
Unveiling the Molecular Mechanisms Driving the Capsaicin-Induced Immunomodulatory Effects on PD-L1 Expression in Bladder and Renal Cancer Cell Lines

 ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. MTT Assay
2.4. ROS Analysis
2.5. Mitochondrial Transmembrane Potential (∆Ψm) Assay
2.6. Annexin V and PI Staining
2.7. Western Blot Analysis
2.8. RT Profiler PCR Array
2.9. Confocal Laser Scanning Microscopy Analysis
2.10. Oxidized Protein Analysis
2.11. TRPV1 Silencing
2.12. Statistical Analysis
3. Results
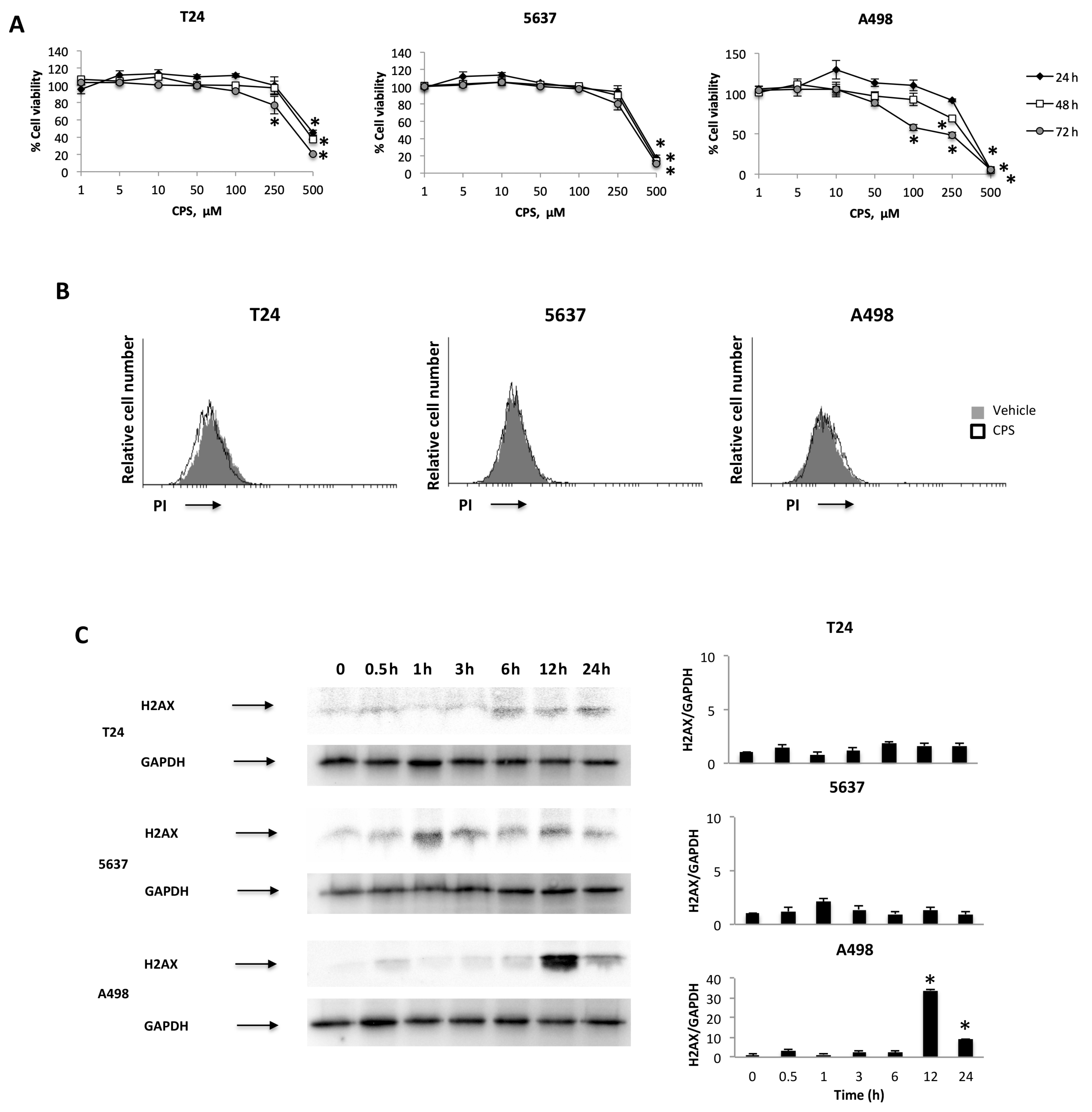
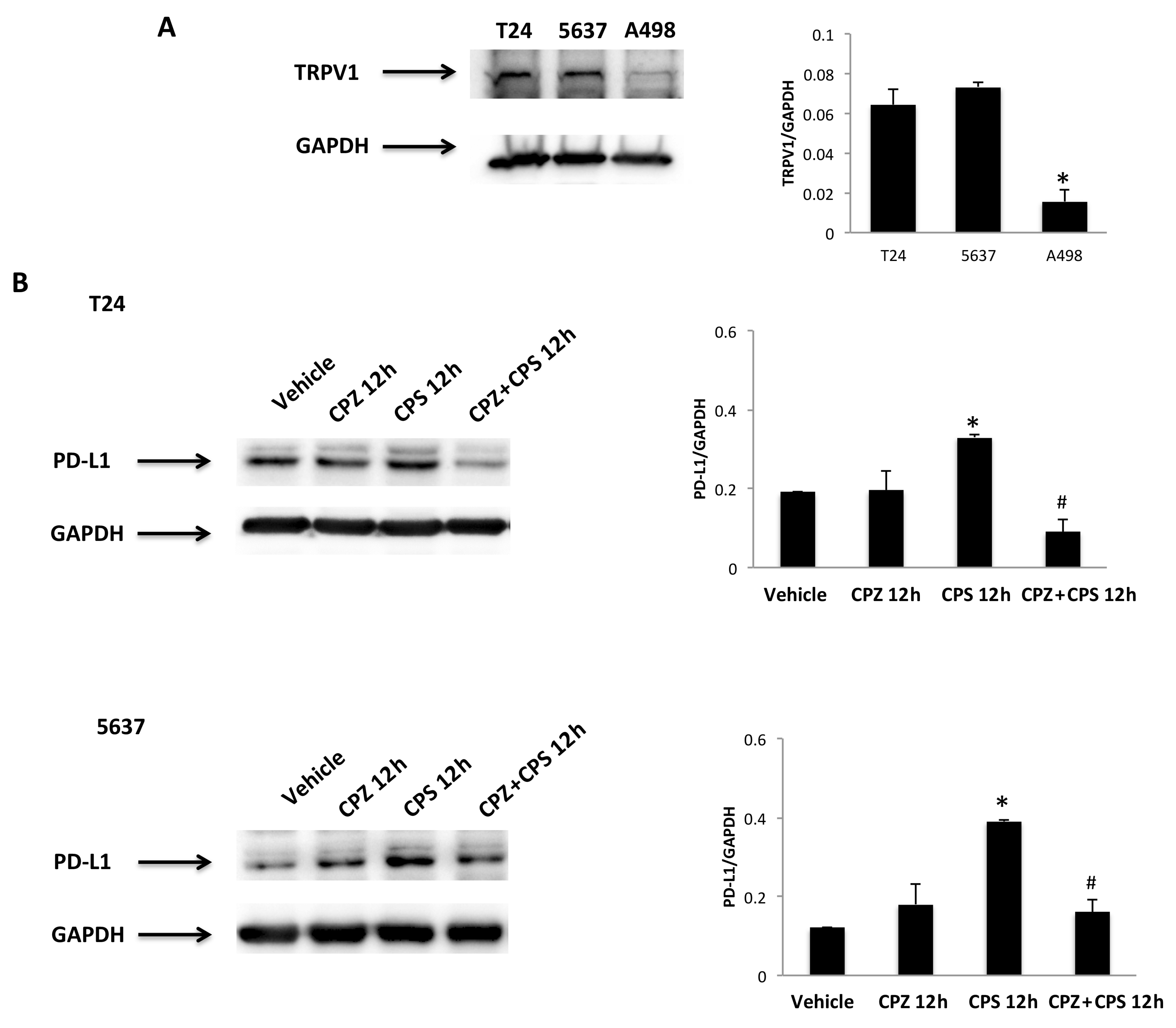
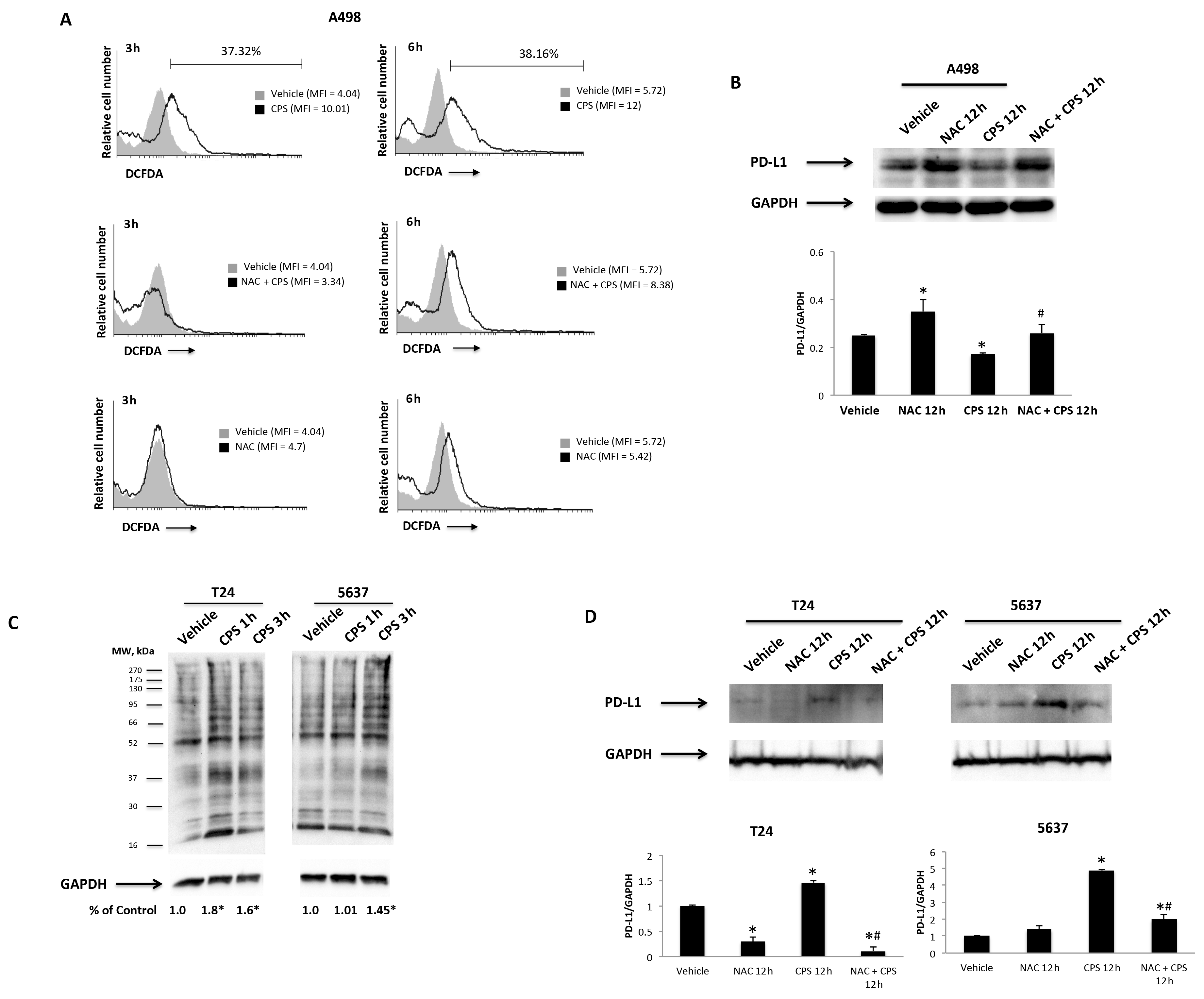
3.1. CPS Induces DNA Damage in Living 5637, T24 and A498 Cancer Cell Lines
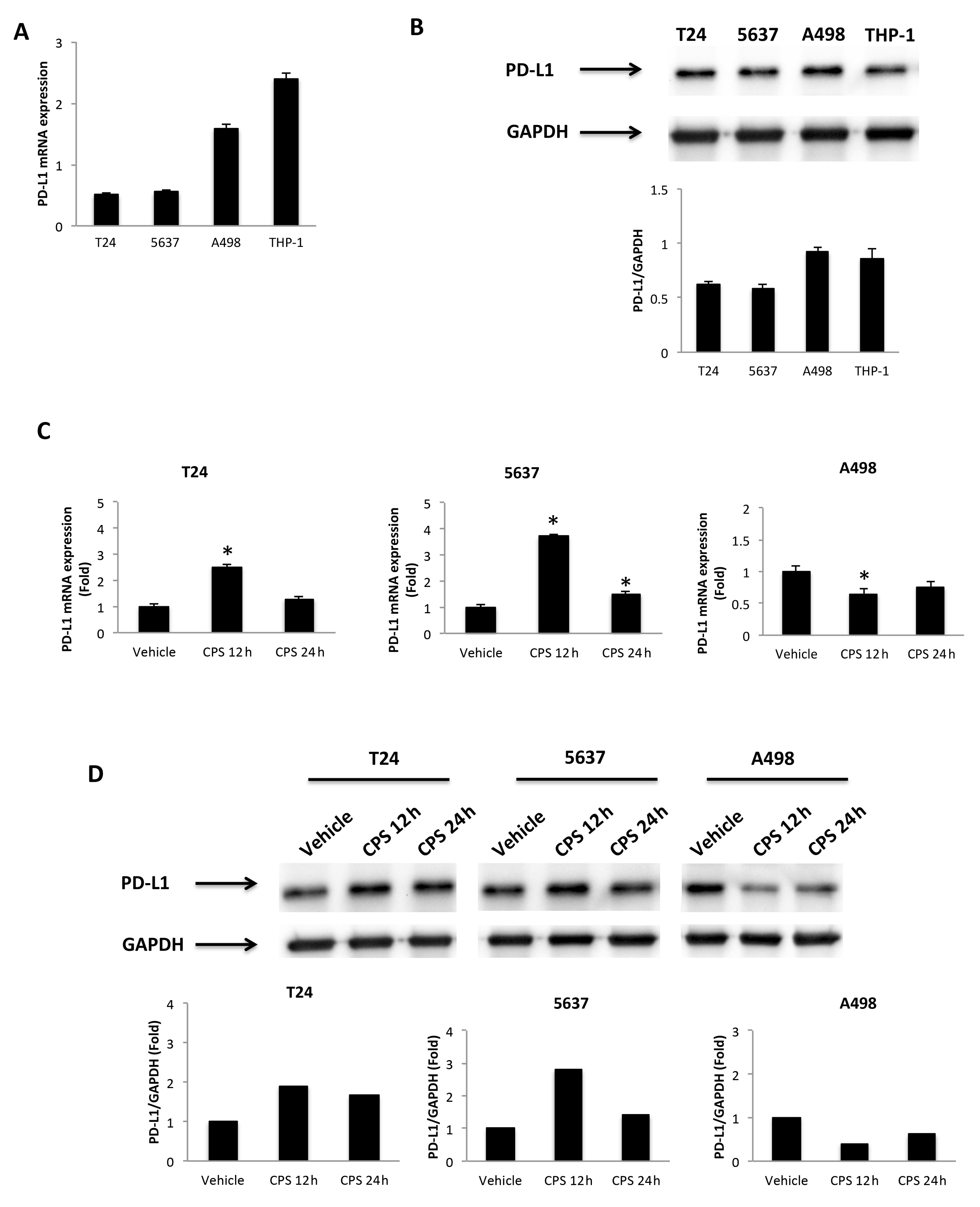
3.2. CPS-Induced DNA Damage Modulates the PD-L1 mRNA and Protein Expression in T24, 5637 and A498 Cells
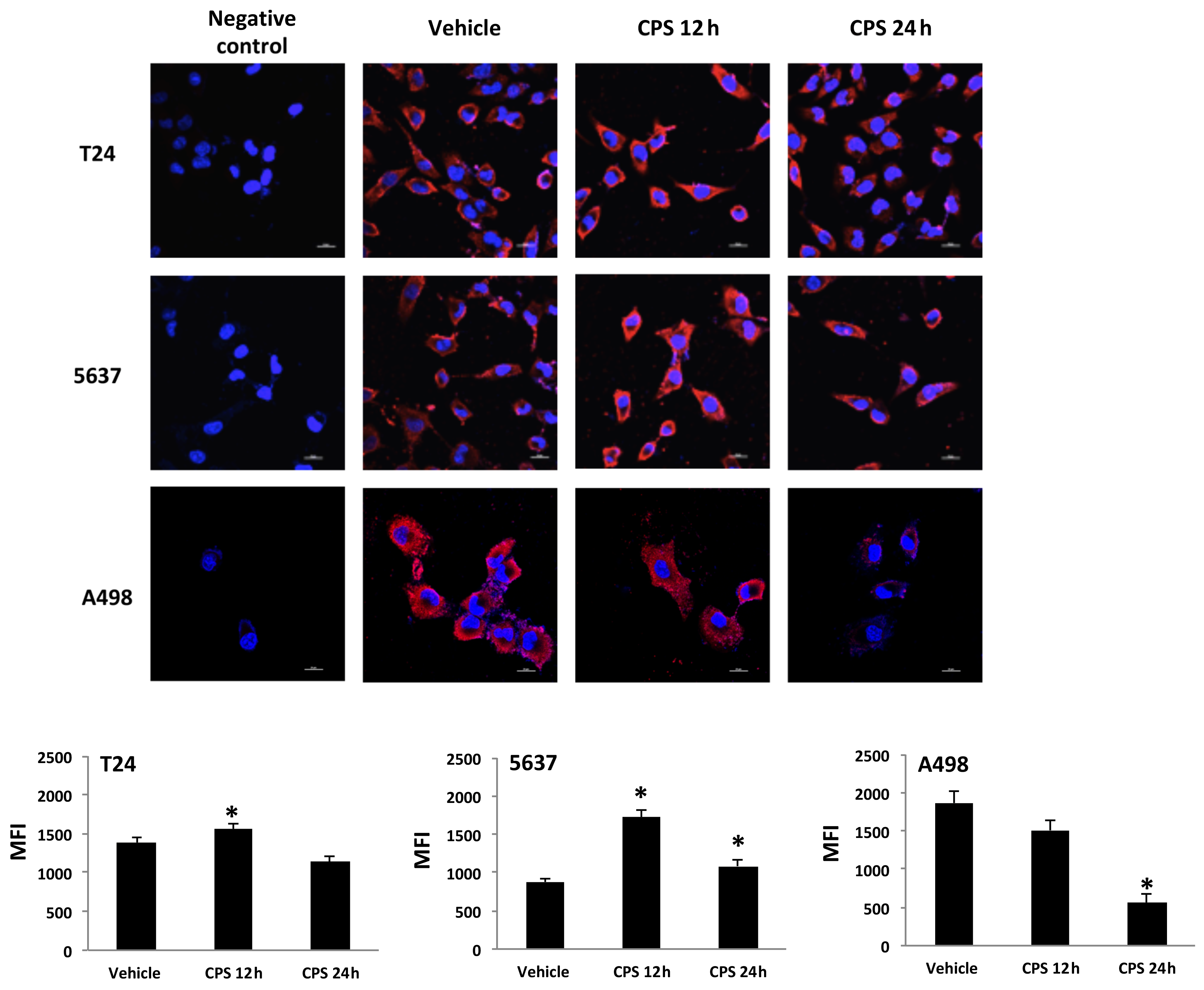
3.3. Distribution of PD-L1 Protein in CPS-Treated T24, 5637 and A498 Cells
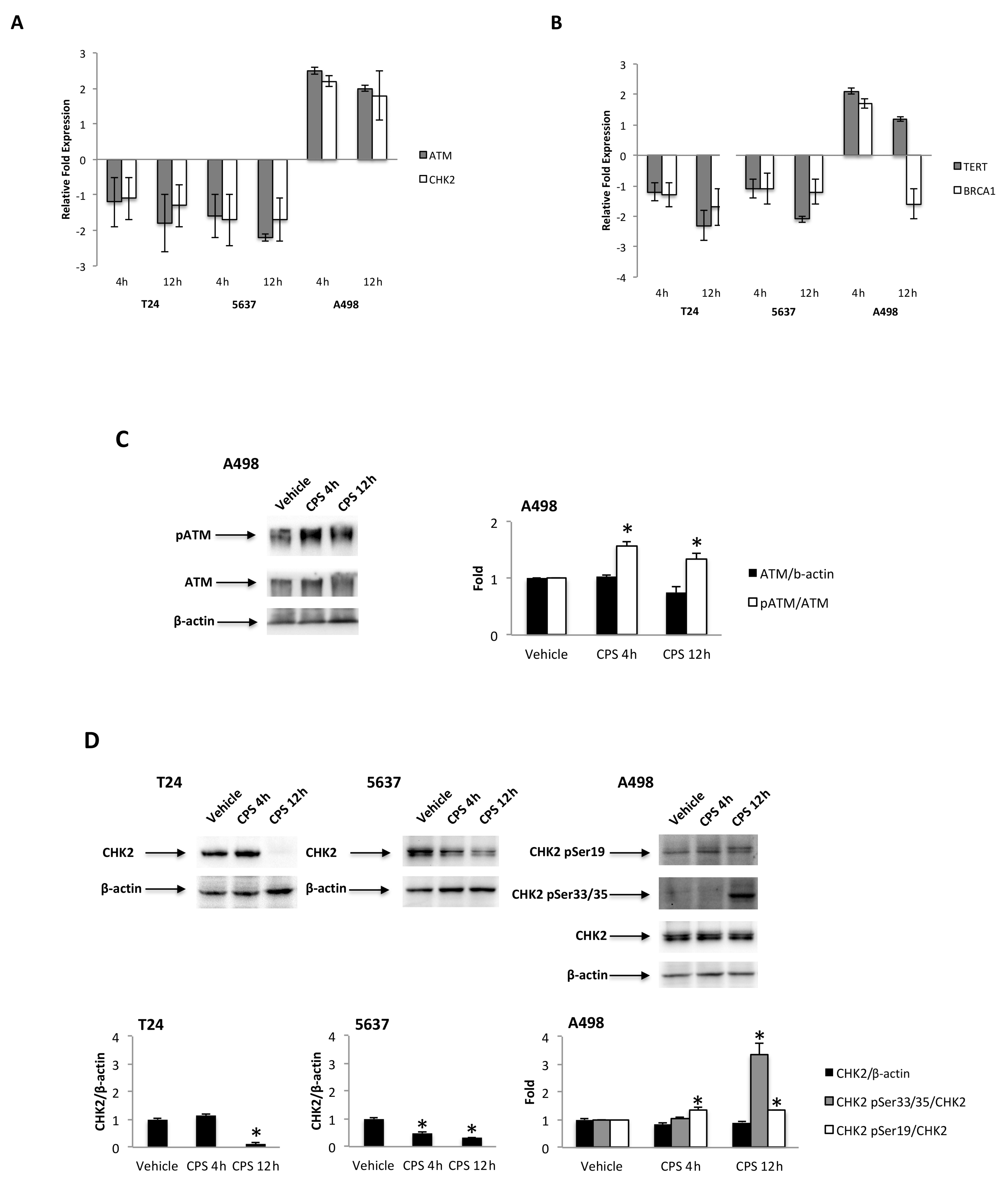
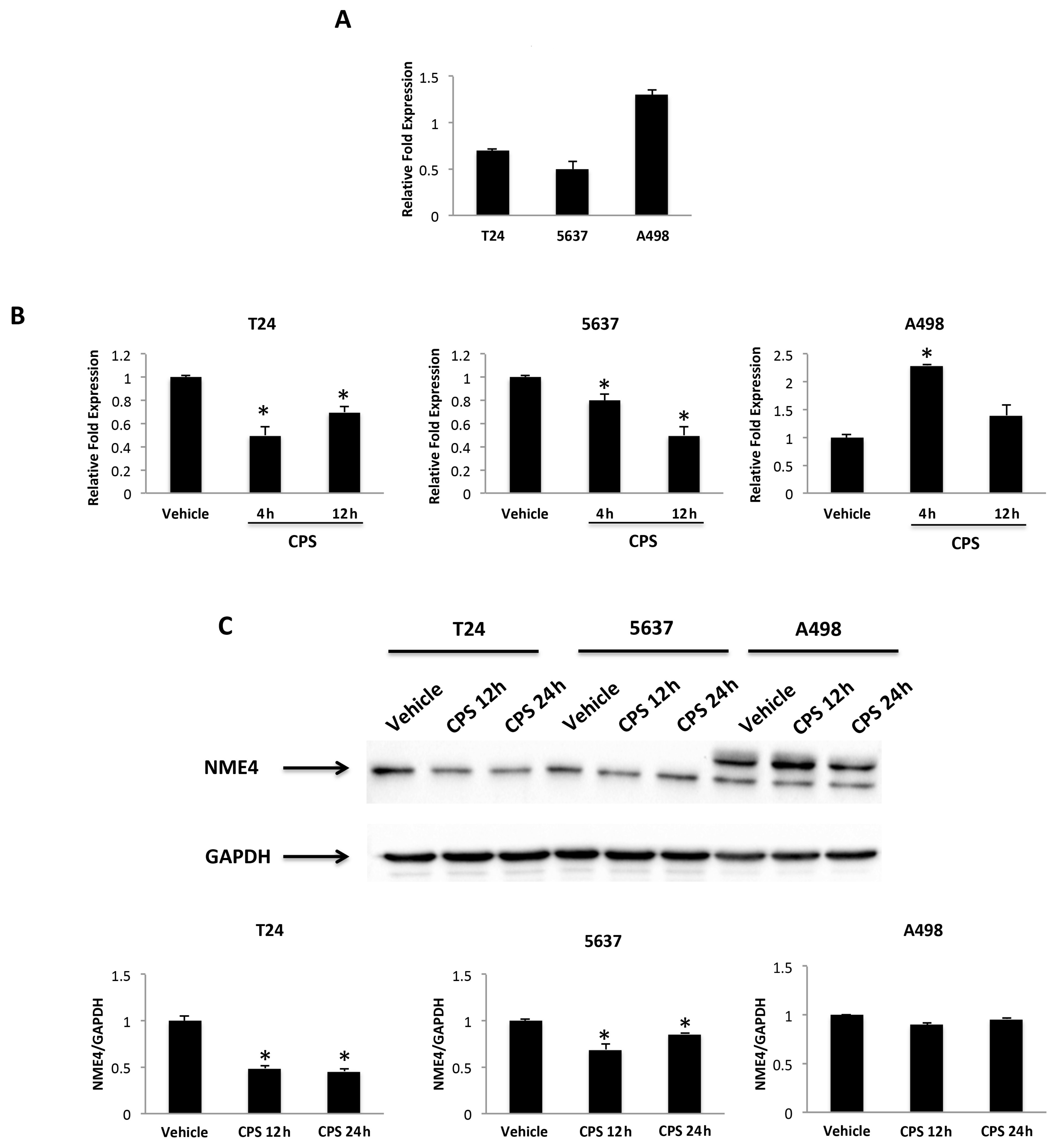
3.4. CPS-Induced DSB DNA Damage Is Associated with p53/ATM/CHK2 and BRCA1/hTERT mRNA Expression in T24, 5637 and A498 Cells
3.5. TRPV1- or ROS-Dependent Signals Induced in CPS-Treated T24, 5637 and A498 Cells
3.6. Transcriptional Factors, Oncogenes, Cytokines and Growth Factors Are Associated Gene-Promoting Drivers in CPS-Induced Modulation of PD-L1 Expression in 5637, T24 and A498 Cells
3.7. Nucleoside Diphosphate Kinase (NDPK)/Nm23-H4 Played a Role in the CPS-Induced PD-L1 Protein Expression Modulation in T24 and 5637 Cells
3.8. CPS-Induced PD-L1 Modulation Requires Stat1/Stat3 Activation in 5637, T24 and A498 Cells
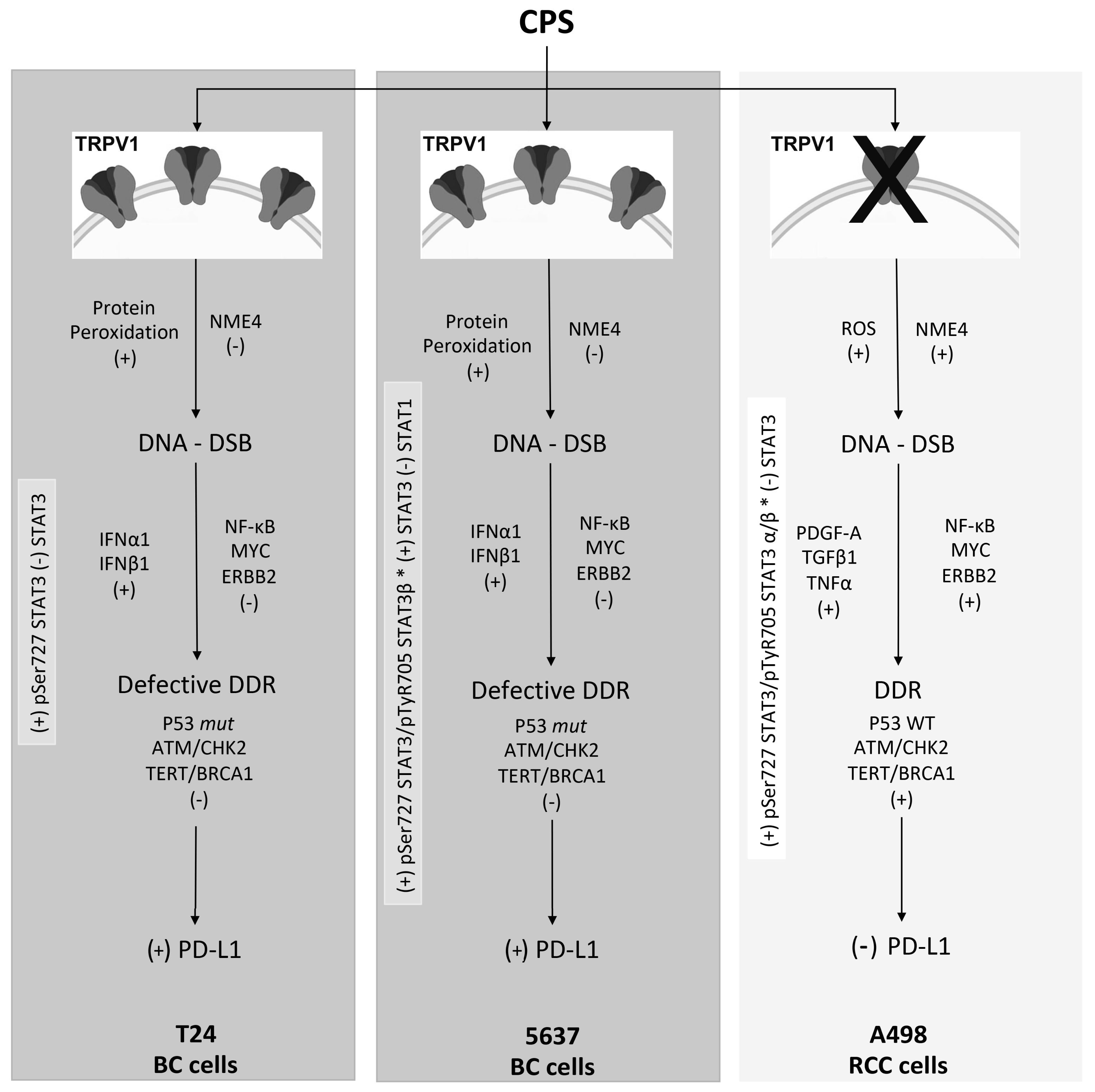
