
Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C

 ,
,  , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Annotation of DNA and RNA Variants and Transcripts
2.3. Variant Collection and Filtering
2.4. Minigene Construction and Mutagenesis
2.5. Minigene Splicing Assays
2.6. ACMG–AMP Clinical Classification of RAD51C Genetic Variants
3. Results
3.1. In Silico Analysis
3.2. Functional STUDY
3.3. Transcript Analysis and ACMG/AMP-Based Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Foulkes, W.D. The ten genes for breast (and ovarian) cancer susceptibility. Nat. Rev. Clin. Oncol. 2021, 18, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [PubMed]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Somyajit, K.; Subramanya, S.; Nagaraju, G. RAD51C: A novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 2010, 31, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- Vaz, F.; Hanenberg, H.; Schuster, B.; Barker, K.; Wiek, C.; Erven, V.; Neveling, K.; Endt, D.; Kesterton, I.; Autore, F.; et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010, 42, 406–409. [Google Scholar] [CrossRef]
- Radice, P.; de Summa, S.; Caleca, L.; Tommasi, S. Unclassified variants in BRCA genes: Guidelines for interpretation. Ann. Oncol. 2011, 22, i18–i23. [Google Scholar] [CrossRef]
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [Green Version]
- Zelli, V.; Compagnoni, C.; Cannita, K.; Capelli, R.; Capalbo, C.; Nolfi, M.D.V.; Alesse, E.; Zazzeroni, F.; Tessitore, A. Applications of Next Generation Sequencing to the Analysis of Familial Breast/Ovarian Cancer. High-Throughput 2020, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- McAlarnen, L.; Stearns, K.; Uyar, D. Challenges of Genomic Testing for Hereditary Breast and Ovarian Cancers. Appl. Clin. Genet. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Milano, L.; Fierheller, C.T.; Serruya, C.; Revil, T.; Oros, K.K.; Behl, S.; Arcand, S.L.; Nayar, P.; Spiegelman, D.; et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers 2022, 14, 2251. [Google Scholar] [CrossRef] [PubMed]
