
A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Immunoprecipitation of HLA-I Complexes
2.3. LC-MS/MS Analysis of HLA-I Peptides
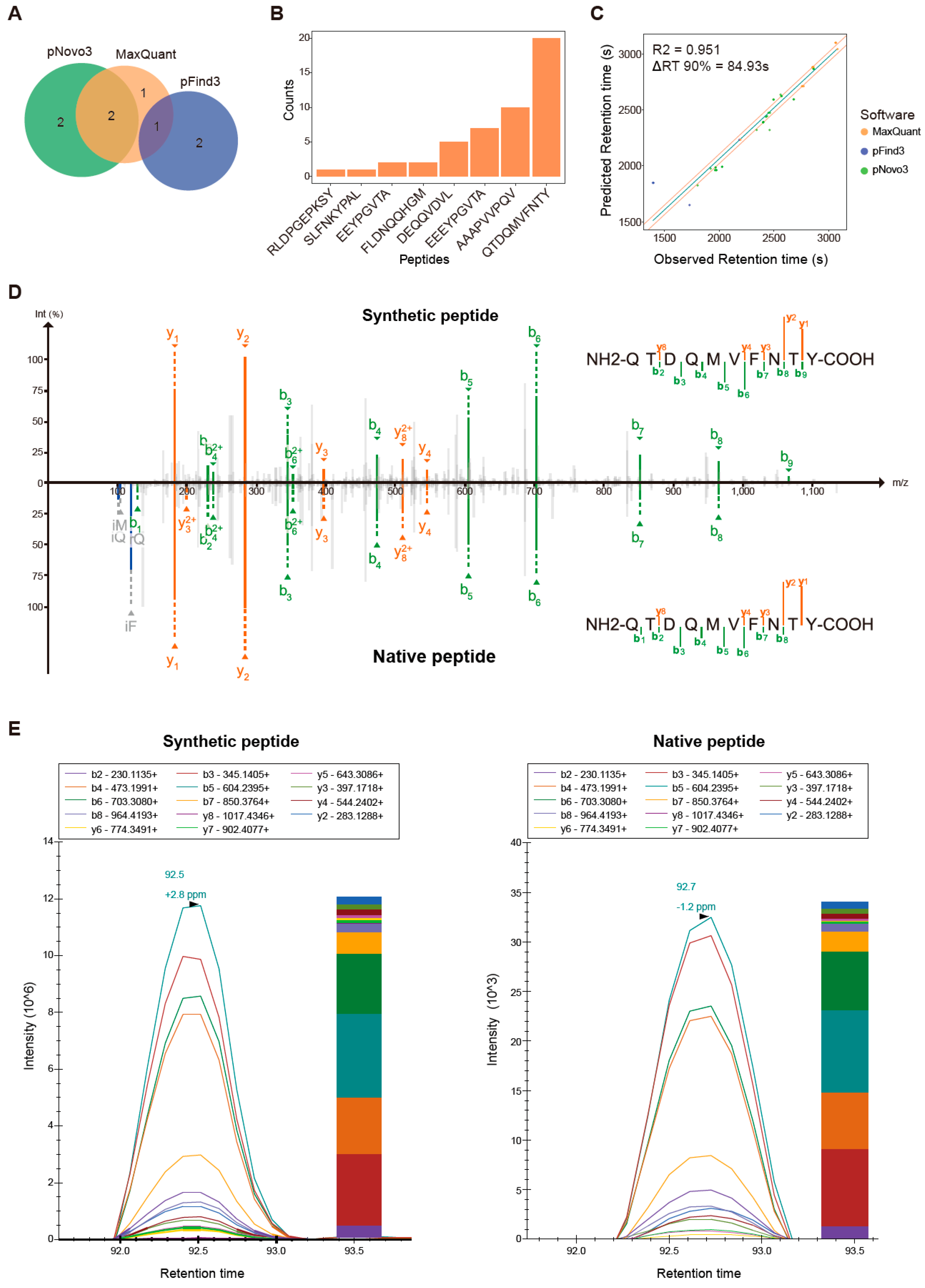
2.4. Immunopeptide Validation Using Parallel Reaction Monitoring Assay
2.5. Calling Mutations from Whole Exome Sequencing (WES) Data
2.6. Construction of Individualized Protein Database
2.7. Analysis of LC-MS/MS Data
2.8. Retention Time Prediction
2.9. Affinity Prediction and Clustering of the Immunopeptidome
2.10. Analysis of Non-Canonical Peptide Traceability
2.11. Gene Functional Annotation for Canonical and Cis-Spliced Peptide Candidates
3. Results
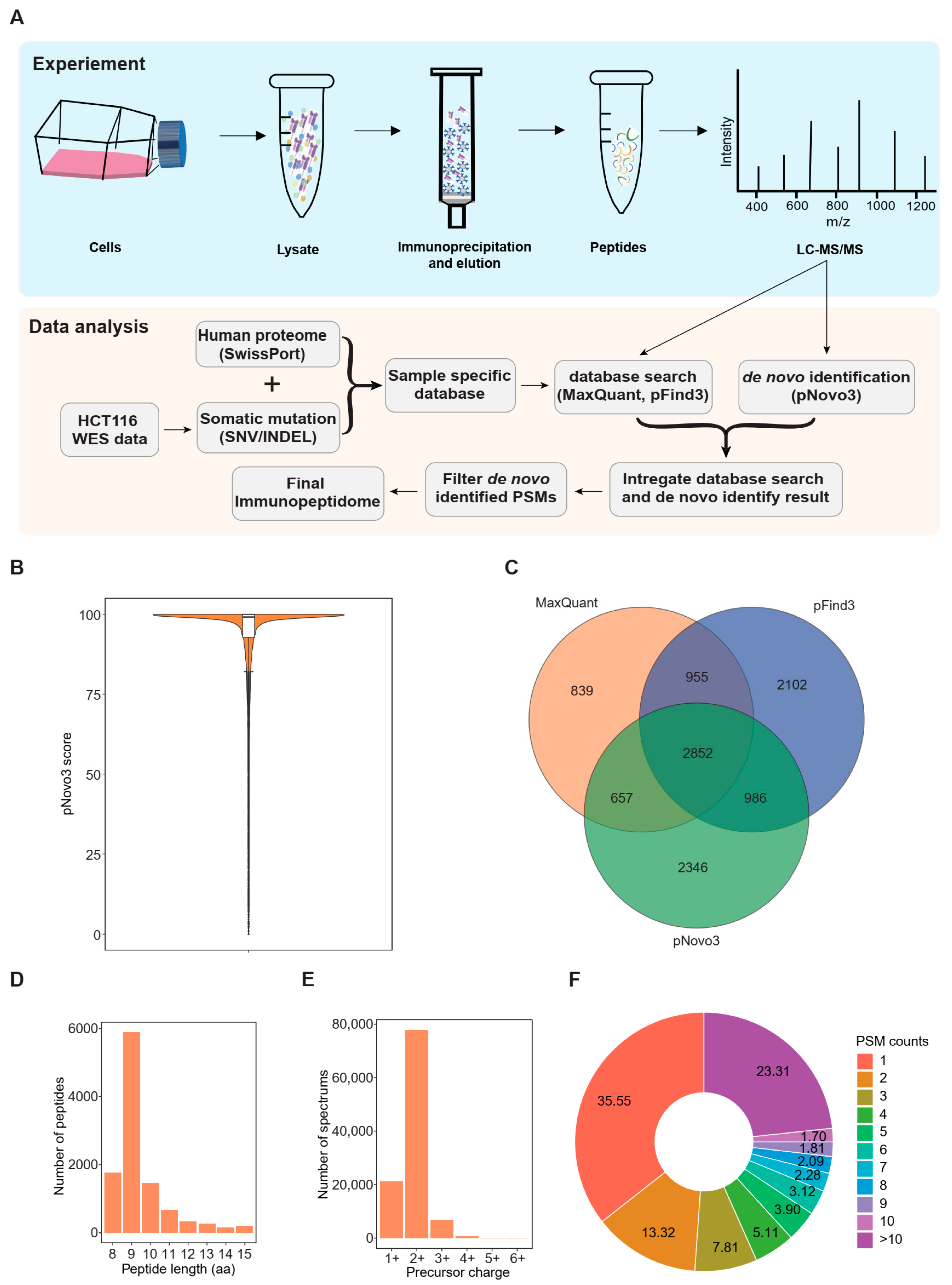
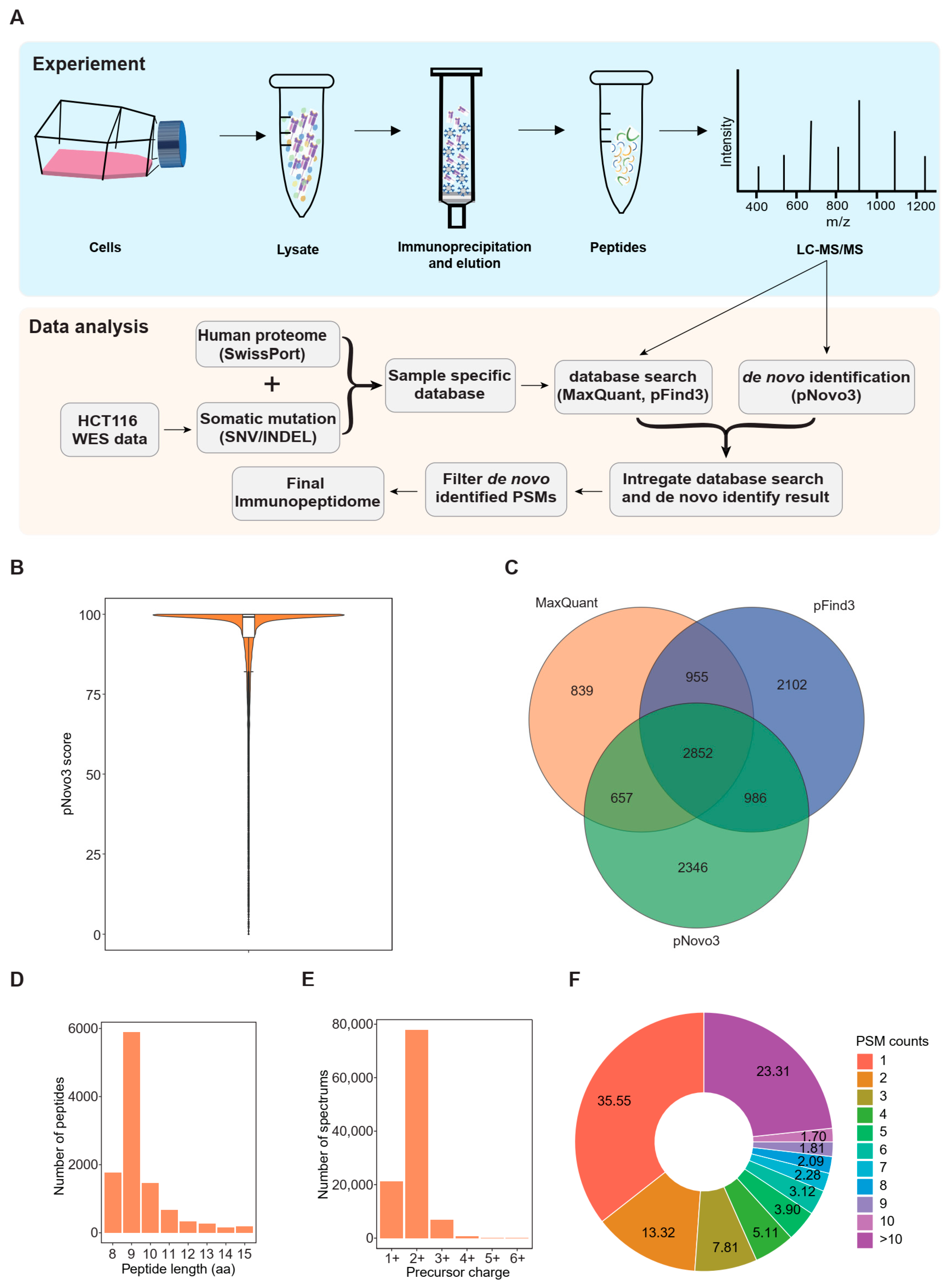
3.1. An Integrated Proteogenomic Approach Identifies the Immunopeptidome in a Colorectal Cancer Cell Line
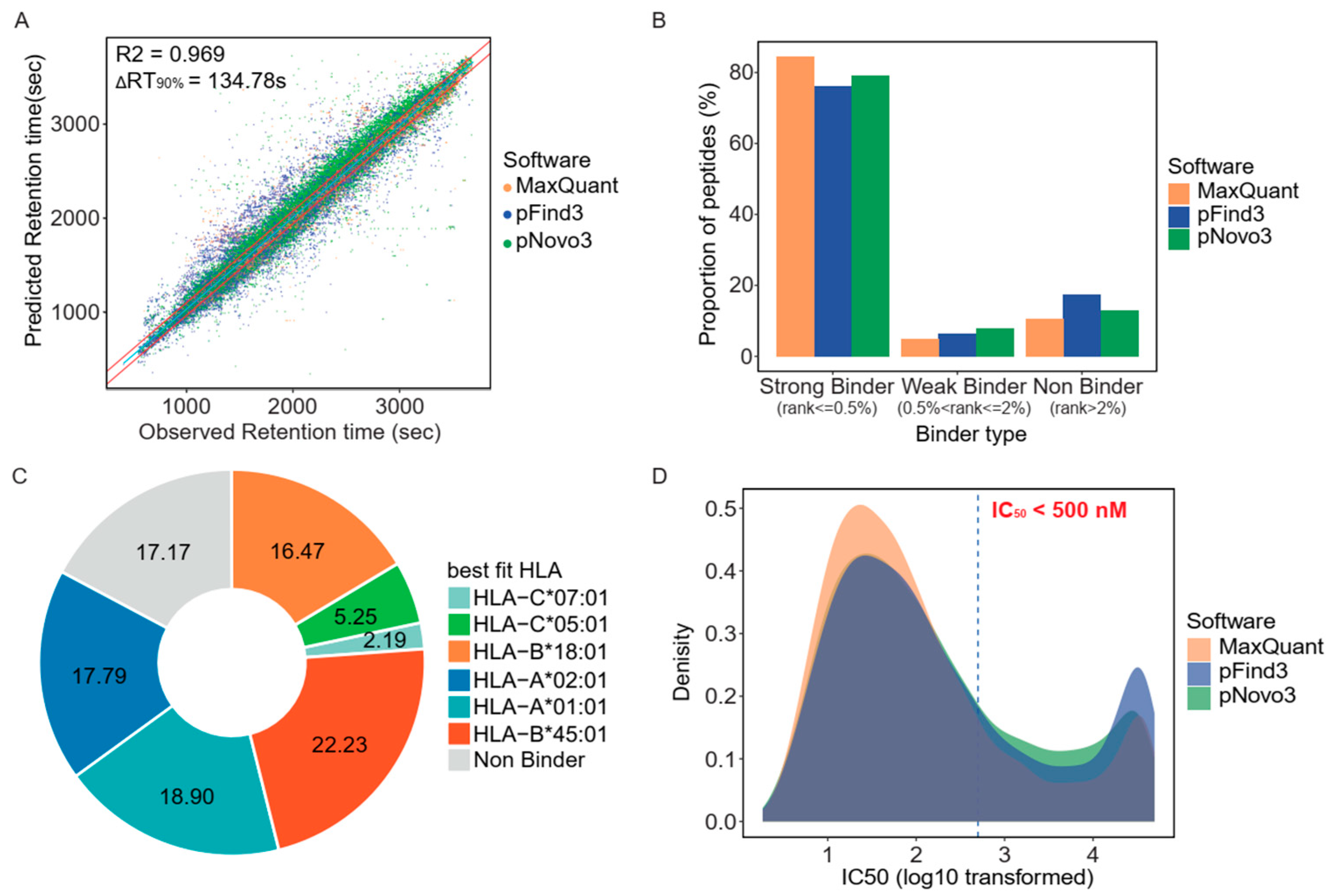
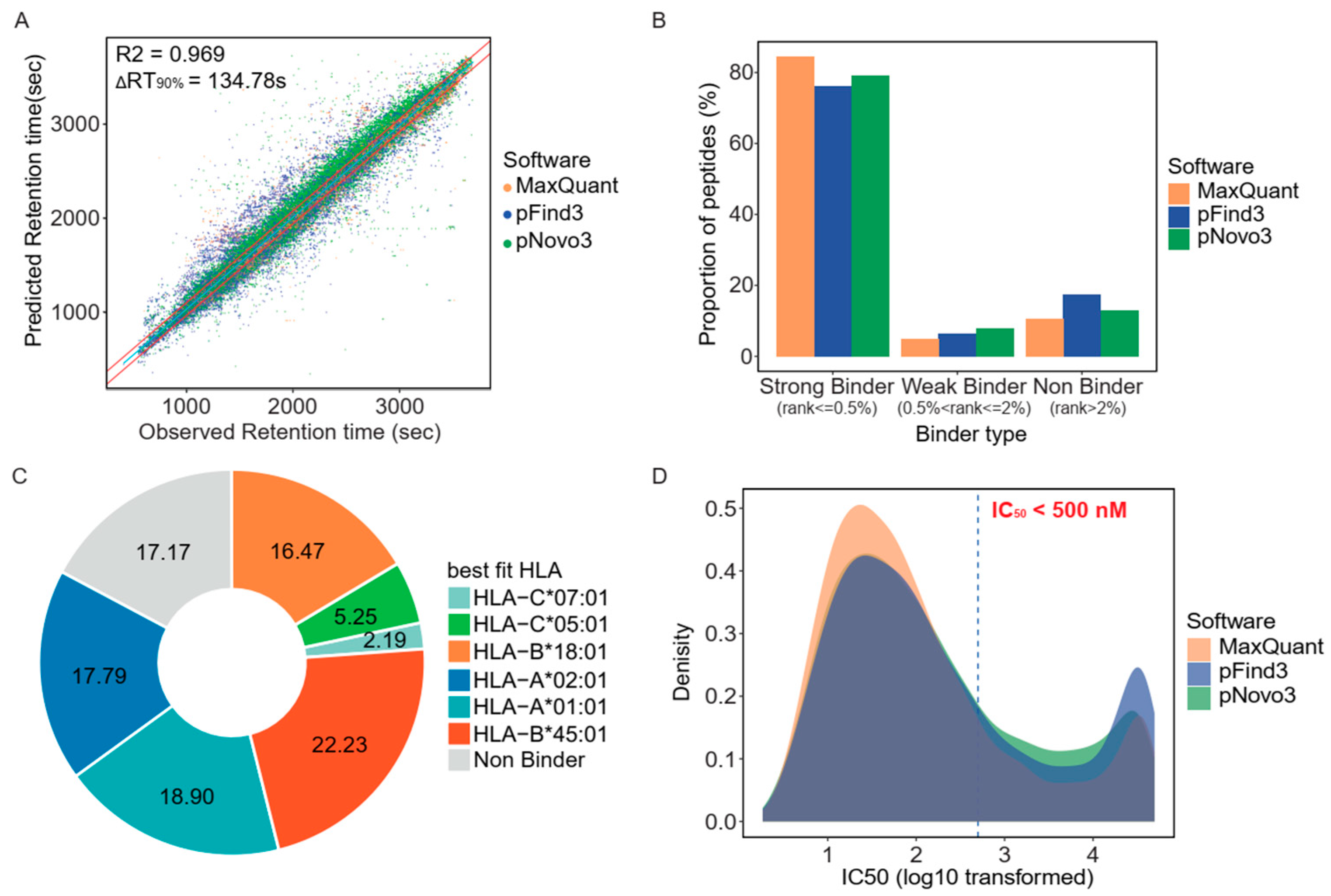
3.2. Retention Time and HLA-Binding Affinity Predictions Confirmed the Accuracy of the Identified Immunopeptidome
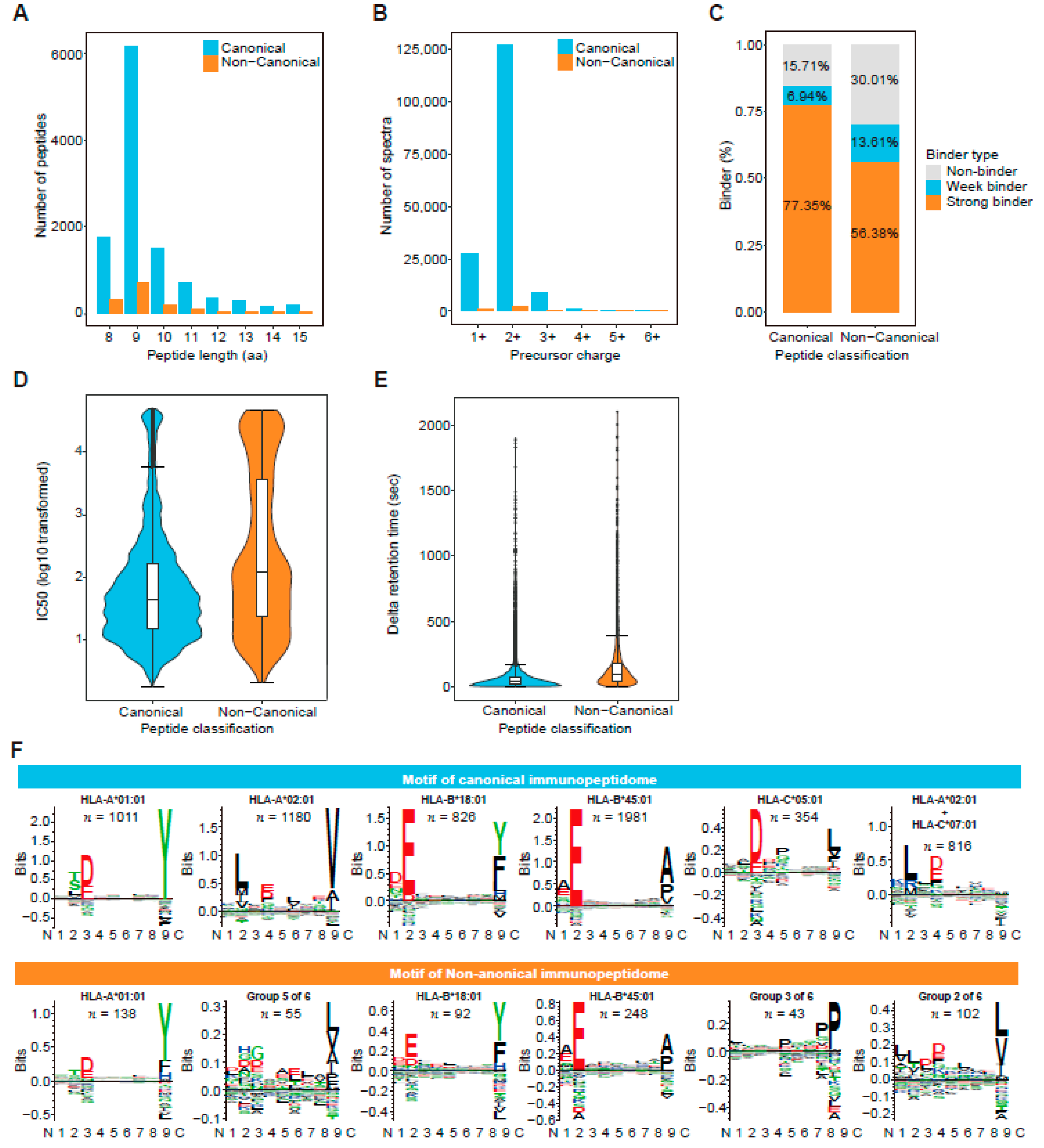
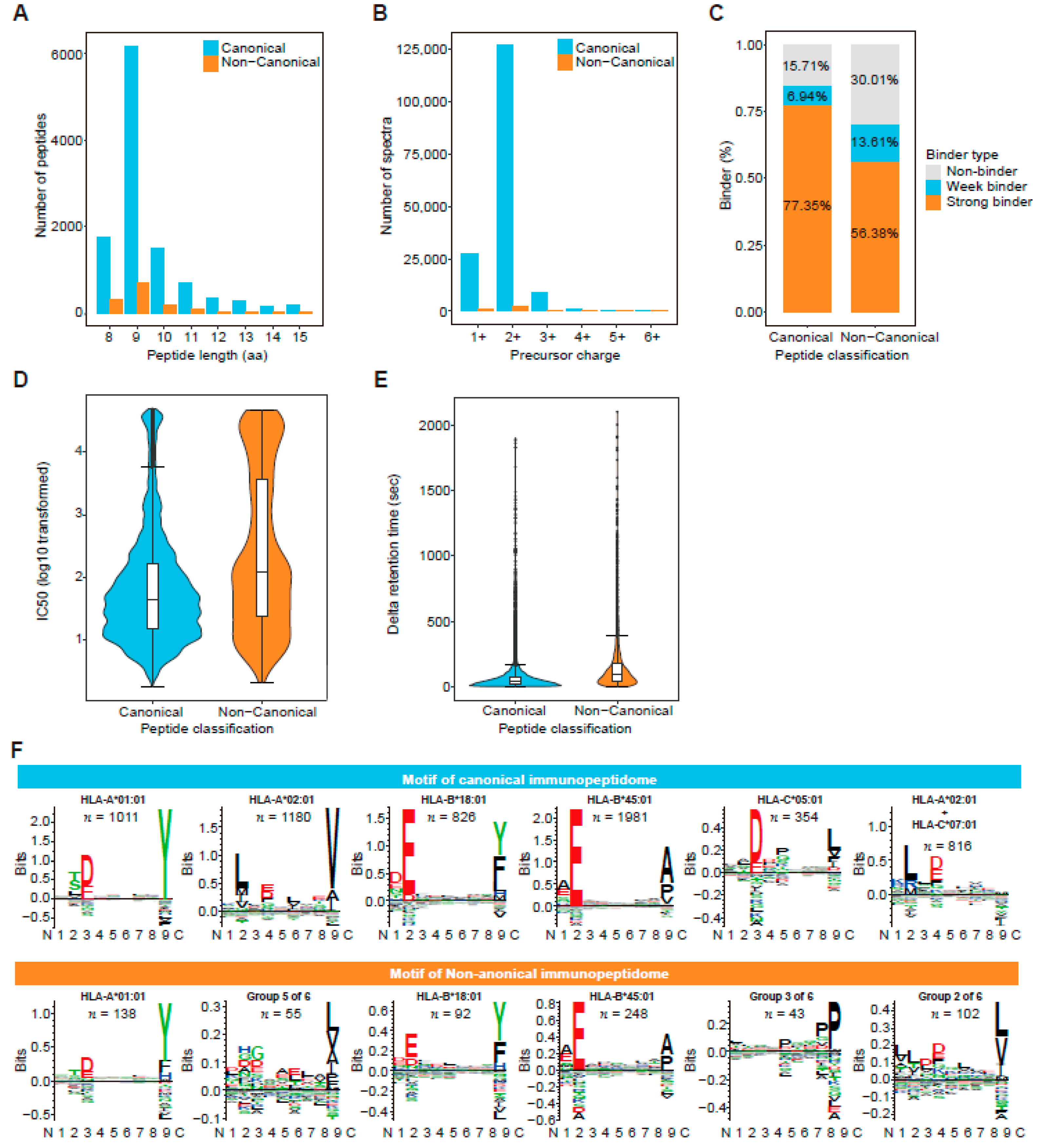
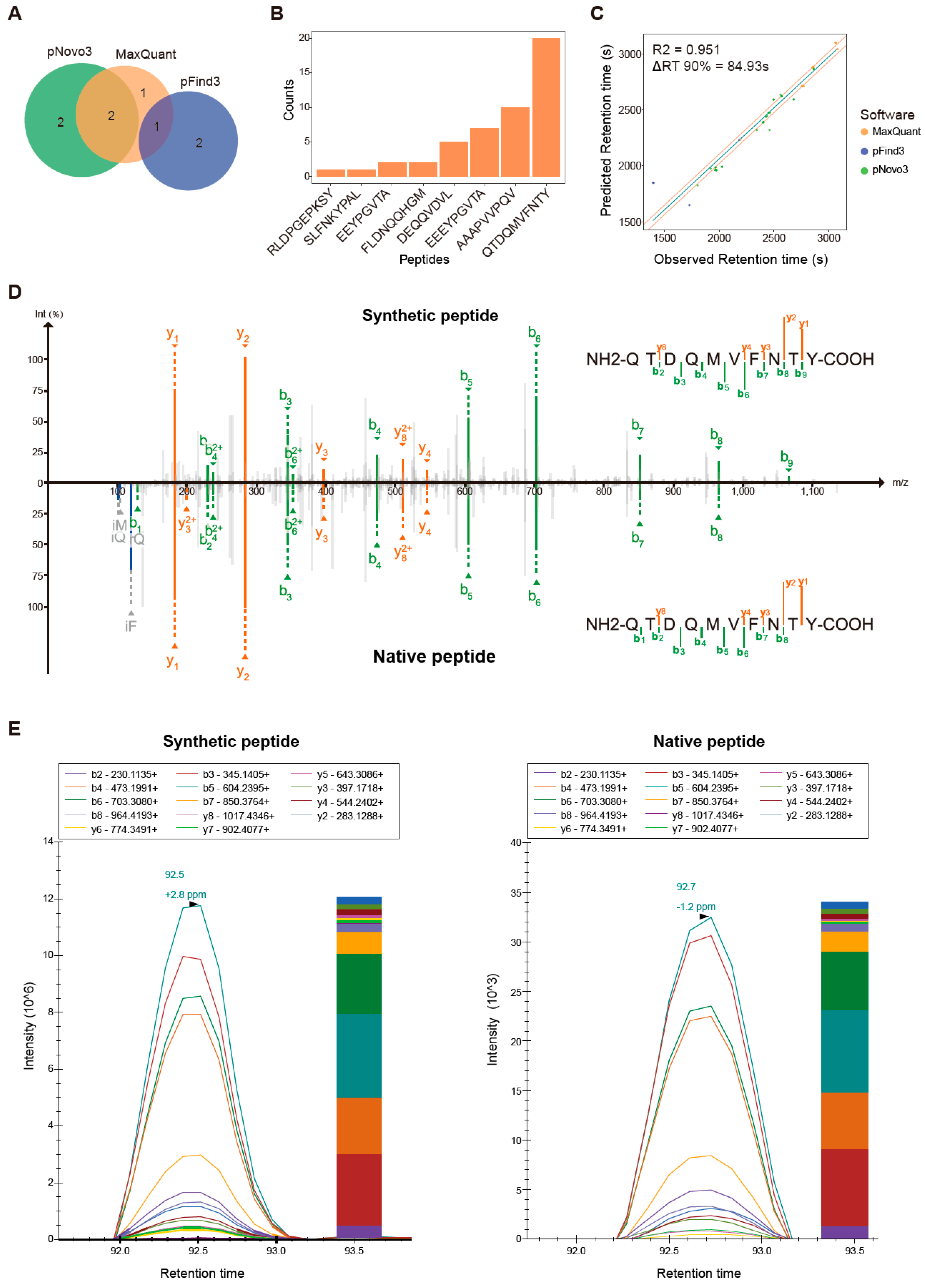
3.3. De Novo Identification Enlarges the Immunopeptidome Landscape of Cancer Cell Lines
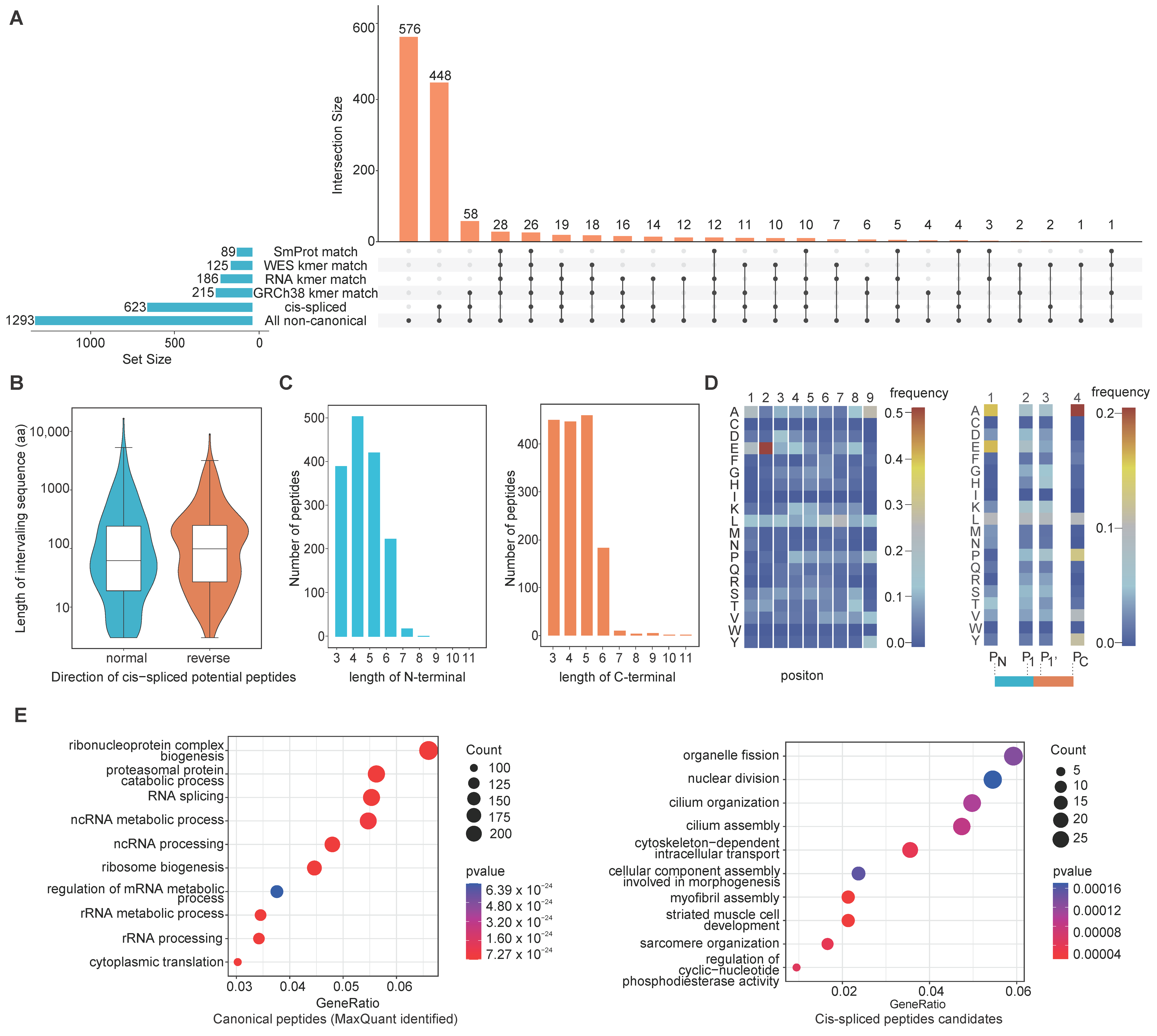
3.4. Traceability Analysis Shows That Some Non-Canonical Peptides Result from Different Omics-Level Variations