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder Cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.E.; Smith, A.B.; Meyer, A.-M.; Kuo, T.-M.; Tyree, S.; Kim, W.Y.; Milowsky, M.I.; Pruthi, R.S.; Millikan, R.C. Trends in Stage-Specific Incidence Rates for Urothelial Carcinoma of the Bladder in the United States: 1988 to 2006. Cancer 2014, 120, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Inman, B.A.; Harrison, M.R.; George, D.J. Novel Immunotherapeutic Strategies in Development for Renal Cell Carcinoma. Eur. Urol. 2013, 63, 881–889. [Google Scholar] [CrossRef]
- Meskawi, M.; Sun, M.; Trinh, Q.-D.; Bianchi, M.; Hansen, J.; Tian, Z.; Rink, M.; Ismail, S.; Shariat, S.F.; Montorsi, F.; et al. A Review of Integrated Staging Systems for Renal Cell Carcinoma. Eur. Urol. 2012, 62, 303–314. [Google Scholar] [CrossRef]
- Govindarajan, V.S.; Sathyanarayana, M.N. Capsicum—Production, Technology, Chemistry, and Quality. Part V. Impact on Physiology, Pharmacology, Nutrition, and Metabolism; Structure, Pungency, Pain, and Desensitization Sequences. Crit. Rev. Food Sci. Nutr. 1991, 29, 435–474. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The Two Faces of Capsaicin. Cancer Res. 2011, 71, 2809–2814. [Google Scholar] [CrossRef]
- Blumberg, P.M. To Not Be Hot When TRPV1 Is Not. Temperature 2015, 2, 166–167. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Ni, Y.-H.; Zhao, C.-R.; Qiao, Z.; Yu, H.-X.; Wang, L.-Y.; Sun, J.-Y.; Du, C.; Zhang, J.-H.; Dong, L.-Y.; et al. Capsaicin Enhances the Antitumor Activity of Sorafenib in Hepatocellular Carcinoma Cells and Mouse Xenograft Tumors through Increased ERK Signaling. Acta Pharmacol. Sin. 2018, 39, 438–448. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, D.; Huang, J.; Hu, Y.; Xu, Y. Application of Capsaicin as a Potential New Therapeutic Drug in Human Cancers. J. Clin. Pharm. Ther. 2020, 45, 16–28. [Google Scholar] [CrossRef]