- Sanz, D.J.; Acedo, A.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Esteban-Cardeñosa, E.; Lastra, E.; Pagani, F.; Miner, C.; Velasco, E.A. A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin. Cancer Res. 2010, 16, 1957–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhine, C.L.; Cygan, K.J.; Soemedi, R.; Maguire, S.; Murray, M.F.; Monaghan, S.F.; Fairbrother, W.G. Hereditary cancer genes are highly susceptible to splicing mutations. PLoS Genet. 2018, 14, e1007231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Asselta, R.; Duga, S.; Velasco, E.A.; Buratti, E. Editorial: RNA Splicing and Backsplicing: Disease and Therapy. Front. Genet. 2020, 11, 626835. [Google Scholar] [CrossRef]
- Tournier, I.; Vezain, M.; Martins, A.; Charbonnier, F.; Baert-Desurmont, S.; Olschwang, S.; Wang, Q.; Buisine, M.P.; Soret, J.; Tazi, J.; et al. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum. Mutat. 2008, 29, 1412–1424. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef] [Green Version]
- Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Llovet, P.; Díez-Gómez, B.; Caloca, M.J.; Pérez-Segura, P.; Fraile-Bethencourt, E.; Colmena, M.; Carvalho, S.; et al. Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene. Cancers 2020, 12, 3771. [Google Scholar] [CrossRef]
- Valenzuela-Palomo, A.; Bueno-Martínez, E.; Sanoguera-Miralles, L.; Lorca, V.; Fraile-Bethencourt, E.; Esteban-Sánchez, A.; Gómez-Barrero, S.; Carvalho, S.; Allen, J.; García-Álvarez, A.; et al. Splicing predictions, minigene analyses, and ACMG-AMP clinical classification of 42 germline PALB2 splice-site variants. J. Pathol. 2022, 256, 321–334. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Garibay, G.R.; Acedo, A.; García-Casado, Z.; Gutiérrez-Enríquez, S.; Tosar, A.; Romero, A.; Garre, P.; Llort, G.; Thomassen, M.; Díez, O.; et al. Capillary Electrophoresis Analysis of Conventional Splicing Assays: IARC Analytical and Clinical Classification of 31 BRCA2 Genetic Variants. Hum. Mutat. 2014, 35, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Acedo, A.; Hernández-Moro, C.; Curiel-García, Á.; Díez-Gómez, B.; Velasco, E.A. Functional classification of BRCA2 DNA variants by splicing assays in a large minigene with 9 exons. Hum. Mutat. 2015, 36, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno-Martínez, E.; Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Lorca, V.; Gómez-Sanz, A.; Carvalho, S.; Allen, J.; Infante, M.; Pérez-Segura, P.; Lázaro, C.; et al. Rad51d aberrant splicing in breast cancer: Identification of splicing regulatory elements and minigene-based evaluation of 53 dna variants. Cancers 2021, 13, 2845. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Díez-Gómez, B.; Velásquez-Zapata, V.; Acedo, A.; Sanz, D.J.; Velasco, E.A. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017, 13, e1006691. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Harrison, S.M.; Boucher, K.M.; Biesecker, L.G. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum. Mutat. 2020, 41, 1734–1737. [Google Scholar] [CrossRef]
