Simple Summary
Galectins are a family of β-galactoside binding proteins whose levels are altered in various stages of different types of cancer. This study provides an analytical comparison of 50 frequently mutated genes in two common cancers and the serum levels of the galectin proteins. The goal is the revelation of potential relationships between the mutation status of these genes and serum levels of galectins. We found that mutations in the KIT gene (which codes for the proto-oncogene c-KIT protein) are associated with increased circulating levels of certain galectins. We also found that patient samples originating from brain tissue have a higher likelihood of having a mutation in the KIT gene. Understanding the relationship between cancer-critical gene mutations and serum galectin levels could provide a feasible and non-invasive avenue to better understand the tumor’s unique genetic profile.
Abstract
To investigate a potential role for galectins as biomarkers that enable diagnosis or prognostication of breast or non-small cell lung cancer, the serum levels of galectins -1, -3, -7, -8, and -9 of cancer patients determined by ELISA assays were compared to the mutation status of 50 known cancer-critical genes, which were determined using multiplex PCR in tumors of the same patients. Mutations in the KIT proto-oncogene, which codes for the c-Kit protein, a receptor tyrosine kinase, correlated with higher levels of galectins -1, -3, -8, and -9 in breast cancer patients and galectin-1 in non-small cell lung cancer patients. Mutations in the KIT gene were more likely found in brain metastases from both of these primary cancers. The most common KIT mutation in our panel was p.M541L, a missense mutation in the transmembrane domain of the c-Kit protein. These results demonstrate an association between KIT oncogenic signaling and elevated serum galectins in patients with metastatic disease. Changes in protein trafficking and the glycocalyx composition of cancer cells may explain the observed alterations in galectin expression. This study can be useful for the targeted selection of receptor tyrosine kinase and galectin inhibitor anti-cancer treatments.
Keywords:
galectin; KIT gene; mutation; c-Kit; CD117; breast cancer; non-small cell lung cancer; ELISA; cancer hotspot panel 1. Introduction
Lung cancer has one of the lowest 5-year survival rates of any cancer in the United States at 21% and is the greatest cause of cancer deaths in both men and women. Although the 5-year relative survival rate of breast cancer is 90% for all subtypes, its high incidence still results in tens of thousands of deaths annually in the United States [1]. Once metastasis occurs, the survival rate greatly drops, with a majority (66.7%) of all solid tumor cancer deaths being caused by metastasis [2]. Sixteen to twenty percent of those diagnosed with lung cancer, and 5.1% with breast cancer, develop metastasis to the brain [3,4]. The high incidence and metastatic ability make these cancers of primary interest for study.
1.1. Cancer-Critical Genes
Multiple genes in the human genome, when mutated, enable the development and progression of neoplastic tissue. Oncogenes are genes that, when mutated, create a product with a gain-of-function (GOF) that allows it to contribute to the dysregulation of the cell. Conversely, tumor suppressor genes lose their ability to protect the cell from dysregulated growth and proliferation when they are mutated. This paper refers to both groups of genes collectively as “cancer critical” genes.
A recent comprehensive study of 9423 tumor exomes identified 299 cancer driver genes [5]. This study evaluates 50 of those cancer-critical genes, providing a broad screening of genes commonly mutated in multiple cellular pathways. The nine involved pathways are the RTK/RAS/MAP, TGFβ, PI3K, Wnt, GPCR, p53, JAK/STAT, Notch, and the cell cycle pathway. These genes and their respective pathways are highlighted in Figure 1. The graphic is not comprehensive of all the possible cancer-critical genes or the full signaling pathways, it is instead designed to highlight the genes used in this study and their potential contribution to unregulated proliferation.
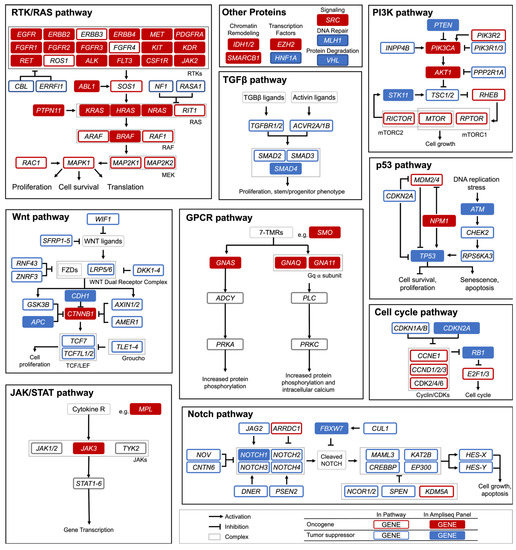
Figure 1.
Signaling Pathways with Cancer Critical Genes. Hotspot panel genes in this study are shaded in red (oncogenes), and blue (tumor suppressor genes). RTK: Receptor Tyrosine Kinase; RAS: Rat Sarcoma; TGFβ: Transforming Growth Factor Beta; PI3K: Phosphoinositide 3-kinase; GPCR: G-Protein Coupled Receptor; JAK: Janus Kinase; STAT: Signal Transducer and Activator of Transcription Proteins. Figure modified from Sanchez-Vega et al. [6].
Seven genes were not a part of any canonical cancer-causing pathway or were members of multiple pathways (e.g., SRC); these are grouped together as “Other” in Figure 1. These genes are involved in DNA repair, genomic stability, epigenetic modification, etc.
The KIT gene codes for c-Kit, a class III receptor tyrosine kinase (RTK) which binds to extracellular Stem Cell Factor (SCF) and activates the PI3K, JAK/STAT, and MAPK pathways in hematopoietic cells, resulting in proliferation and differentiation [7,8,9]. This RTK is also highly expressed by glandular and myoepithelial breast cells [10]. Furthermore, c-Kit is known to play roles in several cancers via gain-of-function and loss-of-function mutations; most notably in gastrointestinal stromal tumors (GIST), and also in melanoma and thyroid carcinoma [11,12,13,14]. Several mutations in c-KIT have been associated with the development of cancers. These mutations are typically missense mutations that usually result in gain-of-function or an otherwise unknown result [15].
The incidence of brain metastasis in breast cancer patients is possibly increasing and is high in patients with hormone receptor-negative tumors with human epidermal growth factor receptor 2 (HER2) overexpression [16,17,18,19,20,21]. Similarly, patients with specific molecular subtypes of NSCLC have an increased predisposition for brain metastases, such as those with an epidermal growth factor receptor (EGFR) mutation, or anaplastic lymphoma kinase (ALK) rearrangement [22,23,24].
Targeted cancer therapy offers more precise cancer treatment with fewer cytotoxic effects on non-cancer cells [25]. This level of therapy requires knowing the cancer’s specific genetic makeup to identify druggable targets. For example, osimertinib, a tyrosine kinase inhibitor, is a targeted therapy for patients with NSCLC with specific sensitizing mutations (p.Thr790Met and p.Leu858Arg) in the EGFR gene [26,27,28,29,30,31,32]. A more thorough understanding of the cellular biology of cancer will reveal the therapeutic targets involved in growth and is a promising strategy for reducing mortality from cancer and its metastases.
1.2. Galectins and Their Role in Cancer
Galectins (formerly known as S-type lectins) are a family of lectin proteins which share a domain with high-affinity binding for β-galactoside sugars. Galectins are divided into three subfamilies based on their structures: prototypical, chimeric, and tandem-repeat [33]. Among other functions, galectins are players in the innate immune system, triggering immune responses as well as resolving inflammation [34]. Further, galectins modulate adaptive immune responses, such as Gal-9 or Gal-1, acting to dampen activated T cell responses [35]. Galectins also have several functions outside of the immune system. They interact with cellular proteins via binding to protein glycosylation sites [36,37]. They can form lattice networks with cell membrane receptors and modulate the functions and transportation of the receptors [38]. Galectins have intracellular interactions as well and enhance oncogenic signals and promote tumor proliferation [39].
Galectin-1, -3, and -9 have been implicated in cancer progression, metastasis, and angiogenesis [40,41,42,43,44]. Galectins-7 and -8 have less studied properties in cancer but are known to support metastatic breast cancer and colon cancer, respectively [45,46]. Their understudied status warrants their inclusion in this study.