- Braga Ferreira, L.G.; Faria, J.V.; dos Santos, J.P.S.; Faria, R.X. Capsaicin: TRPV1-Independent Mechanisms and Novel Therapeutic Possibilities. Eur. J. Pharmacol. 2020, 887, 173356. [Google Scholar] [CrossRef]
- Amantini, C.; Ballarini, P.; Caprodossi, S.; Nabissi, M.; Morelli, M.B.; Lucciarini, R.; Cardarelli, M.A.; Mammana, G.; Santoni, G. Triggering of Transient Receptor Potential Vanilloid Type 1 (TRPV1) by Capsaicin Induces Fas/CD95-Mediated Apoptosis of Urothelial Cancer Cells in an ATM-Dependent Manner. Carcinogenesis 2009, 30, 1320–1329. [Google Scholar] [CrossRef]
- Liu, T.; Wang, G.; Tao, H.; Yang, Z.; Wang, Y.; Meng, Z.; Cao, R.; Xiao, Y.; Wang, X.; Zhou, J. Capsaicin Mediates Caspases Activation and Induces Apoptosis through P38 and JNK MAPK Pathways in Human Renal Carcinoma. BMC Cancer 2016, 16, 790. [Google Scholar] [CrossRef]
- Lin, M.-H.; Lee, Y.-H.; Cheng, H.-L.; Chen, H.-Y.; Jhuang, F.-H.; Chueh, P.J. Capsaicin Inhibits Multiple Bladder Cancer Cell Phenotypes by Inhibiting Tumor-Associated NADH Oxidase (TNOX) and Sirtuin1 (SIRT1). Molecules 2016, 21, 849. [Google Scholar] [CrossRef]
- Islam, A.; Su, A.J.; Zeng, Z.-M.; Chueh, P.J.; Lin, M.-H. Capsaicin Targets TNOX (ENOX2) to Inhibit G1 Cyclin/CDK Complex, as Assessed by the Cellular Thermal Shift Assay (CETSA). Cells 2019, 8, 1275. [Google Scholar] [CrossRef]
- Islam, A.; Yang, Y.-T.; Wu, W.-H.; Chueh, P.J.; Lin, M.-H. Capsaicin Attenuates Cell Migration via SIRT1 Targeting and Inhibition to Enhance Cortactin and β-Catenin Acetylation in Bladder Cancer Cells. Am. J. Cancer Res. 2019, 9, 1172–1182. [Google Scholar]
- Yang, Z.-H.; Wang, X.-H.; Wang, H.-P.; Hu, L.-Q.; Zheng, X.-M.; Li, S.-W. Capsaicin Mediates Cell Death in Bladder Cancer T24 Cells through Reactive Oxygen Species Production and Mitochondrial Depolarization. Urology 2010, 75, 735–741. [Google Scholar] [CrossRef]