- Bueno-Martínez, E.; Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Esteban-Sánchez, A.; Lorca, V.; Llinares-Burguet, I.; Allen, J.; García-Álvarez, A.; Pérez-Segura, P.; Durán, M.; et al. Minigene-based splicing analysis and ACMG/AMP-based tentative classification of 56 ATM variants. J. Pathol. 2022; in press. [Google Scholar]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Tambini, C.E.; Thacker, J. Identification of Functional Domains in the RAD51L2 (RAD51C) Protein and Its Requirement for Gene Conversion. J. Biol. Chem. 2003, 278, 45445–45450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.A.; Sawicka, D.; Barsky, D.; Albala, J.S. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res. 2004, 32, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gayarre, J.; Martín-Gimeno, P.; Osorio, A.; Paumard, B.; Barroso, A.; Fernández, V.; De La Hoya, M.; Rojo, A.; Caldés, T.; Palacios, J.; et al. Characterisation of the novel deleterious RAD51C p.Arg312Trp variant and prioritisation criteria for functional analysis of RAD51C missense changes. Br. J. Cancer 2017, 117, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.D.; Mensaert, K.; López-Perolio, I.; Tserpelis, D.; Xenakis, M.; Lattimore, V.; Walker, L.C.; Kvist, A.; Vega, A.; Gutiérrez-Enríquez, S.; et al. Targeted RNA-seq successfully identifies normal and pathogenic splicing events in breast/ovarian cancer susceptibility and Lynch syndrome genes. Int. J. Cancer 2019, 145, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Makhnoon, S.; Shirts, B.H.; Bowen, D.J. Patients’ perspectives of variants of uncertain significance and strategies for uncertainty management. J. Genet. Couns. 2019, 28, 313–325. [Google Scholar] [CrossRef]
- Truty, R.; Ouyang, K.; Rojahn, S.; Garcia, S.; Colavin, A.; Hamlington, B.; Freivogel, M.; Nussbaum, R.L.; Nykamp, K.; Aradhya, S. Spectrum of splicing variants in disease genes and the ability of RNA analysis to reduce uncertainty in clinical interpretation. Am. J. Hum. Genet. 2021, 108, 696. [Google Scholar] [CrossRef]