In breast cancer, galectins have several roles. Galectins-1 and -3 specifically have been implicated in the progression of lesions into metastatic disease through their roles in cell-to-cell and cell-to-extracellular matrix interactions [47]. Galectin-7 has been found to have interactions with p53 that can induce chemoresistance [45,48]. Increased levels of galectin-8 were shown to lead to lower survival rates [49]. Lastly, galectin-9 has increased expression in breast cancer, and its interactions with Tim-3 may provide an escape from cytotoxic T cells [50,51].
In regard to NSCLC, galectin-1 is overexpressed in these cell lines and in tissue samples from lung cancer patients [52,53]. Knockdown of Gal-1 in lung adenocarcinoma results in reduced tumor growth in vivo and inhibited migration, invasion, and colony formation in vitro [53]. Galectin-3 is also more highly expressed in NSCLC and augments tumorigenesis, invasion, metastasis, and tumor immunity [54]. Galectins -7 and -8 have been shown to have higher mRNA expression levels in NSCLC [55]; however, no studies have examined their roles. Galectin-9 expression in NSCLC is found to be a favorable prognostic marker due to interactions between tumor-infiltrating lymphocytes and tumor cells [55,56].
The tissue levels of galectins are well known to be altered in breast and lung cancer [42,52,57,58,59,60,61,62]. This results in measurably altered serum levels of these galectins [52,63,64,65]. The mechanism for the altered serum expression of galectins in cancer patients remains unclear, however, there are potential explanations. Galectins are secreted in a currently enigmatic non-classical pathway and their trafficking is controlled at different points within the cell [66,67,68,69,70]. The abnormal cellular processes of cancer cells quite possibly result in the dysregulation of the processes involved in galectin secretion. Additionally, while normal cellular glycosylation is required for proper functioning, cancer cells have deviant glycosylation [71,72,73]. This could disturb the type and number of glycoconjugates for which galectins bind. The alterations in both trafficking and the glycomic profile, in turn, could lead to altered galectin levels in these neoplastic tissues, resulting in their demonstrably different serum levels.
As such, levels of galectin-3 have been especially implicated as an emerging biomarker for neoplasms [39,44,74,75,76,77]. Other galectins also have the potential to serve as biomarkers for diseases, such as cancer [77,78,79,80].
Given galectin’s dysregulation in the cancer environment and a possible therapeutic target, several galectin inhibitors have been developed [41,78,81,82]. GR-MD-02 (a galectin-3 inhibitor) is currently in clinical trials to evaluate its usefulness in cancer patients with melanoma, NSCLC, and squamous cell head and neck cancer (NCT02117362, NCT02575404).
Galectins interact with proteins commonly mutated in cancer. Galectin-1 and -3 have interactions with the RAS family of proteins [53,83,84,85,86] and EGFR and TGFβ receptors [87]. Thus, the interplay between galectin levels and mutations in these cancer-critical genes has clinical importance.
Finally, galectins are known players in cancer metastasis. Galectins -1 and -3 are particularly well studied in this aspect. Galectin-3 has been identified as a metastasis-related protein as early as 1998 [88]. Galectin-1 is upregulated in more advanced breast cancers of higher TNM stages and correlates with metastasis to regional lymph nodes [58,89]. Molecularly, the lectin interacts with laminin and fibronectin to promote aggregation [90,91]. Galectin-1 is able to upregulate MMP-2 and MMP-9 and reorganize cytoskeletal elements by activating Cdc42 to increase the amount of filopodia in oral squamous cell carcinoma cells [92]. Knockdown of galectin-1 reduced prostate cancer migration by suppressing androgen receptors and Akt signaling [93].
While many studies show changes in galectin levels during cancer, no comprehensive work has been done to correlate galectin levels with cancer-critical gene mutations in cancer patients [65,77,78,80,94,95]. This paper seeks to provide an initial exploration into serum galectin levels and their correlation with cancer-critical gene mutations in breast and non-small cell lung cancer patients.
2. Materials and Methods
2.1. Patient Samples
Seventy-four cancer patient serum samples were obtained from the Prisma Health Cancer Institute (PHCI) biorepository (Greenville, SC, USA). The collection years ranged from 2012–2018. The PHCI biorepository houses inventory, including live cryopreserved, snap-frozen, and formalin-fixed paraffin-embedded tissues, and blood (whole blood, plasma, and serum). Patient donor permission was obtained via participant informed consent prior to the collection and storage of specimens. The biorepository standard operating procedures include specimen handling and tracking (i.e., collection, processing, storage) and facilities management and operations (i.e., equipment maintenance and monitoring). The PHCI biorepository has been acknowledged in various publications, having provided all specimen types in its inventory for previously conducted research projects [65,96,97].
Thirty-five samples were from breast cancer patients (F:M 1:0, median age 60.6, min-max 33–87) and the other 39 were from NSCLC patients (F:M 17:22, median age 65.1, min-max 47–79). Ten samples were obtained of stages I, II, and III of breast and lung cancer, 5 of stage IV breast cancer, and 9 of stage IV lung cancer. In the breast cancer samples, 31 were ductal, 2 were lobular and 2 were coded non-specifically as “adenocarcinoma” histology. In the lung cancer samples, 24 were adenocarcinoma, 13 were squamous cell and 2 were large cell histology. The samples from the patients contained a random mix of primary tumors and metastatic tissue.
Patient information was collected from the PHCI database. The information included demographic data, such as age, race, gender, and smoking status, as well as tumor data, including TNM staging, grade, histology, site, and cancer stage. This information is available in Supplemental Materials (Tables S1 and S2)..
2.2. Galectin Profiling
The patient’s serum was used to determine the circulating galectin levels using an enzyme-linked immunosorbent assay (ELISA) [65]. This study used a subset of the data described by Blair et. al. (2021). Galectin-1, -3, and -9 concentrations were obtained using the ELISA kits from R&D Systems (Minneapolis, MN, USA). Galectin-7 and -8 concentrations were determined using the ELISA kits from Invitrogen (Carlsbad, CA, USA). Each sample was assayed four times. ELISA kit quality control information can be found in Supplemental Materials as Table S3.
2.3. Cancer HotSpot Panel
Mutation information of the patient’s tumors was provided by the PHCI. The cancer hotspot panel screening was performed by Precision Genetics (Greenville, SC, USA). The Ion Ampliseq Cancer Hotspot Panel, v2 (Life Technologies Corporation, Carlsbad, CA, USA), was used to determine the mutation status of 50 genes in the patients. This panel amplifies 207 amplicons, which cover approximately 2800 mutations in the Catalogue of Somatic Mutations in Cancer (COSMIC) from 50 oncogenes and tumor suppressor genes.
Oncogenes: ABL1, AKT1, ALK, BRAF, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MPL, NPM1, NRAS, PDGFRA, PIK3CA, PTPN11, RET, SMARCB1, SMO, SRC.
Tumor Suppressors: APC, ATM, CDH1, CDKN2A, FBXW7, HNF1A, MLH1, NOTCH1, PTEN, RB1, SMAD4, STK11, TP53, VHL.
The AmpliSeq Cancer Hotspot panel (v2), from which all variants were identified, was validated as a laboratory-developed test (LDT) under the Clinical Laboratory Improvement Amendments (CLIA) guidelines. The accuracy of all variant calls was validated at 99.8%. The sensitivity of the variants was detected at a lower limit of 5% allele frequency down to 30% tumor content (cell admixture). The precision of variant detection was shown to be 99.8% between operators and 98.9% within the operator. False variant calls, a measure of specificity, were less than 1% from the CLIA validation.
Each sequencing run had minimum criteria for variant calls. Coverage across the entire panel must be greater than 90% at 300X for the sequencing run to be analyzed further. A minimum read depth of 100X and 5% allele frequency must be observed for individual variants to be reported. Finally, homopolymer indels and variants within 10 bp of amplicon ends were filtered to minimize the likelihood of false positives.