3.5. Proteogenomics Integrating HCT116 Cell Line-Specific SNV/INDEL Mutation Information Identifies 8 Mutation-Bearing Neo-Epitopes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Coukos, G.; Bassani-Sternberg, M. Identification of Tumor Antigens with Immunopeptidomics. Nat. Biotechnol. 2022, 40, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef]
- Gupta, R.G.; Li, F.; Roszik, J.; Lizée, G. Exploiting Tumor Neoantigens to Target Cancer Evolution: Current Challenges and Promising Therapeutic Approaches. Cancer Discov. 2021, 11, 1024–1039. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Blaeschke, F.; Paul, M.C.; Schuhmann, M.U.; Rabsteyn, A.; Schroeder, C.; Casadei, N.; Matthes, J.; Mohr, C.; Lotfi, R.; Wagner, B.; et al. Low Mutational Load in Pediatric Medulloblastoma Still Translates into Neoantigens as Targets for Specific T-Cell Immunotherapy. Cytotherapy 2019, 21, 973–986. [Google Scholar] [CrossRef]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834.e13. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Capietto, A.H.; Jhunjhunwala, S.; Delamarre, L. Characterizing Neoantigens for Personalized Cancer Immunotherapy. Curr. Opin. Immunol. 2017, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical Potential of Mass Spectrometry-Based Proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [Green Version]
- Hirama, T.; Tokita, S.; Nakatsugawa, M.; Murata, K.; Nannya, Y.; Matsuo, K.; Inoko, H.; Hirohashi, Y.; Hashimoto, S.; Ogawa, S.; et al. Proteogenomic Identification of an Immunogenic HLA Class I Neoantigen in Mismatch Repair-Deficient Colorectal Cancer Tissue. JCI Insight 2021, 6, e146356. [Google Scholar] [CrossRef]