- Nix, P.; Mundt, E.; Coffee, B.; Goossen, E.; Warf, B.M.; Brown, K.; Bowles, K.; Roa, B. Interpretation of BRCA2 Splicing Variants: A Case Series of Challenging Variant Interpretations and the Importance of Functional RNA Analysis. Fam. Cancer 2021, 1, 7–19. [Google Scholar] [CrossRef]
- Ma, S.L.; Vega-Warner, V.; Gillies, C.; Sampson, M.G.; Kher, V.; Sethi, S.K.; Otto, E.A. Whole Exome Sequencing Reveals Novel PHEX Splice Site Mutations in Patients with Hypophosphatemic Rickets. PLoS ONE 2015, 10, e0130729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acedo, A.; Sanz, D.J.; Durán, M.; Infante, M.; Pérez-Cabornero, L.; Miner, C.; Velasco, E.A. Comprehensive splicing functional analysis of DNA variants of the BRCA2 gene by hybrid minigenes. Breast Cancer Res. 2012, 14, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Caloca, M.J.; Gómez-Barrero, S.; Velasco, E.A. Minigene Splicing Assays Identify 12 Spliceogenic Variants of BRCA2 Exons 14 and 15. Front. Genet. 2019, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Goina, E.; Acedo, A.; Buratti, E.; Velasco, E.A. Mis-splicing in breast cancer: Identification of pathogenic BRCA2 variants by systematic minigene assays. J. Pathol. 2019, 248, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Roca, X.; Olson, A.J.; Rao, A.R.; Enerly, E.; Kristensen, V.N.; Børresen-Dale, A.-L.; Andresen, B.S.; Krainer, A.R.; Sachidanandam, R. Features of 5′-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res. 2008, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Madsen, P.P.; Kibæk, M.; Roca, X.; Sachidanandam, R.; Krainer, A.R.; Christensen, E.; Steiner, R.D.; Gibson, K.M.; Corydon, T.J.; Knudsen, I.; et al. Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3+3A>G mutation that causes exon skipping. Hum. Genet. 2005, 118, 680–690. [Google Scholar] [CrossRef]
- Parada, G.E.; Munita, R.; Cerda, C.A.; Gysling, K. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 2014, 42, 10564–10578. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-H.; Tang, X.-Y.; Boulling, A.; Zou, W.-B.; Masson, E.; Fichou, Y.; Raud, L.; Le Tertre, M.; Deng, S.-J.; Berlivet, I.; et al. First estimate of the scale of canonical 5′ splice site GT > GC variants capable of generating wild-type transcripts. Hum. Mutat. 2019, 40, 1856–1873. [Google Scholar] [CrossRef]
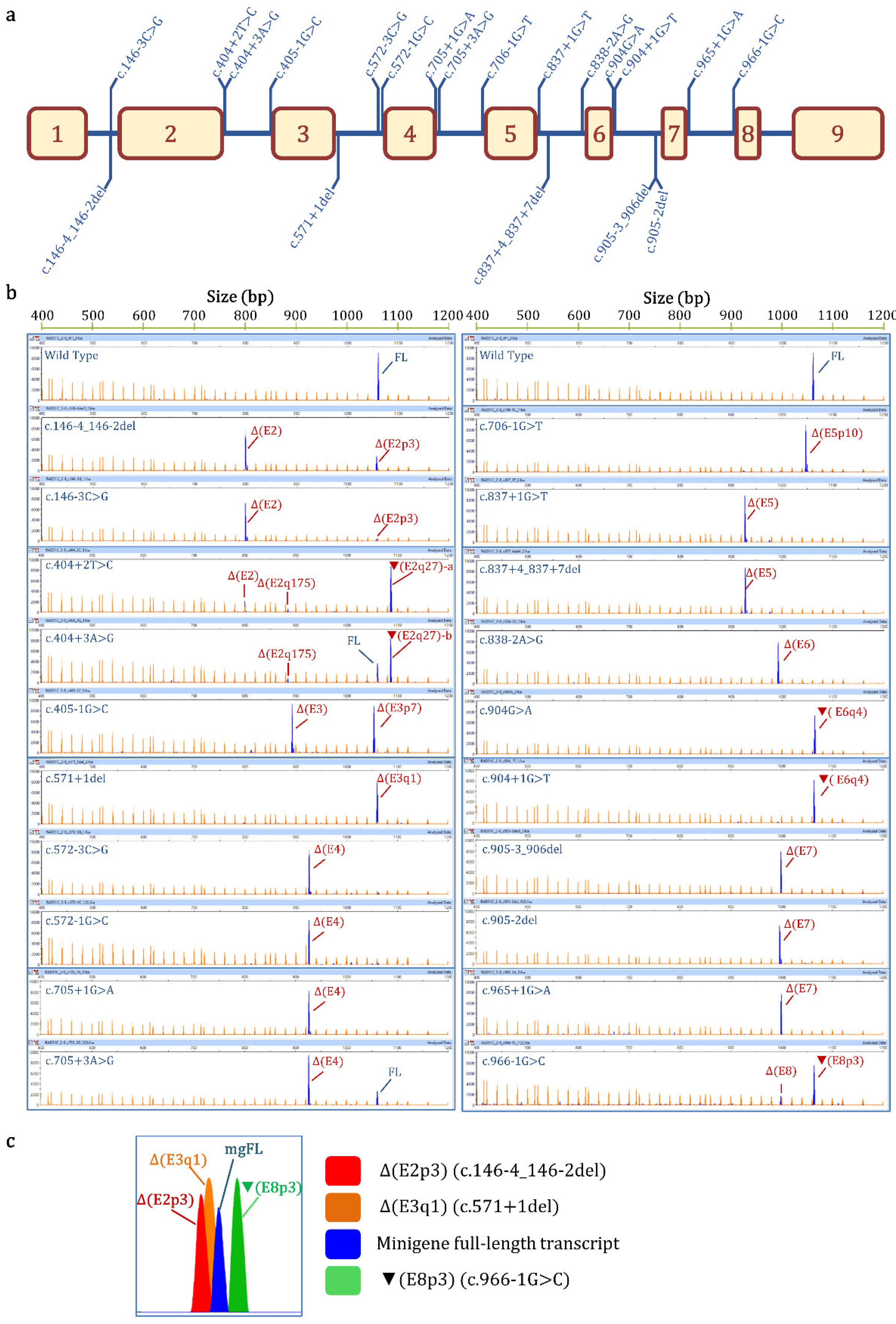

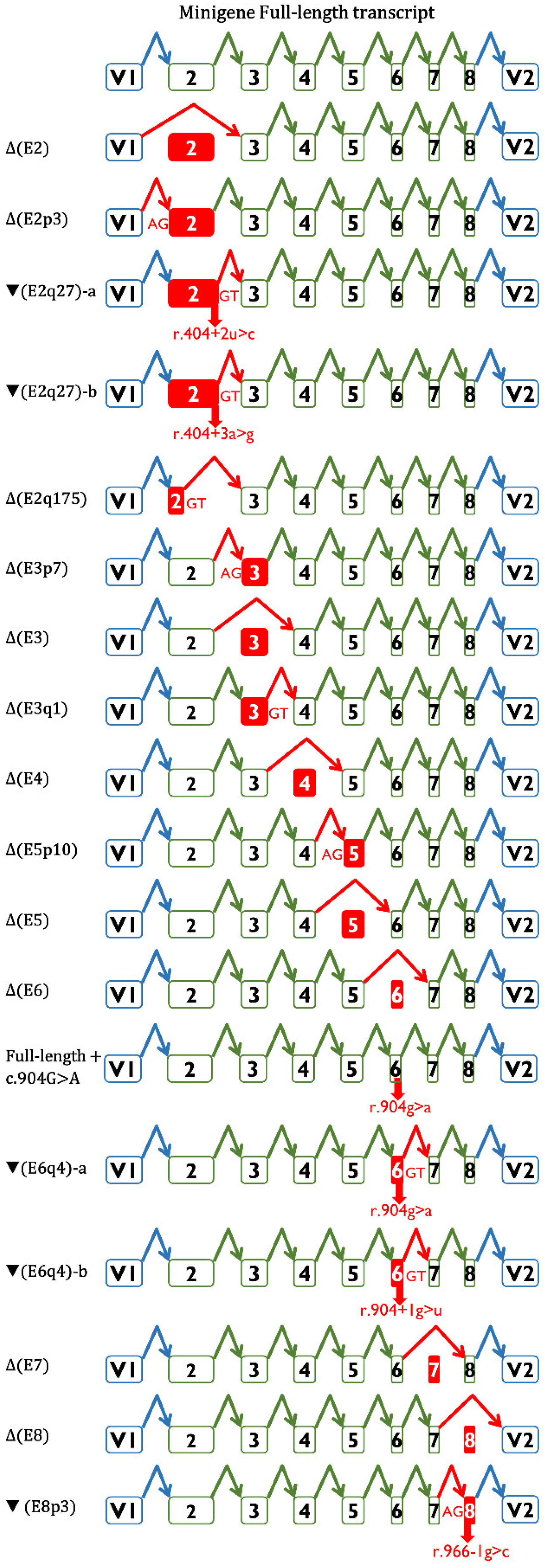

{kind=link}
{kind=link}
{kind=link}
| Variant (HGVS) 1 | Bioinformatics Summary 2 | Transcripts 3 | |||