Each sequencing run included the AcroMetrix Oncology Hotspot Control, which is designed to control the hundreds of amplicons targeted by next-generation sequencing (NGS) panels. It contains over 500 mutations from the COSMIC database and has five variant types of varying nucleotide lengths. The 53 genes represented in the AcroMetrix Oncology Hotspot Control are: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, FOXL2, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MAP2K1, MET, MLH1, MPL, MSH6, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL.
This control was calibrated using the analysis parameters detailed in the CLIA validation. The resulting analysis yielded 351 detected variants, and these variants served as the reference set for quality control of each sequencing run. As a quality control measure for variant detection, a minimum of 344 variants (95%) must be identified from each sequencing run for variants from clinical specimens to be reported. A detailed quality control log was maintained, which documented the results from each run and was a part of the routine CLIA compliance.
2.4. Data Analysis
All statistical analyses were performed using JMP® software by the SAS Institute (Cary, NC, USA). The distributions of the serum galectin levels were analyzed for normality. The distributions for each galectin in a mutated gene were compared to those of patients with a non-mutated version of the same gene by t-test.
Contingency analyses were performed on the mutation status of genes against other categorical variables, such as tissue site and histology. The odds ratios were calculated for the KIT mutations and brain metastases in both cancers. Both cancers were analyzed separately. Values of p less than 0.05 were considered statistically significant.
3. Results
The levels of circulating galectins -1, -3, -7, -8 and -9 in breast and lung cancer patients were revealed by an ELISA assay of patient serum [65]. Tumor tissues from the same patients were analyzed for mutations in 50 cancer-critical genes by multiplex polymerase chain reaction (PCR). The mutation status of these genes was compared to the circulating levels of galectins in the cancer patients.
3.1. Serum Galectin Levels
Table 1 and Table 2 contain the serum galectin levels for the cancer patient groups. Some samples were excluded from further analysis due to the reliability of the results.

Table 1.
Breast Cancer Patient Serum Galectin Levels.
Table 2.
Lung Cancer Patient Serum Galectin Levels.
3.2. Mutation Frequency
Table 3 ranks the genes by frequency of mutation among breast cancer patients as well as the specific mutation. PIK3CA and TP53 were the most mutated genes in this group patients.
Table 3.
Gene Mutation Frequency in Breast Cancer Patients.
Table 4 provides the mutations and their frequencies in the lung cancer patient sample group. TP53 and KDR (VEGF2) were the most mutated genes in this group.
Table 4.
Gene Mutation Frequency in Lung Cancer Patients.
3.3. Associations with Galectins
The screening of galectin levels by gene mutations found several associations between the serum galectin levels and cancer-critical gene mutations. Most notable are the associations with multiple galectin levels and the KIT gene. Figure 2 shows a heat map of the t-test results of comparing the serum galectin levels in patients with a mutated gene to patients with a wild-type gene.
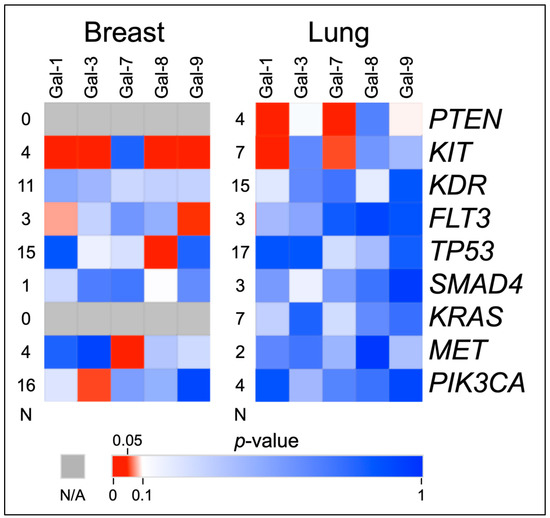
Figure 2.
Heat Map of Mutated versus Wild-Type Galectin Levels T-Tests. This figure shows the p-values of t-test comparisons of the serum galectin levels in mutated vs. non-mutated genes in breast and lung cancer patients. Levels of galectin-1, -3, -8, and -9 are significantly elevated when KIT is mutated in breast cancer samples. Levels of galectin-1 and -7 are significantly elevated when PTEN and KIT are mutated in lung cancer samples. “N” indicates the number of mutated samples in the comparison.
Figure 2 shows that despite the small number of mutations in the KIT gene, they correlated with increases in several galectins in both cancers. These comparisons are charted in Figure 3 and Figure 4. Additionally, PTEN was found to have some correlations with galectin levels in NSCLC patients, as seen in Figure 4.
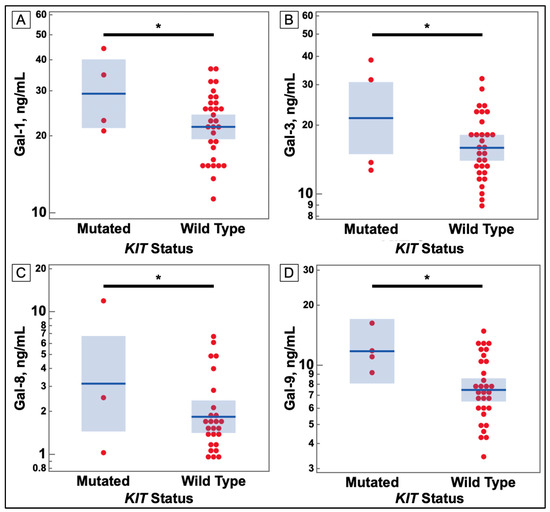
Figure 3.
Comparison of Serum Galectin Levels of Breast Cancer Patients KIT Gene Mutation Status in Tumor Sample. (A) Serum levels of galectin-1, as determined by ELISA, were significantly higher in breast cancer patients with a mutation in the KIT gene. (B) Serum levels of galectin-3, as determined by ELISA, were significantly higher in breast cancer patients with a mutation in the KIT gene. (C) Serum levels of galectin-8, as determined by ELISA, were significantly higher in breast cancer patients with a mutation in the KIT gene. (D) Serum levels of galectin-9, as determined by ELISA, were significantly higher in breast cancer patients with a mutation in the KIT gene. (* p-value ≤ 0.05).
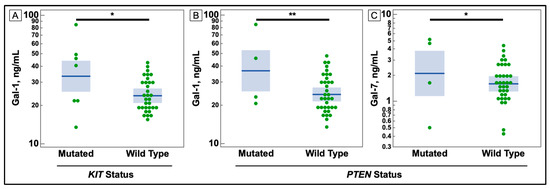
Figure 4.
Comparison of Serum Galectin Levels of Non-small Cell Lung Cancer Patients with KIT and PTEN Mutations in Tumor Sample. (A) Serum levels of galectin-1, as determined by ELISA, were significantly higher in non-small cell lung cancer patients with a mutation in the KIT gene. (B,C) Serum levels of galectins -1, and -7 were elevated in patients with a mutation in the PTEN gene. (* p-value ≤ 0.05, ** ≤ 0.01).
3.4. Associations with Brain Metastases
Figure 5 shows the contingency analysis of the presence of a KIT mutation at the site of the tissue biopsy of the tumor. Tumor samples taken from the brains of cancer patients were significantly more likely to have a mutation in the KIT gene.
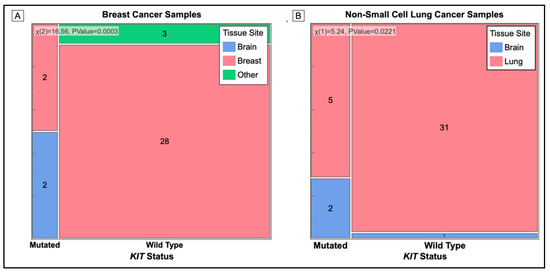
Figure 5.
Mosaic Plot of Cancer Tissue Biopsy Site (Primary and Metastatic) used for Mutation Analysis. Cell sizes are proportional to the number of samples in that category. Mutation status is on the X-axis and color represents the tissue sample of origin. (A) All KIT mutations occur in biopsy samples of breast cancer taken from the brain. The p-value = 0.0003. (B) Biopsy samples taken from the brain have a higher number of KIT mutations than samples from the lung. The p-value = 0.0221.