- Yarmarkovich, M.; Marshall, Q.F.; Warrington, J.M.; Premaratne, R.; Farrel, A.; Groff, D.; Li, W.; di Marco, M.; Runbeck, E.; Truong, H.; et al. Cross-HLA Targeting of Intracellular Oncoproteins with Peptide-Centric CARs. Nature 2021, 599, 477–484. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic Neoantigens Derived from Gene Fusions Stimulate T Cell Responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding Regions Are the Main Source of Targetable Tumor-Specific Antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Ehx, G.; Larouche, J.D.; Durette, C.; Laverdure, J.P.; Hesnard, L.; Vincent, K.; Hardy, M.P.; Thériault, C.; Rulleau, C.; Lanoix, J.; et al. Atypical Acute Myeloid Leukemia-Specific Transcripts Generate Shared and Immunogenic MHC Class-I-Associated Epitopes. Immunity 2021, 54, 737–752.e10. [Google Scholar] [CrossRef]
- Paes, W.; Leonov, G.; Partridge, T.; Chikata, T.; Murakoshi, H.; Frangou, A.; Brackenridge, S.; Nicastri, A.; Smith, A.G.; Learn, G.H.; et al. Contribution of Proteasome-Catalyzed Peptide Cis-Splicing to Viral Targeting by CD8+ T Cells in HIV-1 Infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24748–24759. [Google Scholar] [CrossRef] [Green Version]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A Few Good Peptides: MHC Class I-Based Cancer Immunosurveillance and Immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, J.; Kowalewski, D.J.; Backert, L.; Gwinner, F.; Dorner, S.; Priemer, M.; Tsou, C.