- Qian, K.; Wang, G.; Cao, R.; Liu, T.; Qian, G.; Guan, X.; Guo, Z.; Xiao, Y.; Wang, X. Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways. Molecules 2016, 21, 1406. [Google Scholar] [CrossRef]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin Triggers Autophagic Cell Survival Which Drives Epithelial Mesenchymal Transition and Chemoresistance in Bladder Cancer Cells in an Hedgehog-Dependent Manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef]
- Beltran, J.; Ghosh, A.K.; Basu, S. Immunotherapy of Tumors with Neuroimmune Ligand Capsaicin. J. Immunol. 2007, 178, 3260–3264. [Google Scholar] [CrossRef]
- Bertin, S.; Aoki-Nonaka, Y.; de Jong, P.R.; Nohara, L.L.; Xu, H.; Stanwood, S.R.; Srikanth, S.; Lee, J.; To, K.; Abramson, L.; et al. The Ion Channel TRPV1 Regulates the Activation and Proinflammatory Properties of CD4+ T Cells. Nat. Immunol. 2014, 15, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Morelli, M.B.; Amantini, C.; Santoni, M.; Nabissi, M.; Marinelli, O.; Santoni, A. “Immuno-Transient Receptor Potential Ion Channels”: The Role in Monocyte- and Macrophage-Mediated Inflammatory Responses. Front. Immunol. 2018, 9, 1273. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Park, J.W.; Kurata, T.; Erickson, K.L. Modulation of Select Immune Responses by Dietary Capsaicin. International journal for vitamin and nutrition research. Internationale Zeitschrift fur Vitamin- und Ernahrungsforschung. J. Int. Vitaminol. Nutr. 1998, 68, 114–119. [Google Scholar]
- D’Eliseo, D.; Manzi, L.; Velotti, F. Capsaicin as an Inducer of Damage-Associated Molecular Patterns (DAMPs) of Immunogenic Cell Death (ICD) in Human Bladder Cancer Cells. Cell Stress Chaperones 2013, 18, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Basu, S. Tumor Macrophages as a Target for Capsaicin Mediated Immunotherapy. Cancer Lett. 2012, 324, 91–97. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Kawahara, T.; Ishiguro, Y.; Ohtake, S.; Kato, I.; Ito, Y.; Ito, H.; Makiyama, K.; Kondo, K.; Miyoshi, Y.; Yumura, Y.; et al. PD-1 and PD-L1 Are More Highly Expressed in High-Grade Bladder Cancer than in Low-Grade Cases: PD-L1 Might Function as a Mediator of Stage Progression in Bladder Cancer. BMC Urol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, S.-D.; McCrudden, C.; Chan, K.-W.; Lin, Y.; Kwok, H.-F. The Prognostic Significance of PD-L1 in Bladder Cancer. Oncol. Rep. 2015, 33, 3075–3084. [Google Scholar] [CrossRef]
- Möller, K.; Fraune, C.; Blessin, N.C.; Lennartz, M.; Kluth, M.; Hube-Magg, C.; Lindhorst, L.; Dahlem, R.; Fisch, M.; Eichenauer, T.; et al. Tumor Cell PD-L1 Expression Is a Strong Predictor of Unfavorable Prognosis in Immune Checkpoint Therapy-Naive Clear Cell Renal Cell Cancer. Int. Urol. Nephrol. 2021, 53, 2493–2503. [Google Scholar] [CrossRef]
- Xylinas, E.; Robinson, B.D.; Kluth, L.A.; Volkmer, B.G.; Hautmann, R.; Küfer, R.; Zerbib, M.; Kwon, E.; Thompson, R.H.; Boorjian, S.A.; et al. Association of T-Cell Co-Regulatory Protein Expression with Clinical Outcomes Following Radical Cystectomy for Urothelial Carcinoma of the Bladder. Eur. J. Surg. Oncol. 2014, 40, 121–127. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.; et al. MPDL3280A (Anti-PD-L1) Treatment Leads to Clinical Activity in Metastatic Bladder Cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Tseng, P.-C.; Chen, C.-L.; Satria, R.D.; Wang, Y.-T.; Lin, C.-F. Different Induction of PD-L1 (CD274) and PD-1 (CD279) Expression in THP-1-Differentiated Types 1 and 2 Macrophages. J. Inflamm. Res. 2021, 14, 5241–5249. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA Damage-Inducible Phosphorylation of P53 at N-Terminal Sites Including a Novel Site, Ser20, Requires Tetramerization. EMBO J. 1999, 18, 1815–1823. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced Phosphorylation of P53 by ATM in Response to DNA Damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Chen, S.; Wang, G.; Makrigiorgos, G.M.; Price, B.D. Stable SiRNA-Mediated Silencing of ATM Alters the Transcriptional Profile of HeLa Cells. Biochem. Biophys. Res. Commun. 2004, 317, 1037–1044. [Google Scholar] [CrossRef]
- Bailly, C. Regulation of PD-L1 Expression on Cancer Cells with ROS-Modulating Drugs. Life Sci. 2020, 246, 117403. [Google Scholar] [CrossRef]
- Yang, F.; Zheng, J. Understand Spiciness: Mechanism of TRPV1 Channel Activation by Capsaicin. Protein Cell 2017, 8, 169–177. [Google Scholar] [CrossRef]
- Birder, L.A.; Nakamura, Y.; Kiss, S.; Nealen, M.L.; Barrick, S.; Kanai, A.J.; Wang, E.; Ruiz, G.; de Groat, W.C.; Apodaca, G.; et al. Altered Urinary Bladder Function in Mice Lacking the Vanilloid Receptor TRPV1. Nat. Neurosci. 2002, 5, 856–860. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Liu, X.-Y.; Zhuo, D.-X.; Huang, H.-B.; Zhang, F.-B.; Liao, S.-F. Decreased Expression of TRPV1 in Renal Cell Carcinoma: Association with Tumor Fuhrman Grades and Histopathological Subtypes. Cancer Manag. Res. 2018, 10, 1647–1655. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wigner, P.; Szymańska, B.; Bijak, M.; Sawicka, E.; Kowal, P.; Marchewka, Z.; Saluk-Bijak, J. Oxidative Stress Parameters as Biomarkers of Bladder Cancer Development and Progression. Sci. Rep. 2021, 11, 15134. [Google Scholar] [CrossRef] [PubMed]
- Vidal, N.; Cavaille, J.P.; Graziani, F.; Robin, M.; Ouari, O.; Pietri, S.; Stocker, P. High Throughput Assay for Evaluation of Reactive Carbonyl Scavenging Capacity. Redox Biol. 2014, 2, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhao, X.H.; Zhang, R.H. NME4 Regulates TLR3 Activation and Type I IFN Production through Autophagy-Dependent TRAF6 Degradation. J. Immunol. 2019, 202, 22–64. [Google Scholar]