Table 5 shows the odds ratio between having a KIT mutation and brain metastasis. Our sample population did not contain a breast cancer sample with a brain metastasis and wild-type KIT and therefore, no ratio could be calculated for the group.
Table 5.
Odds Ratio of KIT Mutation and Brain Metastasis in Breast and Lung Cancer Patients.
In summary, in breast cancer patients, we find that PIK3CA and TP53 were the most mutated genes while TP53 and KDR (VEGF2) were the most mutated genes in the lung cancer patients. Levels of galectins -1, -3, -8, and -9 were elevated in patients with mutations in the KIT gene. Simultaneously, samples from a brain metastasis of breast and lung cancer patients had more KIT gene mutations than samples from the primary tumor.
4. Discussion
Galectins -1, -3, -8, and -9 were found to be at higher levels in sera of breast cancer patients with a mutation in the KIT gene than other cancer patients without the mutation.
The most common KIT mutation in our panel was p.Met541Leu (rs3822214). This mutation occurs in the transmembrane region of the protein and has not been implicated as a mutation of clinical concern [98]. Since the mutation occurs in the transmembrane region, some have theorized that the mutation is loss-of-function and impairs the insertion of the receptor into the membrane [99]. However, studies have shown that the p.Met541Leu mutation increases the RTK’s affinity for its ligand, SCF [100,101]. One study found that chronic myelogenous leukemia (CML) patients with this mutation had altered white blood cell counts and overall survival [101].
Our study joins others in finding increasing potential clinical significance for this missense mutation [102]. We investigated the allele frequency of the mutation in these patients and found it indicates a heterozygous germline mutation. This is supported by studies which find that this mutation is common (8.1% allele frequency) in the Caucasian population [103]. For comparison, the mutation appears in 8.57% of our breast cancer patients and 15.38% of the lung cancer patients for 12.61% overall.
Galectins and RTKs, such as c-Kit, are known to have an abundant number of interactions [104]. There is no literature to indicate specific interactions between galectins -1, -3, -8, and -9 and c-Kit, although it is known that galectins do interact with other members of this class of RTKs, such as platelet-derived growth factor receptor (PDGFR) via spatial organization and trafficking [105,106,107].
The association between the c-Kit mutation and increased levels of certain galectins is interesting. There are a variety of possible interpretations of this finding (Figure 6). The mutation could lead to altered receptor glycosylation, which would in turn affect the galectin serum levels. Galectin expression could be upregulated by the GOF c-Kit mutations via the activated intracellular pathways. The nature of the interaction is of interest and worthy of future studies.
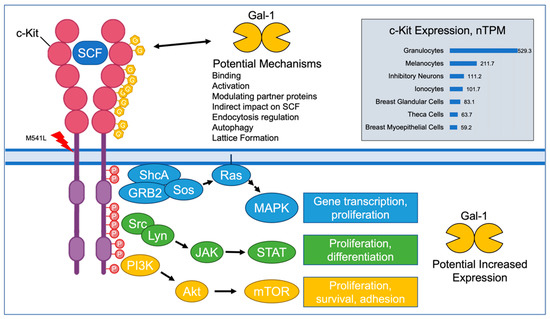
Figure 6.
Potential Interactions between c-Kit and Galectin-1. c-Kit is expressed in immune cells, melanocytes, neurons, and breast tissue cells. Galectin-1 can bind to appropriate glycosylation sites on c-Kit with a variety of possible results affecting c-Kit functioning. The downstream effects of increased c-Kit activity could include increased transcription of galectin-1 levels. (SCF: stem cell factor; nTPM: normalized transcriptions per million; Gal-1: galectin-1) [10,104,108,109].
A query of the TCGA database via UALCAN shows that galectins -1, and -3, have decreased expression in breast invasive carcinoma, suggesting that the observed increase in these galectins could be of a non-tumor origin. The database also shows that galectins -8 and -9 have increased expression in breast cancer tissue [110].
This study also found that tissue samples taken from the metastasis in patients’ brains were more likely to have a mutated KIT gene. It is unclear why a mutated c-Kit protein would result in this outcome and, in fact, one study has shown that the loss of c-Kit expression has been associated with advanced stages of breast cancer [111]. It is possible that the mutation reduces the stability of the c-Kit protein.
4.1. Impact of Findings
These findings serve to further enhance the understanding of the role of galectins in the cancer setting. Serum levels of certain galectins are known to be increased in cancer [65]. Our study shows that certain galectins could have increased serum levels when certain cancer-critical genes are mutated in the tumor sample, indicating a potential relationship.
Additionally, given the high frequency of the p.Met541Leu c-Kit mutation in the general population, its cause for concern in other studies [101,102] and its correlation with brain metastasis in cancer patients of this study, the p.Met541Leu mutation is a potential marker for more aggressive cancer and has promise for future studies.
The practical application of this research is the discovery of further molecular changes correlated with specific tumor mutations. The Ampliseq hotspot panel provides a gene panel that can be used to investigate many genes of interest, not only in breast and lung cancers but in other cancers and diseases as well.
Further investigations could find blood serum markers that better correlate with the mutation status of cancer-critical genes. This approach has applications in both diagnostics and treatment, as the mutation status of specific proteins often translates to their response to cancer treatments. For example, p.Met541Leu KIT-expressing cells have been shown to have increased sensitivity to imatinib, a c-Kit inhibitor [112]. This is a practical goal, as cancer treatment is tailored to specific mutations and the galectin levels can be targeted by galectin inhibitors.
4.2. Study Limitations
Our sample size of 35 breast cancer samples and 39 lung cancer samples reflects the availability of the hotspot panel sequencing data and the pilot nature of this study. Due to the method of sample selection, a traditional power calculation was not performed. The size of the sampling does limit the generalizability of the study. However, we view this work as an exploratory study and a way to find and flag potential genes and gene mutations of interest.
Additionally, we did not control for other patient variables, such as comorbidities and detailed treatment, due to the boundaries of our approved research scope. In regard to the treatment information, our previous work found that the galectin levels in treated versus untreated or not recently treated for this sample group had no observable differences [65]. As such, our study should be interpreted accordingly, as a heterogeneous pool of cancer patients representing the population from which they were obtained.
5. Conclusions
Based on our findings, we propose areas for future studies. The first is a mechanistic analysis of potential binding between galectins and glycosylated c-Kit protein. Second, is the establishment of the role of c-Kit in the regulation of expression and secretion of galectins. Third, is the investigation into the relationship between mutated c-Kit proteins and metastatic brain tumors. Further, c-KIT and its ligand, SCF, are known to be expressed preferentially in small cell lung cancers [113]. As small cell lung cancers were not examined in this study, the next step would be to examine the c-Kit mutation status and galectin levels in SCLC to determine if there is a correlation. Finally, the concept of a hotspot gene panel to find correlations between the mutations and circulating biochemical markers can be expanded to cover more cancer types and molecular markers. As a result, these studies should not only provide new insight into the key aspects of c-Kit and galectin interactions but may also provide an important framework to create rational approaches to prevent the development of metastasis in other cancers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers14112781/s1, Table S1: Breast Cancer Patient Sample Statistics; Table S2: Lung Cancer Patient Sample Statistics; Table S3: ELISA quality control measurements.
Author Contributions
Conceptualization, A.V.B. and W.J.E.; methodology, A.V.B. and A.T.F.; validation, A.T.F. and J.A.E.; formal analysis, A.T.F. and A.M.S.; investigation, A.T.F., B.B.B., A.M.S., C.R.F., W.J.E. and A.V.B.; resources, A.V.B., B.B.B., J.C.M. and W.J.E.; data curation, A.T.F. and A.M.S.; writing—original draft preparation, A.T.F., A.M.S., A.P.M. and J.C.S.; writing—review and editing, A.T.F., J.A.E., C.R.F. and A.V.B.; visualization, A.T.F.; supervision, A.V.B.; project administration, A.V.B.; funding acquisition, A.V.B., J.C.M. and W.J.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Prisma Health Cancer Institute Philanthropy Grant and Sargent Foundation. The APC was funded by the Biomedical Sciences Department at the University of South Carolina School of Medicine Greenville.