-C.; Hoffgaard, F.; Römer, M.; Schuster, H.; et al. Pitfalls in HLA Ligandomics-How to Catch a Li(e)Gand. Mol. Cell. Proteom. 2021, 20, 100110. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Hinojosa, S.; Grant, M.; Panigrahi, A.; Zhang, H.; Caisova, V.; Bollard, C.M.; Rood, B.R. Proteogenomic Discovery of Neoantigens Facilitates Personalized Multi-Antigen Targeted T Cell Immunotherapy for Brain Tumors. Nat. Commun. 2021, 12, 6689. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Müller, M.; Pak, H.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated Proteogenomic Deep Sequencing and Analytics Accurately Identify Non-Canonical Peptides in Tumor Immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Y.; Yang, Y.; Yang, Y.; Li, H.; Dong, X.; Wang, H.; Xie, Z.; Zhao, Q. An Integrated Approach for Discovering Noncanonical MHC-I Peptides Encoded by Small Open Reading Frames. J. Am. Soc. Mass Spectrom. 2021, 32, 2346–2357. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Vaudel, M.; Zhang, B.; Ren, Y.; Wen, B. PDV: An Integrative Proteomics Data Viewer. Bioinformatics 2019, 35, 1249–1251. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Regier, A.A.; Farjoun, Y.; Larson, D.E.; Krasheninina, O.; Kang, H.M.; Howrigan, D.P.; Chen, B.J.; Kher, M.; Banks, E.; Ames, D.C.; et al. Functional Equivalence of Genome Sequencing Analysis Pipelines Enables Harmonized Variant Calling across Human Genetics Projects. Nat. Commun. 2018, 9, 4038. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019, 861054. [Google Scholar] [CrossRef] [Green Version]