|---|---|---|---|---|---|
| Canonical | PTC | In-Frame | Uncharacterized | ||
| Wild-type | 98.6% ± 0.2% | 1106 nt (1.4% ± 0.2%) | |||
| c.146-4_146-2del | [−]3′SS (9.5→ −0.8) [+]3′SS (5.9) 3 nt downstream | - | Δ(E2): 73.8% ± 0.8% | Δ(E2p3): 25.1% ± 0.4% | 657nt (1.1% ± 0.9%) |
| c.146-3C>G | [−]3′SS (9.5→ 1.9) | - | Δ(E2): 94.8% ± 0.9% | Δ(E2p3): 5.2% ± 0.9% | |
| c.404+2T>C | [−]5′SS (4.8→ −3.0) Cr. 5′SS (5.4) 27nt downstream | - | ▼(E2q27)-a: 77.2% ± 1.3% Δ(E2): 16.7% ± 0.3% (E2q175): 4.7% ± 0.1% | 657 nt (1.4% ± 1.2%) | |
| c.404+3A>G | [−]5′SS (4.8→ 0.6) Cr. 5′SS (5.4) 27 nt downstream | 26.3% ± 0.4% | ▼(E2q27)-b: 66.4% ± 1.6% Δ(E2q175): 5.4% ± 0.3% | 657 nt (1.9% ± 1.6%) | |
| c.405-1G>C | [−]3′SS (7.7→ −0.4) [+]3′SS (4.2) 7 nt downstream | - | Δ(E3p7): 48.9 ± 1.6% Δ(E3): 48.2% ± 1.2% | 813 nt (2.9% ± 0.3%) | |
| c.571+1del | [−]5′SS (10.5→ −14.1) [+]5′SS (11.1) 1 nt upstream | - | Δ(E3q1): 98.4% ± 1.4% | 800 nt (1.6% ± 1.4%) | |
| c.572-3C>G | [−]3′SS (7.4→ −1.4) | - | Δ(E4): 94.7% ± 0.3% | 1063 nt (3.0% ± 0.2%) 1008 nt (2.3% ± 0.1%) | |
| c.572-1G>C | [−]3′SS (7.4→ −0.6) | - | Δ(E4): 93.8% ± 0.0% | 1008 nt (3.2% ± 0.0%) 1063 nt (1.5% ± 0.0%) 976 nt (1.5% ± 0.0%) | |
| c.705+1G>A | [−]5′SS (9.1→ 0.9) | - | Δ(E4): 100% | ||
| c.705+3A>G | [↓]5′SS (9.1→ 4.6) [+]5′SS (6.1) 2 nt downstream | 21.3% ± 1.3% | Δ(E4): 78.7% ± 1.3% | ||
| c.706-1G>T | [−]3′SS (11.1→ 2.5) [+]3′SS (4.3) 10 nt downstream | - | Δ(E5p10): 100% | ||
| c.837+1G>T | [−]5′SS (8.6→ 0.1) | - | Δ(E5): 95.3% ± 0.4% | 976 nt (4.7% ± 0.4) | |
| c.837+4_837+7del | [−]5′SS (8.6→ −8.9) | - | Δ(E5): 100% | ||
| c.838-2A>G | [−]3′SS (10.2→ 2.2) | - | Δ(E6): 98.4% ± 1.4% | 590 nt (1.6% ± 0.4) | |
| c.904G>A (p.Gly302Arg) | [−]5′SS (5.6→ 1) Cr. 5′SS (6.2) 4 nt downstream | 2.4% ± 0.1% | ▼(E6q4)-a: 97.6% ± 0.1% | ||
| c.904+1G>T | [−]5′SS (5.6→ −3.0) Cr. 5′SS (6.2) 4 nt downstream | - | ▼(E6q4)-b: 100% | ||
| c.905-3_906del | [−]3′SS (8.2→ −8.6) [+]3′SS (4.5) 7 nt downstream | - | Δ(E7): 100% | ||
| c.905-2del | [−]3′SS (8.2→ 2.1) | - | Δ(E7): 100% | ||
| c.965+1G>A | [−]5′SS (8.7→ 0.5) | - | Δ(E7): 100% | ||
| c.966-1G>C | [−]3′SS (7.3→ −0.8) [↑] Cr. 3′SS (6.8) 3 nt upstream | - | Δ(E8): 20.6% ± 0.1% | ▼(E8p3): 79.4% ± 0.1% | |
| HGVS 1 | ClinVar Accession | PVS1 2 | PP3/BP4 3 | PVS1_O/BP7_O mgR51C_ex2-8 4 | PM2 5 | pSAD-based ACMG/AMP-like Classification 6 | ClinVar Classification 7 |
|---|---|---|---|---|---|---|---|