Institutional Review Board Statement
Ethical review and approval were waived for this study, which is nonhuman subject research and therefore does not require IRB approval. The study was approved by the Tissue Utilization Committee per the IRB-approved biorepository protocol and SOPs (IRB #Pro00069834).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study is available in this article.
Acknowledgments
The authors thank the Prisma Health Cancer Institute’s biorepository staff for the patient samples. We thank Rebecca Russ-Sellers for her biostatistical help. We also thank Guy Benian for his review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
PCR, Polymerase Chain Reaction; ELISA, Enzyme-Linked Immunosorbent Assay; HER2, Human Epidermal Growth Factor Receptor 2; NSCLC, Non-Small Cell Lung Cancer; EGFR, Epidermal Growth Factor Receptor; ALK, Anaplastic Lymphoma Kinase; RTK, Receptor Tyrosine Kinase; RAS, Rat Sarcoma; MAP, Mitogen Activated Protein; TGFβ, Transforming Growth Factor Beta; PI3K, Phosphoinositide 3-kinase; Wnt, Wingless-Related Integration Site; GPCR, G-Protein Coupled Receptor; p53, Cellular Tumor Antigen p53; JAK, Janus Kinase; STAT, Signal Transducer and Activator of Transcription Proteins; Tim-3, T-cell Immunoglobulin Mucin 3; GR-MD-02, Belapectin; TNM, Tumor Node Metastasis; MMP-2, Matrix Metalloproteinase 2; MMP-9, Matrix Metalloproteinase 9; Cdc42, Cell Division Control Protein 42; Akt, Protein Kinase B; PHCI, Prisma Health Cancer Institute; ABL1, Tyrosine-Protein Kinase ABL1; Akt1, RAC-alpha Serine/Threonine Protein Kinase; BRAF, Serine/Threonine Protein Kinase BRAF; CSF1R, Colony Stimulating Factor 1 Receptor; CTNNB1, Catenin-beta 1; ERBB2, Human Epidermal Growth Factor Receptor 2; ERBB4, Receptor Tyrosine-Protein Kinase ERBB4; EZH2, Enhancer of Zeste Homolog 2; FGFR1, Fibroblast Growth Factor Receptor 1; FGFR2, Fibroblast Growth Factor Receptor 2; FGFR3, Fibroblast Growth Factor Receptor 3; FLT3, Fetal Liver Tyrosine Kinase 3; GNA11, Guanine Nucleotide-Binding Protein Subunit Alpha 11; GNAQ, Guanine Nucleotide-Binding Protein Subunit Alpha; GNAS, Guanine Nucleotide-Binding Protein Alpha Stimulating; HRAS, GTPase HRAS; IDH1, Isocitrate Dehydrogenase 1; IDH2, Isocitrate Dehydrogenase 2; JAK2, Tyrosine-Protein Kinase JAK2; JAK3, Tyrosine-Protein Kinase JAK3; KDR, Vascular Endothelial Growth Factor Receptor 2; KIT, Mast/Stem Cell Growth Factor Receptor KIT; KRAS, GTPase KRAS; MET, Hepatocyte Growth Factor Receptor; MPL, Thrombopoietin Receptor; NPM1, Nucleophosmin 1; NRAS, GTPase NRAS; PDGFRA, Platelet-Derived Growth Factor Receptor A; PIK3CA, Phosphatidylinositol 4;5-Bisphosphate 3-Kinase Catalytic Subunit Alpha Isoform; PTPN11, Tyrosine-Protein Phosphatase Non-Receptor Type 11; RET, Proto-Oncogene Tyrosine-Protein Kinase Receptor RET; SMARCB1, SWI/SNF-Related Matrix-Associated Actin-Dependent Regulator of Chromatin Subfamily B Member 1; SMO, Smoothened; SRC, Proto-Oncogene Tyrosine-Protein Kinase SRC; APC, Adenomatous Polyposis Coli Protein; ATM, Ataxia Telangiectasia Mutated; CDH1, Cadherin 1; CDKN2A, Cyclin Dependent Kinase Inhibitor 2A; FBXW7, F-box WD Repeat-Containing Protein 7; HNF1A, Hepatocyte Nuclear Factor 1 Homeobox A; MLH1, MutL Homolog 1; NOTCH1, Neurogenic Locus Notch Homolog 1; PTEN, Phosphatase and Tensin Homolog; RB1, RB Transcriptional Corepressor 1; SMAD4, Mothers Against Decapentaplegic Homolog 4; STK11, Serine/Threonine Kinase 11; TP53, EKC/KEOPS Complex Subunit TP53RK; VHL, Von Hippel Lindau Disease Tumor Suppressor; SCF, Stem Cell Factor; GIST, Gastrointestinal Stromal Tumors; CML, Chronic Myelogenous Leukemia; nTPM, Normalized Transcriptions per Million; mRNA, Messenger Ribonucleic Acid; Gal-1, Galectin-1; Gal-3, Galectin-3; Gal-7, Galectin-7; Gal-8, Galectin-8; Gal-9, Galectin-9; GOF, Gain of Function; TCGA, The Cancer Genome Atlas; UALCAN, The University of Alabama at Birmingham Cancer Data Analysis Portal; SCLC, Small Cell Lung Cancer.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schouten, L.J.; Rutten, J.; Huveneers, H.A.M.; Twijnstra, A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 2002, 94, 2698–2705. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence Proportions of Brain Metastases in Patients Diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.E18. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, H.; Figueira, M.; Socorro, S. The stem cell factor (SCF)/c-KIT signalling in testis and prostate cancer. J. Cell Commun. Signal. 2017, 11, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Wandzioch, E.; Edling, C.E.; Palmer, R.H.; Carlsson, L.; Hallberg, B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood 2004, 104, 51–57. [Google Scholar] [CrossRef]
- Linnekin, D.; Mou, S.; DeBerry, C.S.; Weiler, S.R.; Keller, J.R.; Ruscetti, F.W.; Longo, D.L. Stem Cell Factor, the JAK-STAT Pathway and Signal Transduction. Leuk. Lymphoma 1997, 27, 439–444. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Ashman, L.K.; Griffith, R. Therapeutic targeting of c-KIT in cancer. Expert Opin. Investig. Drugs 2012, 22, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, S.E.; Trent, J.C.; Stemke-Hale, K.; Lazar, A.J.; Pricl, S.; Pavan, G.M.; Fermeglia, M.; Gopal, Y.N.V.; Yang, D.; Podoloff, D.A.; et al. Activity of dasatinib against L576P KIT mutant melanoma: Molecular, cellular, and clinical correlates. Mol. Cancer Ther. 2009, 8, 2079–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating c-KIT Mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Pentikäinen, O.T.; Singh, P.K. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. et Biophys. Acta 2021, 1876, 188631. [Google Scholar] [CrossRef]
- Slimane, K.; Andre, F.; Delaloge, S.; Dunant, A.; Perez, A.; Grenier, J.; Massard, C.; Spielmann, M. Risk factors for brain relapse in patients with metastatic breast cancer. Ann. Oncol. 2004, 15, 1640–1644. [Google Scholar] [CrossRef]
- Crivellari, D.; Pagani, O.; Veronesi, A.; Lombardi, D.; Nolè, F.; Thürlimann, B.; Hess, D.; Borner, M.; Bauer, J.; Martinelli, G.; et al. High incidence of central nervous system involvement in patients with metastatic or locally advanced breast cancer treated with epirubicin and docetaxel. Ann. Oncol. 2001, 12, 353–356. [Google Scholar] [CrossRef]
- Miller, K.D.; Weathers, T.; Haney, L.G.; Timmerman, R.; Dickler, M.; Shen, J.; Jr, G.W.S. Occult central nervous system involvement in patients with metastatic breast cancer: Prevalence, predictive factors and impact on overall survival. Ann. Oncol. 2003, 14, 1072–1077. [Google Scholar] [CrossRef]
- Tham, Y.-L.; Sexton, K.; Kramer, R.; Hilsenbeck, S.; Elledge, R. Primary breast cancer phenotypes associated with propensity for central nervous system metastases. Cancer 2006, 107, 696–704. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P. Brain Metastases: The HER2 Paradigm. Clin. Cancer Res. 2007, 13, 1648–1655. [Google Scholar] [CrossRef]