- Lowy-Gallego, E.; Fairley, S.; Zheng-Bradley, X.; Ruffier, M.; Clarke, L.; Flicek, P.; Church, D.M.; Rendon, A.; England, G. Variant Calling on the GRCh38 Assembly with the Data from Phase Three of the 1000 Genomes Project. Wellcome Open Res. 2019, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.; Sirotkin, K. DbSNP—Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Chi, H.; Liu, C.; Yang, H.; Zeng, W.F.; Wu, L.; Zhou, W.J.; Wang, R.M.; Niu, X.N.; Ding, Y.H.; Zhang, Y.; et al. Comprehensive Identification of Peptides in Tandem Mass Spectra Using an Efficient Open Search Engine. Nat. Biotechnol. 2018, 36, 1059–1061. [Google Scholar] [CrossRef]
- Yang, H.; Chi, H.; Zeng, W.F.; Zhou, W.J.; He, S.M. PNovo 3: Precise de Novo Peptide Sequencing Using a Learning-to-Rank Framework. Bioinformatics 2019, 35, i183–i190. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Li, K.; Zhang, Y.; Zhang, B. Cancer Neoantigen Prioritization through Sensitive and Reliable Proteogenomics Analysis. Nat. Commun. 2020, 11, 1759. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Alvarez, B.; Nielsen, M. GibbsCluster: Unsupervised Clustering and Alignment of Peptide Sequences. Nucleic Acids Res. 2017, 45, W458–W463. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.C.F.; Nielsen, M. Seq2Logo: A Method for Construction and Visualization of Amino Acid Binding Motifs and Sequence Profiles Including Sequence Weighting, Pseudo Counts and Two-Sided Representation of Amino Acid Enrichment and Depletion. Nucleic