- Tokarska-Schlattner, M.; Boissan, M.; Munier, A.; Borot, C.; Mailleau, C.; Speer, O.; Schlattner, U.; Lacombe, M.-L. The Nucleoside Diphosphate Kinase D (NM23-H4) Binds the Inner Mitochondrial Membrane with High Affinity to Cardiolipin and Couples Nucleotide Transfer with Respiration. J. Biol. Chem. 2008, 283, 26198–26207. [Google Scholar] [CrossRef]
- Zheng, S.; Liu, Q.; Liu, T.; Yang, L.; Zhang, Q.; Shen, T.; Zhang, X.; Han, X.; Lu, X. NME4 Modulates PD-L1 Expression via the STAT3 Signaling Pathway in Squamous Cell Carcinoma. Biochem. Biophys. Res. Commun. 2020, 526, 29–34. [Google Scholar] [CrossRef]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Bartkova, J.; Guldberg, P.; Grønbaek, K.; Koed, K.; Primdahl, H.; Møller, K.; Lukas, J.; Ørntoft, T.F.; Bartek, J. Aberrations of the Chk2 Tumour Suppressor in Advanced Urinary Bladder Cancer. Oncogene 2004, 23, 8545–8551. [Google Scholar] [CrossRef]
- McNiel, E.A.; Tsichlis, P.N. Analyses of Publicly Available Genomics Resources Define FGF-2-Expressing Bladder Carcinomas as EMT-Prone, Proliferative Tumors with Low Mutation Rates and High Expression of CTLA-4, PD-1 and PD-L1. Signal Transduct. Target. Ther. 2017, 2, 16045. [Google Scholar] [CrossRef]
- Li, H.; Xia, J.; Zhu, F.; Xi, Z.; Pan, C.; Gu, L.; Tian, Y. LPS Promotes the Expression of PD-L1 in Gastric Cancer Cells through NF-κB Activation. J. Cell. Biochem. 2018, 119, 9997–10004. [Google Scholar] [CrossRef]
- Glorieux, C.; Xia, X.; Huang, P. The Role of Oncogenes and Redox Signaling in the Regulation of PD-L1 in Cancer. Cancers 2021, 13, 4426. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC Regulates the Antitumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Rajabi, H.; Kufe, D. MUC1-C Oncoprotein Integrates a Program of EMT, Epigenetic Reprogramming and Immune Evasion in Human Carcinomas. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 117–122. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-{gamma} and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-γ/PD-L1 Axis between T Cells and Tumor Microenvironment: Hints for Glioma Anti-PD-1/PD-L1 Therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef]