| c.146-4_146-2del | VCV000482181.5 | PVS1 | N/A | PVS1_O_P (+1) [Δ(E2): 74%, P_VS+ Δ(E2p3): 26%, P_P] | (0/250,394) PM2_P (+1) | VUS (+2) | LP |
| c.146-3C>G | VCV000484752.4 | N/A | (−79.4%) PP3 | PVS1_O_VS (+8) [Δ(E2): 95% P_VS + Δ(E2p3): 5% P_P] | (0/250,394) PM2_P (+1) | LP (+9) | VUS |
| c.404+2T>C | VCV000182835.10 | PVS1 | N/A | PVS1_O_VS (+8) [▼(E2q27)-a: 77%, P_VS Δ(E2): 17%, P_VS (E2q175): 5%, P_VS] | (1/246,102) PM2_P (+1) | LP (+9) | P/LP |
| c.404+3A>G | VCV000409857.4 | N/A | (−92.5%) PP3 | PVS1_O_N/A BP7_O-N/A [▼(E2q27): 66%, P_VS + Δ(E2q175): 5%, P_VS + FL, 27% B_S] | (0/246,102) PM2_P (+1) | VUS (+1) | VUS |
| c.405-1G>C | VCV000141823.5 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E3p7): 49%, P_VS Δ(E3): 48%, P_VS] | (0/251,476) PM2_P (+1) | LP (+9) | LP |
| c.571+1del | VCV000482176.8 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E3q1): 98%, P_VS] | (1/251,452) PM2_P (+1) | LP (+9) | LP |
| c.572-3C>G | VCV000633386.5 | N/A | (−99.4%) PP3 | PVS1_O_VS (+8) [Δ(E4): 95%, P_VS] | (0/251,198) PM2_P (+1) | LP (+9) | VUS |
| c.572-1G>C | VCV000480497.10 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E4): 94%, P_VS] | (0/251,220) PM2_P (+1) | LP (+9) | P/LP |
| c.705+1G>A | VCV000230577.9 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E4): 100%, P_VS] | (0/251,038) PM2_P (+1) | LP (+9) | LP |
| c.705+3A>G | VCV000241775.7 | N/A | (−31.0%) PP3 | PVS1_O_N/A BP7_O_N/A [Δ(E4): 79%, P_VS + FL: 21%, B_S] | (0/250,946) PM2_P (+1) | VUS (+1) | VUS |
| c.706-1G>T | VCV000452310.4 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E5p10): 100%, P_VS] | (0/282,746) PM2_P (+1) | LP (+9) | LP |
| c.837+1G>T | VCV000241779.3 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E5): 95%, P_VS] | (0/251,374) PM2_P (+1) | LP (+9) | LP |
| c.837+4_837+7del | VCV000128212.8 | N/A | (−100.0%) PP3 | PVS1_O_VS (+8) [Δ(E5): 100%, P_VS] | (0/251,374) PM2_P (+1) | LP (+9) | LP(1);VUS(2) |
| c.838-2A>G | VCV000480508.3 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E6): 98%, P_VS] | (0/250,982) PM2_P (+1) | LP (+9) | LP |
| c.904G>A | VCV000478781.9 | N/A | (−89.3%) PP3 | PVS1_O_VS (+8) [▼(E6q4)-a: 98%, P_VS] | (0/250,832) PM2_P (+1) | LP (+9) | LP(1);VUS(3) |
| c.904+1G>T | VCV000480510.7 | PVS1 | N/A | PVS1_O_VS (+8) [▼(E6q4)-b: 100%, P_VS] | (0/250,832) PM2_P (+1) | LP (+9) | LP |
