
AF8c, a Multi-Kinase Inhibitor Induces Apoptosis by Activating DR5/Nrf2 via ROS in Colorectal Cancer Cells

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Chemicals and Antibodies
2.3. Cell Viability Assay
2.4. Colony Formation
2.5. Western Blot Analysis
2.6. Analysis of Apoptosis
2.7. Determination of Mitochondrial ROS
2.8. Transfection
2.9. RNA Isolation, and Quantitative Real-Time PCR (qRT-PCR)
2.10. Immunoprecipitation (IP)
2.11. Fraction
2.12. Immunofluorescence Staining
2.13. 3D Cell Culture
2.14. In Vivo Tumor Xenograft Study
2.15. Immunohistochemistry
2.16. Terminal Deoxyribonucleotidyl Transferase-Mediated Deoxyuridine Triphosphate Nick-End Labeling (TUNEL) Assay
2.17. Patient-Derived Colorectal Cancer (PDC) Cells
2.18. Statistical Analysis
3. Results
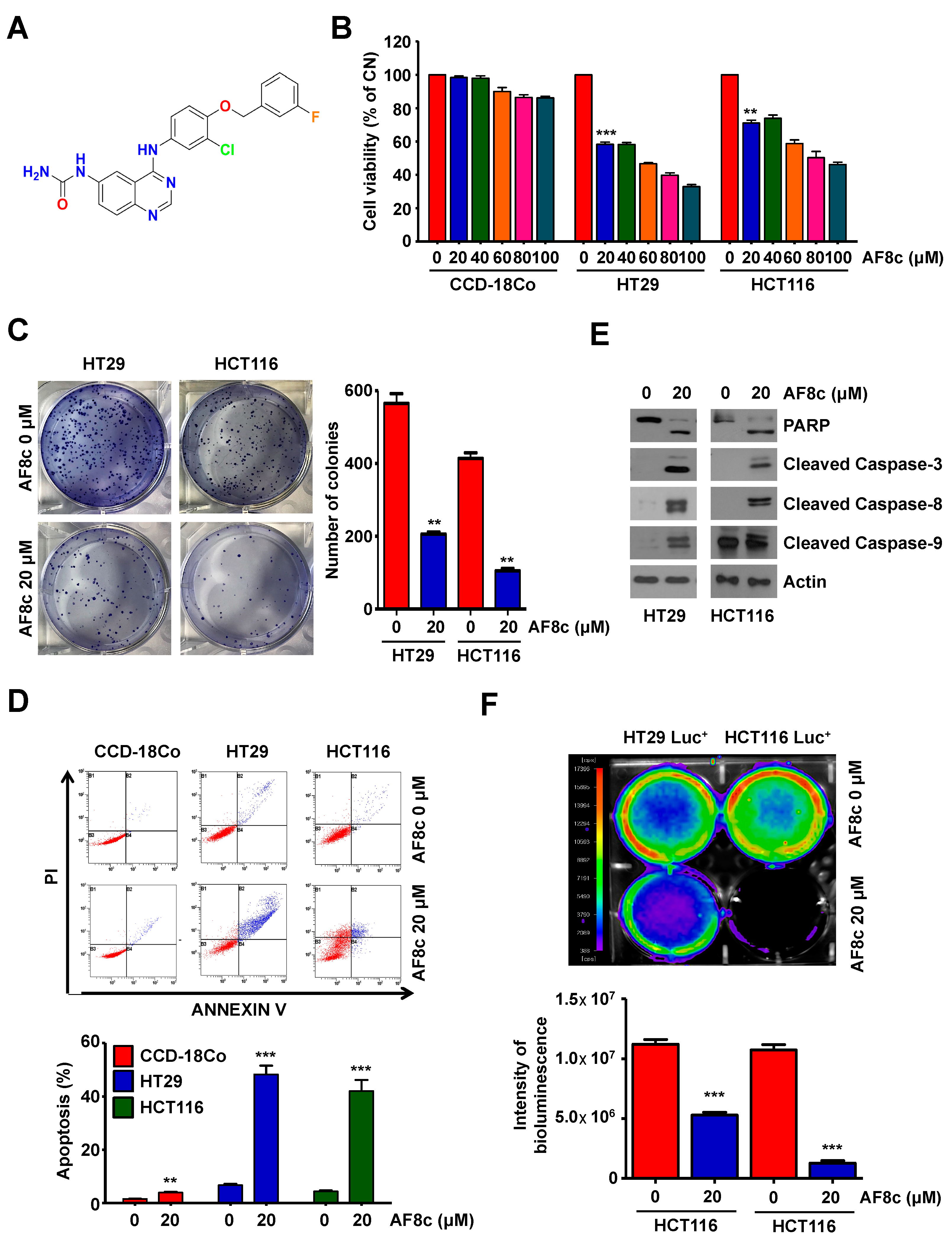
3.1. Treatment with AF8c Inhibits Cell Viability and Induces Apoptosis in Human CRC Cells
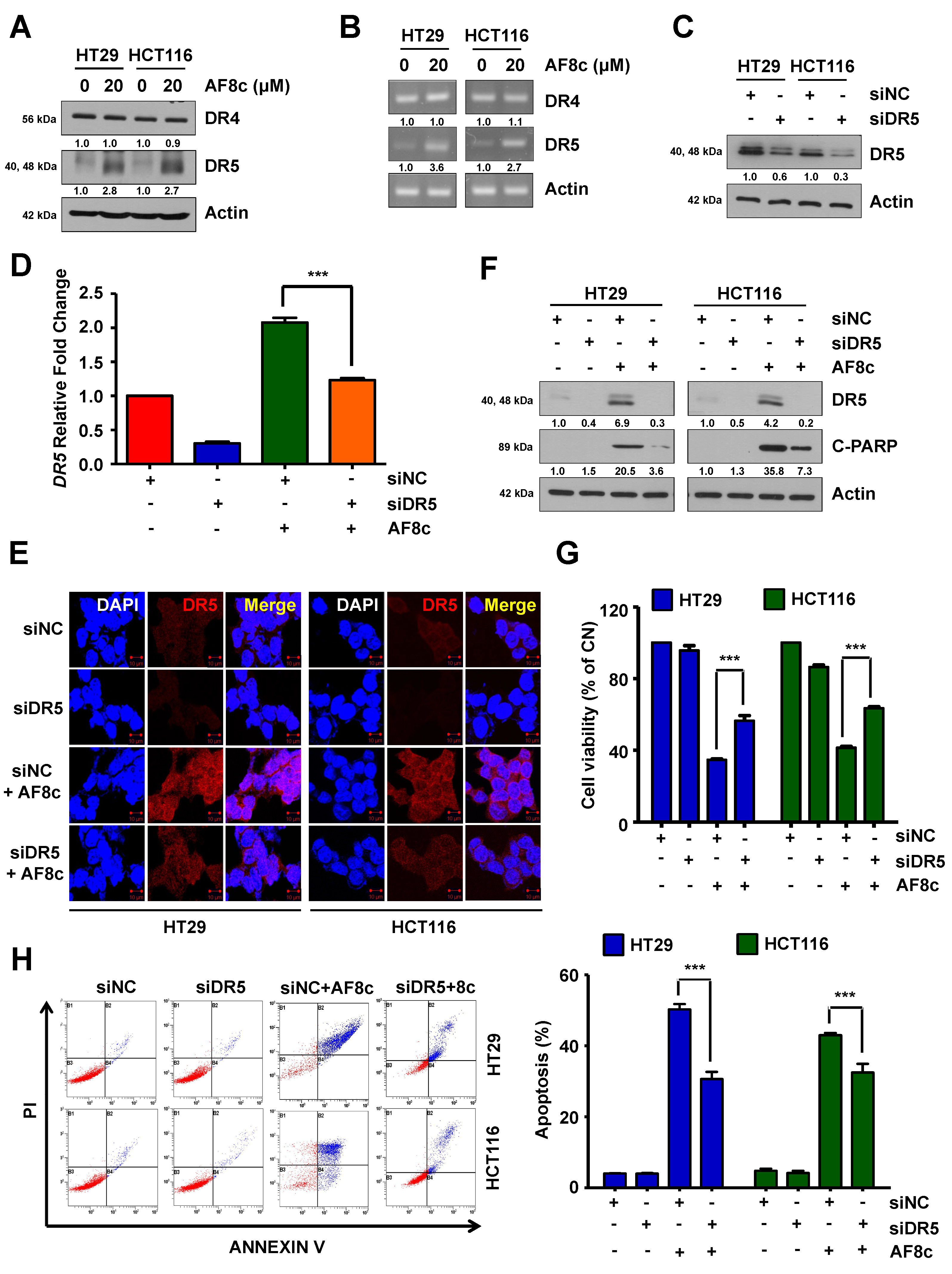
3.2. AF8c-Mediated Upregulation of DR5 Is a Factor for Apoptosis Induction
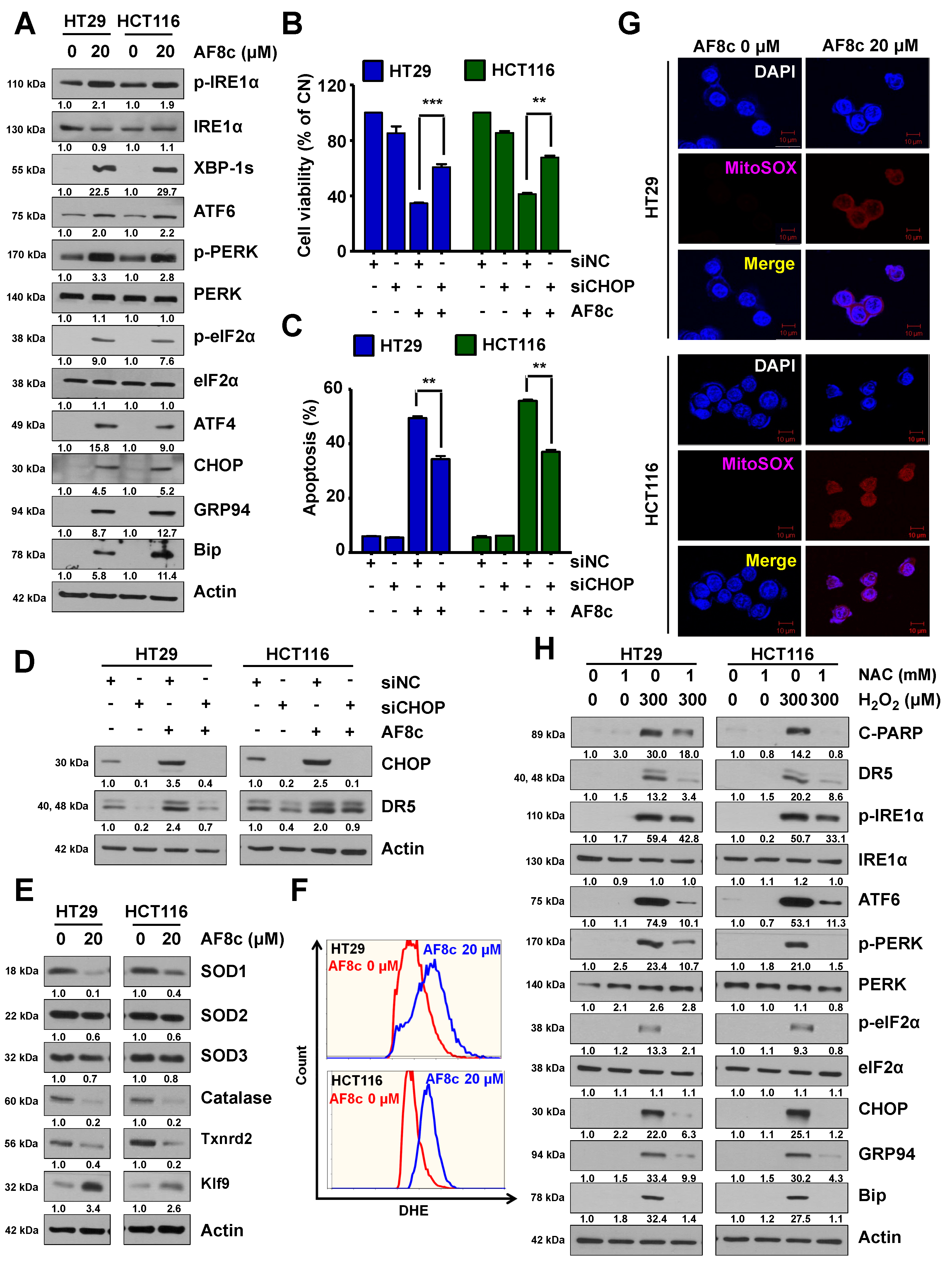
3.3. ER Stress and ROS Leads to Apoptosis in Cells Exposed to AF8c
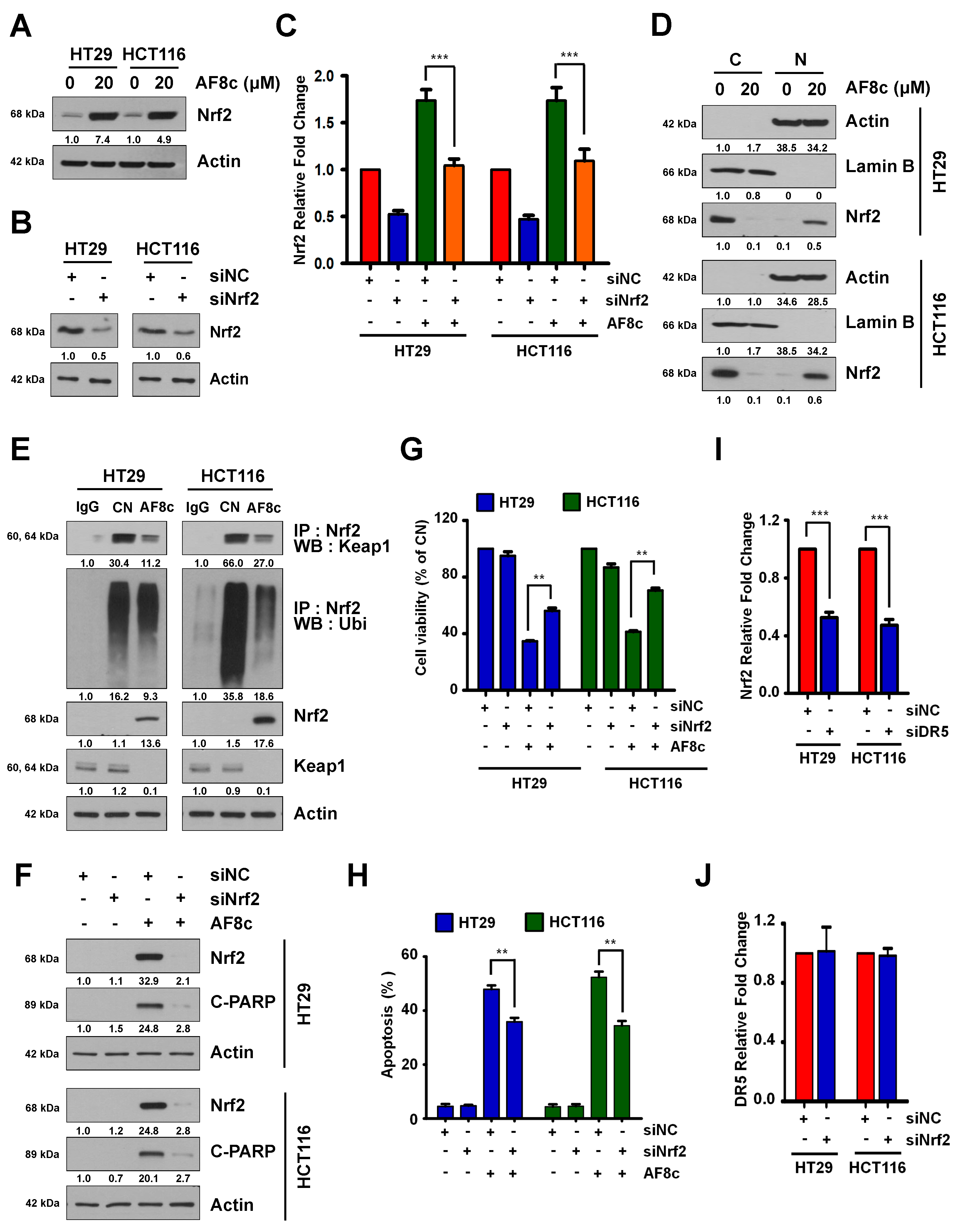
3.4. Activation of DR5-Induced Nrf2 by AF8c Is a Prerequisite for Apoptotic Cell Death
3.5. AF8c Enhances Apoptotic Cell Death in PDC Cells through Activation of DR5 and Nrf2 by Increased ER Stress