Acids Res. 2012, 40, W281–W287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, H.; Chen, X.; Zheng, Y.; Kang, Q.; Hao, D.; Zhang, L.; Song, T.; Luo, H.; Hao, Y.; et al. SmProt: A Reliable Repository with Comprehensive Annotation of Small Proteins Identified from Ribosome Profiling. Genom. Proteom. Bioinform. 2021, 19, 602–610. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Long, J.; He, J.; Li, C.I.; Cai, Q.; Shu, X.O.; Zheng, W.; Li, C. Exome Sequencing Generates High Quality Data in Non-Target Regions. BMC Genom. 2012, 13, 194. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Mishto, M.; Liepe, J. Post-Translational Peptide Splicing and T Cell Responses. Trends Immunol. 2017, 38, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Mansurkhodzhaev, A.; Barbosa, C.R.R.; Mishto, M.; Liepe, J. Proteasome-Generated Cis-Spliced Peptides and Their Potential Role in CD8+ T Cell Tolerance. Front. Immunol. 2021, 12, 614276. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A Large Fraction of HLA Class I Ligands Are Proteasome-Generated Spliced Peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liepe, J.; Sidney, J.; Lorenz, F.K.M.; Sette, A.; Mishto, M. Mapping the MHC Class I-Spliced Immunopeptidome of Cancer Cells. Cancer Immunol. Res. 2019, 7, 62–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.P.; Helm, D.; Rettel, M.; Stein, F.; Hernandez-Sanchez, A.; Urban, K.; Gebert, J.; Kloor, M.; Neu-Yilik, G.; von Knebel Doeberitz, M.; et al. NMD Inhibition by 5-Azacytidine Augments Presentation