| c.905-3_906del | VCV000182846.7 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E7): 100%, P_VS] | (0/282,730) PM2_P (+1) | LP (+9) | P/LP |
| c.905-2del | VCV000230587.7 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E7): 100%, P_VS] | (0/282,730) PM2_P (+1) | LP (+9) | LP |
| c.965+1G>A | VCV000182838.5 | PVS1 | N/A | PVS1_O_VS (+8) [Δ(E7): 100%, P_VS] | (0/ 251118) PM2_P (+1) | LP (+9) | LP |
| c.966-1G>C | VCV000851327.3 | PVS1 | N/A | PVS1_O_P (+1) [Δ(E8): 21%, P_VS + ▼(E8p3): 79%, P_P] | (0/251,358) PM2_P (+1) | VUS (+2) | LP |
| RAD51C Variant | Splicing Motif 1 | Splicing Outcome 2 | Clinical Interpretation |
|---|---|---|---|
| Exon 2 | |||
| c.146-4_146-2del | [±]3′SS | Δ(E2): 73.8%; Δ(E2p3): 25.1% | VUS |
| c.146-3C>T | [−]3′SS | 100% mgFL-transcript | VUS 3 |
| c.146-3C>G | [−]3′SS | Δ(E2): 94.8%; Δ(E2p3): 5.2% | Likely Pathogenic |
| c.404G>A | [−]5′SS | ▼(E2q27): 69.3%; Δ(E2q175): 19.9%; Δ(E2q22): 4.3%; Δ(E2): 2.4% | Likely Pathogenic 3 |
| c.404+2T>C | [−]5′SS | ▼(E2q27): 77.2%; Δ(E2): 16.7%; (E2q175): 4.7% | Likely Pathogenic |
| c.404+3A>G | [−]5′SS | ▼(E2q27): 66.4%; mgFL: 26.3%; Δ(E2q175): 5.4% | VUS |
| Exon 3 | |||
| c.405-6T>A | [±]3′SS/Pyr | ▼(E3p4):95.2%; Δ(E3): 4.8% | Likely Pathogenic 3 |
| c.405-1G>C | [±]3′SS | Δ(E3p7): 48.9%; Δ(E3): 48.2% | Likely Pathogenic |
| c.571+1del | [±]5′SS | Δ(E3q1): 98.4% | Likely Pathogenic |
| c.571+4A>G | [±]5′SS | Δ(E3): 76.5%; ▼(E3q4): 11.6%; FL: 5.4%; Δ(E3q114): 4.0% | Likely Pathogenic 3 |
| c.571+5G>A | [−]5′SS | Δ(E3): 91.5%; Δ(E3q114): 4.8% | Pathogenic 3 |
| Exon 4 | |||
| c.572-3C>G | [−]3′SS | Δ(E4): 94.7% | Likely Pathogenic |
| c.572-1G>C | [−]3′SS | Δ(E4): 93.8% | Likely Pathogenic |
| c.572-1G>T | [−]3′SS | Δ(E4): 93.4% | Likely Pathogenic 3 |
| c.705G>T | [−]5′SS | Δ(E4): 100% | Likely Pathogenic 3 |
| c.705+1G>A | [−]5′SS | Δ(E4): 100% | Likely Pathogenic |
| c.705+3A>G | [−]5′SS | Δ(E4): 78.7%; mgFL: 21.3% | VUS |
| c.705+5G>C | [−]5′SS | mgFL: 51.6%; Δ(E4): 48.4% | VUS 3 |
| Exon 5 | |||
| c.706-2A>C | [±]3′SS | Δ(E5p10): 91.4%; Δ(E5): 4.0%; Δ(E5p52): 1.8% | Likely Pathogenic 3 |
| c.706-2A>G | [±]3′SS | Δ(E5): 65.4%; Δ(E5p10): 33.5% | Pathogenic 3 |
| c.706-1G>T | [±]3′SS | Δ(E5p10): 100% | Likely Pathogenic |
| c.837+1G>T | [−]5′SS | Δ(E5): 95.3% | Likely Pathogenic |
| c.837+2T>C | [−]5′SS | Δ(E5): 89.3%; Δ(E4_5): 2.2% | Likely Pathogenic 3 |
| c.837+4_837+7del | [−]5′SS | Δ(E5): 100% | Likely Pathogenic |
| Exon 6 | |||
