(13):3066. https://doi.org/10.3390/cancers14133066
Chicago/Turabian StyleSilva-Rodríguez, Paula, Daniel Fernández-Díaz, Manuel Bande, María Pardo, Lourdes Loidi, and María José Blanco-Teijeiro. 2022. "GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma" Cancers 14, no. 13: 3066. https://doi.org/10.3390/cancers14133066
APA StyleSilva-Rodríguez, P., Fernández-Díaz, D., Bande, M., Pardo, M., Loidi, L., & Blanco-Teijeiro, M. J. (2022). GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers, 14(13), 3066. https://doi.org/10.3390/cancers14133066