- Gori, S.; Rimondini, S.; De Angelis, V.; Colozza, M.; Bisagni, G.; Moretti, G.; Sidoni, A.; Basurto, C.; Aristei, C.; Anastasi, P.; et al. Central Nervous System Metastases in HER-2–Positive Metastatic Breast Cancer Patients Treated with Trastuzumab: Incidence, Survival, and Risk Factors. Oncol. 2007, 12, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-Y.; Na, I.I.; Kim, C.H.; Park, S.; Baek, H.; Yang, S.H. EGFR Mutation and Brain Metastasis in Pulmonary Adenocarcinomas. J. Thorac. Oncol. 2014, 9, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, I.; Zaorsky, N.; Palmer, J.D.; Mehra, R.; Lu, B. Targeting brain metastases in ALK-rearranged non-small-cell lung cancer. Lancet Oncol. 2015, 16, e510–e521. [Google Scholar] [CrossRef]
- Johung, K.L.; Yeh, N.; Desai, N.B.; Williams, T.M.; Lautenschlaeger, T.; Arvold, N.D.; Ning, M.S.; Attia, A.; Lovly, C.M.; Goldberg, S.; et al. Extended Survival and Prognostic Factors for Patients With ALK-Rearranged Non–Small-Cell Lung Cancer and Brain Metastasis. J. Clin. Oncol. 2016, 34, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non–Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv192–iv237. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Planchard, D.; Lu, S.; Sun, H.; Yamamoto, N.; Kim, D.-W.; Tan, D.; Yang, J.-H.; Azrif, M.; Mitsudomi, T.; et al. Pan-Asian adapted Clinical Practice Guidelines for the management of patients with metastatic non-small-cell lung cancer: A CSCO–ESMO initiative endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann. Oncol. 2019, 30, 171–210. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Han, J.-Y.; Katakami, N.; Kim, H.R.; Hodge, R.; Kaur, P.; Brown, A.P.; Ghiorghiu, D.; et al. CNS Efficacy of Osimertinib in Patients With T790M-Positive Advanced Non–Small-Cell Lung Cancer: Data From a Randomized Phase III Trial (AURA3). J. Clin. Oncol. 2018, 36, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Nakagawa, K.; Cho, B.C.; Cobo, M.; Cho, E.K.; Bertolini, A.; Bohnet, S.; Zhou, C.; Lee, K.H.; Nogami, N.; et al. CNS Response to Osimertinib Versus Standard Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3290–3297. [Google Scholar] [CrossRef] [PubMed]
- Raposo, C.; Canelas, A.; Barros, M. Human Lectins, Their Carbohydrate Affinities and Where to Find Them. Biomolecules 2021, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When Galectins Recognize Glycans: From Biochemistry to Physiology and Back Again. Biochemistry 2011, 50, 7842–7857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, E.; Meggyes, M.; Doba, K.; Barakonyi, A.; Szereday, L. Immune Checkpoint Molecules in Reproductive Immunology. Front. Immunol. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Huergo, S.P.; Blidner, A.G.; A Rabinovich, G. Galectins: Emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr. Opin. Immunol. 2017, 45, 8–15. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Toscano, M.A. Turning ’sweet’ on immunity: Galectin–glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009, 9, 338–352. [Google Scholar] [CrossRef]
- Lau, K.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Raz, A. Biological Modulation by Lectins and Their Ligands in Tumor Progression and Metastasis. Anti-Cancer Agents Med. Chem. 2008, 8, 22–36. [Google Scholar] [CrossRef] [Green Version]
- Markowska, A.I.; Liu, F.-T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, M.A.; Mayoral, C.; Meneses, A.; Villalvazo, L.; Guzmán, A.; Espinosa, B.; Ochoa, J.L.; Zenteno, E.; Guevara, J. Identification of Galectin-3 and Mucin-Type O-Glycans in Breast Cancer and Its Metastasis to Brain. Cancer Investig. 2008, 26, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-W.; Chang, H.-T.; Chen, C.-H.; Lin, S.-W.; Hsu, T.-L.; Wong, C.-H. Galectin-3 Binding Protein and Galectin-1 Interaction in Breast Cancer Cell Aggregation and Metastasis. J. Am. Chem. Soc. 2015, 137, 9685–9693. [Google Scholar] [CrossRef] [PubMed]
- Kocoglu, H.; Topcu, T.O.; Kavgaci, H.; Gunaldi, M.; Akyol, M.; Mentese, A.; Yaman, S.O.; Orem, A.; Ozdemir, F.; Aydin, F. The clinical importance of serum galectin-3 levels in breast cancer patients with and without metastasis. J. Cancer Res. Ther. 2018, 14, S583–S586. [Google Scholar] [CrossRef] [PubMed]
- Campion, C.G.; Labrie, M.; Lavoie, G.; St-Pierre, Y. Expression of Galectin-7 Is Induced in Breast Cancer Cells by Mutant p53. PLoS ONE 2013, 8, e72468. [Google Scholar] [CrossRef] [PubMed]
- Hadari, Y.; Arbel-Goren, R.; Levy, Y.; Amsterdam, A.; Alon, R.; Zakut, R.; Zick, Y. Galectin-8 binding to integrins inhibits cell adhesion and induces apoptosis. J. Cell Sci. 2000, 113, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Kojima, S.; Yamazaki, N.; Fink, C.; Kaltner, H.; Kayser, K.; Gabius, H.-J. Galectins-1 and -3 and their ligands in tumor biology: Non-uniform properties in cell-surface presentation and modulation of adhesion to matrix glycoproteins for various tumor cell lines, in biodistribution of free and liposome-bound galectins and in thei. J. Cancer Res. Clin. Oncol. 1999, 125, 461–474. [Google Scholar] [CrossRef]
- Grosset, A.-A.; Labrie, M.; Gagné, D.; Vladoiu, M.-C.; Gaboury, L.; Doucet, N.; St-Pierre, Y. Cytosolic galectin-7 impairs p53 functions and induces chemoresistance in breast cancer cells. BMC Cancer 2014, 14, 1–10. [Google Scholar] [CrossRef]
- Trebo, A.; Ditsch, N.; Kuhn, C.; Heidegger, H.H.; Zeder-Goess, C.; Kolben, T.; Czogalla, B.; Schmoeckel, E.; Mahner, S.; Jeschke, U.; et al. High Galectin-7 and Low Galectin-8 Expression and the Combination of both are Negative Prognosticators for Breast Cancer Patients. Cancers 2020, 12, 953. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, P.-L.; Hung, J.-Y.; Huang, S.-K.; Chou, S.-H.; Cheng, D.-E.; Jong, Y.-J.; Hung, C.-H.; Yang, C.-J.; Tsai, Y.-M.; Hsu, Y.-L.; et al. Lung Cancer-Derived Galectin-1 Mediates Dendritic Cell Anergy through Inhibitor of DNA Binding 3/IL-10 Signaling Pathway. J. Immunol. 2010, 186, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.-Y.; Tang, S.-J.; Sun, G.-H.; Chou, T.-Y.; Yeh, T.-S.; Yu, S.-L.; Sun, K.-H. Galectin-1 Promotes Lung Cancer Progression and Chemoresistance by Upregulating p38 MAPK, ERK, and Cyclooxygenase-2. Clin. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.-N.; Dang, Y.-F.; Gong, F.-L.; Guo, X.-L. Role and regulation mechanism of Gal-3 in non-small cell lung cancer and its potential clinical therapeutic significance. Chem. Interactions 2019, 309, 108724. [Google Scholar] [CrossRef]