| c.838-2A>G | [−]3′SS | Δ(E6): 98.4% | Likely Pathogenic |
| c.904G>A | [−]5′SS | ▼(E6q4): 97.6%; FL: 2.4% | Likely Pathogenic |
| c.904+1G>T | [−]5′SS | ▼(E6q4): 100% | Likely Pathogenic |
| Exon 7 | |||
| c.905-3_906del | [−]3′SS | Δ(E7): 100% | Likely Pathogenic |
| c.905-3C>G | [−]3′SS | Δ(E7): 98.1%; Δ(E7_8): 1.9% | Likely Pathogenic 3 |
| c.905-2del | [−]3′SS | Δ(E7): 100% | Likely Pathogenic |
| c.905-2A>C | [−]3′SS | Δ(E7): 97.4 % | Pathogenic 3 |
| c.905-2_905-1del | [−]3′SS | Δ(E7): 100% | Pathogenic 3 |
| c.965+1G>A | [−]5′SS | Δ(E7): 100% | Likely Pathogenic |
| c.965+5G>A | [−]5′SS | Δ(E7): 100% | Likely Pathogenic 3 |
| Exon 8 | |||
| c.966-3C>A | [−]3′SS | Δ(E8): 86.8%; ▼(E8p3): 9.7%; FL: 2% | VUS 3 |
| c.966-2A>G | [±]3′SS | Δ(E8): 86.7%; ▼(E8p3):11.0% | VUS 3 |
| c.966-2A>T | [±]3′SS | Δ(E8): 89.1%; ▼(E8p3):5.9% | VUS 3 |
| c.966-1G>C | [±]3′SS | ▼(E8p3): 79.4%; Δ(E8): 20.6% | VUS |
| c.1026+5_1026+7del | [−]5′SS | Δ(E8): 79.5%; Δ(E8q18):13.8%; ▼(E8q41): 3.3% | Pathogenic 3 |
| c.1026+5G>T | [−]5′SS | Δ(E8): 78.0%; Δ(E8q18):18.7%; ▼(E8q44): 1.4% | Likely Pathogenic 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanoguera-Miralles, L.; Bueno-Martínez, E.; Valenzuela-Palomo, A.; Esteban-Sánchez, A.; Llinares-Burguet, I.; Pérez-Segura, P.; García-Álvarez, A.; de la Hoya, M.; Velasco-Sampedro, E.A. Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C. Cancers 2022, 14, 2960. https://doi.org/10.3390/cancers14122960
Sanoguera-Miralles L, Bueno-Martínez E, Valenzuela-Palomo A, Esteban-Sánchez A, Llinares-Burguet I, Pérez-Segura P, García-Álvarez A, de la Hoya M, Velasco-Sampedro EA. Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C. Cancers. 2022; 14(12):2960. https://doi.org/10.3390/cancers14122960
Chicago/Turabian StyleSanoguera-Miralles, Lara, Elena Bueno-Martínez, Alberto Valenzuela-Palomo, Ada Esteban-Sánchez, Inés Llinares-Burguet, Pedro Pérez-Segura, Alicia García-Álvarez, Miguel de la Hoya, and Eladio A. Velasco-Sampedro. 2022. "Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C" Cancers 14, no. 12: 2960. https://doi.org/10.3390/cancers14122960
APA StyleSanoguera-Miralles, L., Bueno-Martínez, E., Valenzuela-Palomo, A., Esteban-Sánchez, A., Llinares-Burguet, I., Pérez-Segura, P., García-Álvarez, A., de la Hoya, M., & Velasco-Sampedro, E. A. (2022). Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C. Cancers, 14(12), 2960. https://doi.org/10.3390/cancers14122960