3.6. AF8c Suppresses Tumor Growth In Vivo by Increasing DR5, Nrf2, and CHOP Expression and Subsequent Apoptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, M.; Xynos, I.D.; Lazaris, A.; Smaro, T.; Kosmas, C.; Dokou, A.; Felekouras, E.; Antoniou, E.; Polyzos, A.; Sarantonis, J.; et al. Predictors of survival in stage IV metastatic colorectal cancer. Anticancer Res. 2010, 30, 653–660. [Google Scholar] [PubMed]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Wu, C.; Goldberg, R.M. Recent therapeutic advances in the treatment of colorectal cancer. Annu. Rev. Med. 2015, 66, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.; Sforza, V.; Ciardiello, D.; Napolitano, S.; Della Corte, C.M.; Morgillo, F.; Raucci, A.; et al. Present and future of metastatic colorectal cancer treatment: A review of new candidate targets. World J. Gastroenterol. 2017, 23, 4675–4688. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Mutlu, A.; Gyulkhandanyan, A.V.; Freedman, J.; Leytin, V. Activation of caspases-9, -3 and -8 in human platelets triggered by BH3-only mimetic ABT-737 and calcium ionophore A23187: Caspase-8 is activated via bypass of the death receptors. Br. J. Haematol. 2012, 159, 565–571. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Kumar, R.; Herbert, P.E.; Warrens, A.N. An introduction to death receptors in apoptosis. Int. J. Surg. 2005, 3, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- van