- Schulkens, I.A.; Heusschen, R.; Boogaart, V.V.D.; Van Suylen, R.-J.; Dingemans, A.-M.C.; Griffioen, A.W.; Thijssen, V.L. Galectin Expression Profiling Identifies Galectin-1 and Galectin-9Δ5 as Prognostic Factors in Stage I/II Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e107988. [Google Scholar] [CrossRef]
- He, Y.; Jia, K.; Dziadziuszko, R.; Zhao, S.; Zhang, X.; Deng, J.; Wang, H.; Hirsch, F.R.; Zhou, C. Galectin-9 in non-small cell lung cancer. Lung Cancer 2019, 136, 80–85. [Google Scholar] [CrossRef]
- Dalotto-Moreno, T.; Croci, D.O.; Cerliani, J.P.; Martinez-Allo, V.C.; Dergan-Dylon, S.; Méndez-Huergo, S.P.; Stupirski, J.C.; Mazal, D.; Osinaga, E.; Toscano, M.; et al. Targeting Galectin-1 Overcomes Breast Cancer-Associated Immunosuppression and Prevents Metastatic Disease. Cancer Res. 2012, 73, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.-J.; Moon, H.-G.; Cho, B.I.; Jeong, C.-Y.; Joo, Y.-T.; Lee, Y.-J.; Hong, S.-C.; Choi, S.-K.; Ha, W.-S.; Kim, J.W.; et al. Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. Int. J. Cancer 2007, 120, 2331–2338. [Google Scholar] [CrossRef]
- Demers, M.; Rose, A.A.; Grosset, A.-A.; Biron-Pain, K.; Gaboury, L.; Siegel, P.M.; St-Pierre, Y. Overexpression of Galectin-7, A Myoepithelial Cell Marker, Enhances Spontaneous Metastasis of Breast Cancer Cells. Am. J. Pathol. 2010, 176, 3023–3031. [Google Scholar] [CrossRef]
- Danguy, A.; Rorive, S.; Decaestecker, C.; Bronckart, Y.; Kaltner, H.; Hadari, Y.R.; Goren, R.; Zich, Y.; Petein, M.; Salmon, I.; et al. Immunohistochemical profile of galectin-8 expression in benign and malignant tumors of epithelial, mesenchymatous and adipous origins, and of the nervous system. Histol. Histopathol. 2001, 16, 861–868. [Google Scholar] [CrossRef]
- Szöke, T.; Kayser, K.; Baumhäkel, J.-D.; Trojan, I.; Furak, J.; Tiszlavicz, L.; Horvath, A.; Szluha, K.; Gabius, H.-J.; Andre, S. Prognostic Significance of Endogenous Adhesion/Growth-Regulatory Lectins in Lung Cancer. Oncology 2005, 69, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassen, R.; Brichory, F.; Caulet-Maugendre, S.; Bidon, N.; Delaval, P.; Desrues, B.; Dazord, L. Expression of Po66-CBP, a type-8 galectin, in different healthy, tumoral and peritumoral tissues. Anticancer Res. 1999, 19, 5429–5433. [Google Scholar]
- Barrow, H.; Guo, X.; Wandall, H.H.; Pedersen, J.W.; Fu, B.; Zhao, Q.; Chen, C.; Rhodes, J.M.; Yu, L.-G. Serum Galectin-2, -4, and -8 Are Greatly Increased in Colon and Breast Cancer Patients and Promote Cancer Cell Adhesion to Blood Vascular Endothelium. Clin. Cancer Res. 2011, 17, 7035–7046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurisci, I.; Tinari, N.; Natoli, C.; Angelucci, D.; Cianchetti, E.; Iacobelli, S. Concentrations of galectin-3 in the sera of normal controls and cancer patients. Clin. Cancer Res. 2000, 6, 1389–1393. [Google Scholar]
- Blair, B.B.; Funkhouser, A.T.; Goodwin, J.L.; Strigenz, A.M.; Chaballout, B.H.; Martin, J.C.; Arthur, C.M.; Funk, C.R.; Edenfield, W.J.; Blenda, A.V. Increased Circulating Levels of Galectin Proteins in Patients with Breast, Colon, and Lung Cancer. Cancers 2021, 13, 4819. [Google Scholar] [CrossRef]
- Gong, H.C.; Honjo, Y.; Nangia-Makker, P.; Hogan, V.; Mazurak, N.; Bresalier, R.; Raz, A. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res. 1999, 59, 6239–6245. [Google Scholar]
- Lipkowitz, M.S.; Leal-Pinto, E.; Cohen, B.E.; Abramson, R.G. Galectin 9 is the sugar-regulated urate transporter/channel UAT. Glycoconj. J. 2002, 19, 491–498. [Google Scholar] [CrossRef]
- Menon, S.; Kang, C.-M.; Beningo, K.A. Galectin-3 secretion and tyrosine phosphorylation is dependent on the calpain small subunit, Calpain 4. Biochem. Biophys. Res. Commun. 2011, 410, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, D.; Fan, Y.; Li, X.; Xue, H.; Ma, Y.; Zhou, Y.; Tai, G. The Two Endocytic Pathways Mediated by the Carbohydrate Recognition Domain and Regulated by the Collagen-like Domain of Galectin-3 in Vascular Endothelial Cells. PLoS ONE 2012, 7, e52430. [Google Scholar] [CrossRef] [Green Version]
- Ideo, H.; Hoshi, I.; Yamashita, K.; Sakamoto, M. Phosphorylation and externalization of galectin-4 is controlled by Src family kinases. Glycobiology 2013, 23, 1452–1462. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, D.H.; Bertozzi, C.R. Glycans in Cancer and Inflammation — Potential for Therapeutics and Diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Tharmalingam, T.; Rudd, P.M. Glycans as cancer biomarkers. Biochim. et Biophys. Acta Gen. Subj. 2011, 1820, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Bresalier, R.S.; Byrd, J.C.; Tessler, D.; Lebel, J.; Koomen, J.; Hawke, D.; Half, E.; Liu, K.-F.; Mazurek, N. A circulating ligand for galectin-3 is a haptoglobin-related glycoprotein elevated in individuals with colon cancer. Gastroenterology 2004, 127, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Kohnoe, S.; Tsujita, E.; Watanabe, A.; Nakashima, H.; Baba, H.; Maehara, Y. Galectin-3 expression is a potent prognostic marker in colorectal cancer. Anticancer Res. 2005, 25, 3117–3121. [Google Scholar]
- Barrow, H.; Rhodes, J.M.; Yu, L.-G. The role of galectins in colorectal cancer progression. Int. J. Cancer 2011, 129, 1–8. [Google Scholar] [CrossRef]
- Sakaki, M.; Oka, N.; Nakanishi, R.; Yamaguchi, K.; Fukumori, T.; Kanayama, H.-O. Serum level of galectin-3 in human bladder cancer. J. Med Investig. 2008, 55, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Gheysen, L.; Soumoy, L.; Trelcat, A.; Verset, L.; Journe, F.; Saussez, S. New Treatment Strategy Targeting Galectin-1 against Thyroid Cancer. Cells 2021, 10, 1112. [Google Scholar] [CrossRef]
- Shimura, T.; Shibata, M.; Gonda, K.; Kofunato, Y.; Okada, R.; Ishigame, T.; Kimura, T.; Kenjo, A.; Kono, K.; Marubashi, S. Significance of Circulating Galectin-3 in Patients with Pancreatobiliary Cancer. Anticancer Res. 2017, 37, 4979–4986. [Google Scholar] [CrossRef] [Green Version]
- Masoodi, M.; Shah, Z.A.; Beigh, A.H.; Ahmad, S.Z.; Mir, A.W.; Yasin, B.; Rasool, R.; Masoodi, K.Z.; Bhat, G.M. Galectin-1 as a predictive biomarker in ovarian cancer. J. Ovarian Res. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2019, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin Targeted Therapy in Oncology: Current Knowledge and Perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 Augments K-Ras Activation and Triggers a Ras Signal That Attenuates ERK but Not Phosphoinositide 3-Kinase Activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Ji, B.; Ramachandran, V.; Wang, H.; Hafley, M.; Logsdon, C.; Bresalier, R. Overexpressed Galectin-3 in Pancreatic Cancer Induces Cell Proliferation and Invasion by Binding Ras and Activating Ras Signaling. PLoS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalom-Feuerstein, R.; Cooks, T.; Raz, A.; Kloog, Y. Galectin-3 Regulates a Molecular Switch from N-Ras to K-Ras Usage in Human Breast Carcinoma Cells. Cancer Res. 2005, 65, 7292–7300. [Google Scholar] [CrossRef] [Green Version]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of Cytokine Receptors by Golgi N-Glycan Processing and Endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Bresalier, R.S.; Mazurek, N.; Sternberg, L.R.; Byrd, J.C.; Yunker, C.K.; Nangia-Makker, P.; Raz, A. Metastasis of human colon cancer is altered by modifying expression of the β-galactoside-binding protein galectin 3. Gastroenterology 1998, 115, 287–296. [Google Scholar] [CrossRef]