of Immunogenic Frameshift-Derived Neoepitopes. iScience 2021, 24, 102389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, G.; Hou, G.; Xiang, H.; Zhang, X.; Huang, Y.; Zhang, X.; Li, B.; Lee, L.J. IntroSpect: Motif-Guided Immunopeptidome Database Building Tool to Improve the Sensitivity of HLA Binding Peptide Identification. Biomolecules 2021, 12, 579. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Van Lehmann, K.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Caesar-Johnson, S.J.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Faridi, P.; Woods, K.; Ostrouska, S.; Deceneux, C.; Aranha, R.; Duscharla, D.; Wong, S.Q.; Chen, W.; Ramarathinam, S.H.; Lim Kam Sian, T.C.C.; et al. Spliced Peptides and Cytokine-Driven Changes in the Immunopeptidome of Melanoma. Cancer Immunol. Res. 2020, 8, 1322–1334. [Google Scholar] [CrossRef]
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the Contribution of Proteasomal Spliced Peptides to the HLA-I Ligandome. Mol. Cell. Proteom. 2018, 17, 2347–2357. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | HLA | IC50 (nM) | Mutation Locus | Gene | Protein | AA Change | Wild Peptides Identified | Reference |
|---|---|---|---|---|---|---|---|---|
| QTDQMVFNTY | HLA-A*01:01 | 15.85 | chr8:23258741 | CHMP7 | Q8WUX9 | p.A324T | Yes | [25,48,53,54] |
| RLDPGEPKSY | HLA-A*01:01 | 1193.67 | chr9:35750732 | RGP1 | Q92546 | p.S110P | No | [53] |
| AAAPVVPQV | HLA-A*02:01 | 175.14 | chr6:149796499 | PCMT1 | P22061 | p.A168V | No | [53] |