Noesel, M.M.; van Bezouw, S.; Salomons, G.S.; Voûte, P.A.; Pieters, R.; Baylin, S.B.; Herman, J.G.; Versteeg, R. Tumor-specific Down-Regulation of the Tumor Necrosis Factor-related Apoptosis-inducing Ligand Decoy Receptors DcR1 and DcR2 Is Associated with Dense Promoter Hypermethylation. Cancer Res. 2002, 62, 2157–2161. [Google Scholar]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Trevino, S.; Rosas-Murrieta, N.H.; Millan-Perez-Pena, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef]
- Moradi-Marjaneh, R.; Hassanian, S.M.; Mehramiz, M.; Rezayi, M.; Ferns, G.A.; Khazaei, M.; Avan, A. Reactive oxygen species in colorectal cancer: The therapeutic impact and its potential roles in tumor progression via perturbation of cellular and physiological dysregulated pathways. J. Cell Physiol. 2019, 234, 10072–10079. [Google Scholar] [CrossRef]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- Gil, H.S.; Lee, J.H.; Farag, A.K.; Hassan, A.H.E.; Chung, K.S.; Choi, J.H.; Roh, E.J.; Lee, K.T. AKF-D52, a Synthetic Phenoxypyrimidine-Urea Derivative, Triggers Extrinsic/Intrinsic Apoptosis and Cytoprotective Autophagy in Human Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 5849. [Google Scholar] [CrossRef]