- Folgueira, M.A.A.K.; Maistro, S.; Katayama, M.L.H.; Roela, R.A.; Mundim, F.G.L.; Nanogaki, S.; de Bock, G.H.; Brentani, M.M. Markers of breast cancer stromal fibroblasts in the primary tumour site associated with lymph node metastasis: A systematic review including our case series. Biosci. Rep. 2013, 33, 921–929. [Google Scholar] [CrossRef]
- Lotan, R.; Belloni, P.N.; Tressler, R.J.; Lotan, D.; Xu, X.-C.; Nicolson, G.L. Expression of galectins on microvessel endothelial cells and their involvement in tumour cell adhesion. Glycoconj. J. 1994, 11, 462–468. [Google Scholar] [CrossRef]
- Brûle, F.V.D.; Califice, S.; Garnier, F.; Fernandez, P.L.; Berchuck, A.; Castronovo, V. Galectin-1 Accumulation in the Ovary Carcinoma Peritumoral Stroma Is Induced by Ovary Carcinoma Cells and Affects Both Cancer Cell Proliferation and Adhesion to Laminin-1 and Fibronectin. Lab. Investig. 2003, 83, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.H.; Hong, T.M.; Cheng, H.W.; Pan, S.H.; Liang, Y.R.; Hong, H.C.; Chiang, W.F.; Wong, T.Y.; Shieh, D.B.; Shiau, A.L.; et al. Galectin-1-Mediated tumor invasion and metastasis, Up-Regulated matrix metalloproteinase expression, and reorganized actin cytoskeletons. Mol. Cancer Res. 2009, 7, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, T.-C.; Liu, R.; Wu, C.-T.; Li, X.; Xiao, W.; Deng, X.; Kiss, S.; Wang, T.; Chen, X.-J.; Carney, R.P.; et al. Targeting Galectin-1 Impairs Castration-Resistant Prostate Cancer Progression and Invasion. Clin. Cancer Res. 2018, 24, 4319–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, M.; Kaur, T.; Kamboj, S.S.; Singh, J. Roles of Galectin-7 in Cancer. Asian Pac. J. Cancer Prev. 2016, 17, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, V.L.; Heusschen, R.; Caers, J.; Griffioen, A.W. Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim. et Biophys. Acta 2015, 1855, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Callahan, S.P.; Brevetta, R.A.; Stenbit, A.E.; Smith, W.M.; Martin, J.C.; Blenda, A.V.; Arce, S.; Edenfield, W.J. Efficacy of therapeutic plasma exchange in the treatment of penn class 3 and 4 cytokine release syndrome complicating COVID-19. Respir. Med. 2020, 175, 106188. [Google Scholar] [CrossRef] [PubMed]
- Shuford, S.; Lipinski, L.; Abad, A.; Smith, A.M.; Rayner, M.; O’Donnell, L.; Stuart, J.; Mechtler, L.L.; Fabiano, A.J.; Edenfield, J.; et al. Prospective prediction of clinical drug response in high-grade gliomas using an ex vivo 3D cell culture assay. Neuro-Oncology Adv. 2021, 3, vdab065. [Google Scholar] [CrossRef]
- rs3822214 RefSNP Report-dbSNP-NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/rs3822214#clinical_significance (accessed on 5 January 2022).
- Murakami, T.; Fukai, K.; Oiso, N.; Hosomi, N.; Kato, A.; Garganta, C.; Barnicoat, A.; Poppelaars, F.; Aquaron, R.; Paller, A.S.; et al. New KIT mutations in patients with piebaldism. J. Dermatol. Sci. 2004, 35, 29–33. [Google Scholar] [CrossRef]
- Foster, R.; Byrnes, E.; Meldrum, C.; Griffith, R.; Ross, G.; Upjohn, E.; Braue, A.; Scott, R.; Varigos, G.; Ferrao, P.; et al. Association of paediatric mastocytosis with a polymorphism resulting in an amino acid substitution (M541L) in the transmembrane domain of c-KIT. Br. J. Dermatol. 2008, 159, 1160–1169. [Google Scholar] [CrossRef]
- Inokuchi, K.; Yamaguchi, H.; Tarusawa, M.; Futaki, M.; Hanawa, H.; Tanosaki, S.; Dan, K. Abnormality of c-kit oncoprotein in certain patients with chronic myelogenous leukemia–potential clinical significance. Leukemia 2002, 16, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Lobl, M.B.; Hass, B.; Clarey, D.; Higgins, S.; Wysong, A. Next-generation sequencing identifies novel single nucleotide polymorphisms in high-risk cutaneous squamous cell carcinoma: A pilot study. Exp. Dermatol. 2020, 29, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Emig, M.; Lohse, P.; Ehninger, G.; Hochhaus, A.; Schackert, H.K. The c-kit (CD117) sequence variation M541L, but not N564K, is frequent in the general population, and is not associated with CML in Caucasians. Leukemia 2005, 20, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Porębska, N.; Poźniak, M.; Matynia, A.; Żukowska, D.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Galectins as modulators of receptor tyrosine kinases signaling in health and disease. Cytokine Growth Factor Rev. 2021, 60, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Obermann, J.; Priglinger, C.S.; Merl-Pham, J.; Geerlof, A.; Priglinger, S.; Götz, M.; Hauck, S.M. Proteome-wide Identification of Glycosylation-dependent Interactors of Galectin-1 and Galectin-3 on Mesenchymal Retinal Pigment Epithelial (RPE) Cells. Mol. Cell. Proteom. 2017, 16, 1528–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastrzebski, K.; Zdżalik-Bielecka, D.; Mamińska, A.; Kalaidzidis, Y.; Hellberg, C.; Miaczynska, M. Multiple routes of endocytic internalization of PDGFRβ contribute to PDGF-induced STAT3 signaling. J. Cell Sci. 2016, 130, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-H.; Chen, Y.-L.; Lee, K.-H.; Chang, C.-C.; Cheng, T.-M.; Wu, S.-Y.; Tu, C.-C.; Tsui, W.-L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Feng, Z.-C.; Riopel, M.; Popell, A.; Wang, R. A survival Kit for pancreatic beta cells: Stem cell factor and c-Kit receptor tyrosine kinase. Diabetologia 2015, 58, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Sousa, L.P.; Mandel-Bausch, E.M.; Tome, F.; Reshetnyak, A.V.; Hadari, Y.; Schlessinger, J.; Lax, I. Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition. Proc. Natl. Acad. Sci. 2016, 113, E4784–E4793. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Takeuchi, H.; Nishizaki, T.; Higashi, H.; Era, S. A loss of c-kit expression is associated with an advanced stage and poor prognosis in breast cancer. Br. J. Cancer 2006, 94, 1874–1878. [Google Scholar] [CrossRef]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potti, A.; Moazzam, N.; Ramar, K.; Hanekom, D.S.; Kargas, S.; Koch, M. CD117 (c-KIT) overexpression in patients with extensive-stage small-cell lung carcinoma. Ann. Oncol. 2003, 14, 894–897. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).