| DEQQVDVL | HLA-B*18:01 | 771.4 | chr20:31605574 | ID1 | P41134 | p.N63D | Yes | - |
| EEEYPGVTA | HLA-B*45:01 | 106.68 | chr22:17726722 | BCL2L13 | Q9BXK5 | p.I216V | Yes | [48] |
| EEYPGVTA | HLA-B*45:01 | 448.13 | chr22:17726722 | BCL2L13 | Q9BXK5 | p.I216V | No | - |
| SLFNKYPAL | HLA-A*02:01 | 11.61 | chrX:115643440 | PLS3 | P13797 | p.N372S | No | - |
| FLDNQQHGM | HLA-C*05:01 | 20.86 | chr1:88804485 | PKN2 | Q16513 | p.R459Q | No | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, H.; Zhang, L.; Bu, F.; Guan, X.; Chen, L.; Zhang, H.; Zhao, Y.; Chen, H.; Zhang, W.; Li, Y.; et al. A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources. Cancers 2022, 14, 3016. https://doi.org/10.3390/cancers14123016
Xiang H, Zhang L, Bu F, Guan X, Chen L, Zhang H, Zhao Y, Chen H, Zhang W, Li Y, et al. A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources. Cancers. 2022; 14(12):3016. https://doi.org/10.3390/cancers14123016
Chicago/Turabian StyleXiang, Haitao, Le Zhang, Fanyu Bu, Xiangyu Guan, Lei Chen, Haibo Zhang, Yuntong Zhao, Huanyi Chen, Weicong Zhang, Yijian Li, and et al. 2022. "A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources" Cancers 14, no. 12: 3016. https://doi.org/10.3390/cancers14123016
APA StyleXiang, H., Zhang, L., Bu, F., Guan, X., Chen, L., Zhang, H., Zhao, Y., Chen, H., Zhang, W., Li, Y., Lee, L. J., Mei, Z., Rao, Y., Gu, Y., Hou, Y., Mu, F., & Dong, X. (2022). A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources. Cancers, 14(12), 3016. https://doi.org/10.3390/cancers14123016