- Wang, X.; Xue, Q.; Wu, L.; Wang, B.; Liang, H. Dasatinib promotes TRAIL-mediated apoptosis by upregulating CHOP-dependent death receptor 5 in gastric cancer. FEBS Open Bio. 2018, 8, 732–742. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Moon, J.H.; Lee, J.H.; Nazim, U.M.; Park, S.Y. Telmisartan generates ROS-dependent upregulation of death receptor 5 to sensitize TRAIL in lung cancer via inhibition of autophagy flux. Int. J. Biochem. Cell Biol. 2018, 102, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Roh, E.J. Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2019, 39, 349–385. [Google Scholar] [CrossRef]
- Dolloff, N.G.; Mayes, P.A.; Hart, L.S.; Dicker, D.T.; Humphreys, R.; El-Deiry, W.S. Off-target lapatinib activity sensitizes colon cancer cells through TRAIL death receptor up-regulation. Sci. Transl. Med. 2011, 3, 86ra50. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-Regulation of the erbB-2 Receptor by Trastuzumab (Herceptin) Enhances Tumor Necrosis Factor-related Apoptosis-inducing Ligand-mediated Apoptosis in Breast and Ovarian Cancer Cell Lines that Overexpress erbB-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Xu, X.M.; He, C.; Hu, X.T.; Fang, B.L. Tumor necrosis factor-related apoptosis-inducing ligand gene on human colorectal cancer cell line HT29. World J. Gastroenterol. 2003, 9, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Shrader, M.; Pino, M.S.; Lashinger, L.; Bar-Eli, M.; Adam, L.; Dinney, C.P.; McConkey, D.J. Gefitinib reverses TRAIL resistance in human bladder cancer cell lines via inhibition of AKT-mediated X-linked inhibitor of apoptosis protein expression. Cancer Res. 2007, 67, 1430–1435. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Farag, A.K.; Viswanath, A.N.I.; Bedair, T.M.; Leem, D.G.; Lee, K.-T.; Pae, A.N.; Roh, E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorganic Med. Chem. Lett. 2015, 25, 5147–5154. [Google Scholar] [CrossRef]
- Nam, E.; Park, C. Maspin suppresses survival of lung cancer cells through modulation of Akt pathway. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2010, 42, 42–47. [Google Scholar] [CrossRef][Green Version]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Chiron, D.; Gomez-Bougie, P.; Descamps, G.; Menoret, E.; Bataille, R.; Moreau, P.; le Gouill, S.; Amiot, M.; Pellat-Deceunynck, C. Cell death via DR5, but not DR4, is regulated by p53 in myeloma cells. Cancer Res. 2012, 72, 4562–4573. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Jayasooriya, R.G.; Park, S.R.; Choi, Y.H.; Choi, I.W.; Kim, G.Y. Caffeic acid phenethyl ester enhances TRAIL-mediated apoptosis via CHOP-induced death receptor 5 upregulation in hepatocarcinoma Hep3B cells. Mol. Cell Biochem. 2016, 418, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Et Biophys. Sin. 2015, 47, 146–147. [Google Scholar] [CrossRef]
- Zlotorynski, E. DR5 unfolds ER stress. Nat. Rev. Mol. Cell Biol. 2014, 15, 498–499. [Google Scholar] [CrossRef]
- Zou, W.; Yue, P.; Khuri, F.R.; Sun, S.Y. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008, 68, 7484–7492. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Farag, A.K.; Yun, H.K.; Jeong, Y.A.; Kim, D.Y.; Jo, M.J.; Park, S.H.; Kim, B.R.; Kim, J.L.; Kim, B.G.; et al. AF8c, a Multi-Kinase Inhibitor Induces Apoptosis by Activating DR5/Nrf2 via ROS in Colorectal Cancer Cells. Cancers 2022, 14, 3043. https://doi.org/10.3390/cancers14133043
Jeong S, Farag AK, Yun HK, Jeong YA, Kim DY, Jo MJ, Park SH, Kim BR, Kim JL, Kim BG, et al. AF8c, a Multi-Kinase Inhibitor Induces Apoptosis by Activating DR5/Nrf2 via ROS in Colorectal Cancer Cells. Cancers. 2022; 14(13):3043. https://doi.org/10.3390/cancers14133043
Chicago/Turabian StyleJeong, Soyeon, Ahmed K. Farag, Hye Kyeong Yun, Yoon A. Jeong, Dae Yeong Kim, Min Jee Jo, Seong Hye Park, Bo Ram Kim, Jung Lim Kim, Bu Gyeom Kim, and et al. 2022. "AF8c, a Multi-Kinase Inhibitor Induces Apoptosis by Activating DR5/Nrf2 via ROS in Colorectal Cancer Cells" Cancers 14, no. 13: 3043. https://doi.org/10.3390/cancers14133043
APA StyleJeong, S., Farag, A. K., Yun, H. K., Jeong, Y. A., Kim, D. Y., Jo, M. J., Park, S. H., Kim, B. R., Kim, J. L., Kim, B. G., Lee, D.-H., Roh, E. J., & Oh, S. C. (2022). AF8c, a Multi-Kinase Inhibitor Induces Apoptosis by Activating DR5/Nrf2 via ROS in Colorectal Cancer Cells. Cancers, 14(13), 3043. https://doi.org/10.3390/cancers14133043