- Yamamoto, R.; Nishikori, M.; Tashima, M.; Sakai, T.; Ichinohe, T.; Takaori-Kondo, A.; Ohmori, K.; Uchiyama, T. B7-H1 Expression Is Regulated by MEK/ERK Signaling Pathway in Anaplastic Large Cell Lymphoma and Hodgkin Lymphoma. Cancer Sci. 2009, 100, 2093–2100. [Google Scholar] [CrossRef]
- Huang, L.; Li, Y.; Du, Y.; Zhang, Y.; Wang, X.; Ding, Y.; Yang, X.; Meng, F.; Tu, J.; Luo, L.; et al. Mild Photothermal Therapy Potentiates Anti-PD-L1 Treatment for Immunologically Cold Tumors via an All-in-One and All-in-Control Strategy. Nat. Commun. 2019, 10, 4871. [Google Scholar] [CrossRef]
- Li, Z.; Deng, J.; Sun, J.; Ma, Y. Hyperthermia Targeting the Tumor Microenvironment Facilitates Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 595207. [Google Scholar] [CrossRef]
- Oikawa, S.; Nagao, E.; Sakano, K.; Kawanishi, S.; Oikawa, S.; Nagao, E.; Sakano, K.; Kawanishi, S. Mechanism of Oxidative DNA Damage Induced by Capsaicin, a Principal Ingredient of Hot Chili Pepper. Free Radic. Res. 2006, 40, 966–973. [Google Scholar] [CrossRef]
- Luqman, S.; Rizvi, S.I. Protection of Lipid Peroxidation and Carbonyl Formation in Proteins by Capsaicin in Human Erythrocytes Subjected to Oxidative Stress. Phytother. Res. 2006, 20, 303–306. [Google Scholar] [CrossRef]
- Lascu, I.; Gonin, P. The Catalytic Mechanism of Nucleoside Diphosphate Kinases. J. Bioenerg. Biomembr. 2000, 32, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, M.-L.; Tokarska-Schlattner, M.; Boissan, M.; Schlattner, U. The Mitochondrial Nucleoside Diphosphate Kinase (NDPK-D/NME4), a Moonlighting Protein for Cell Homeostasis. Lab. Investig. 2018, 98, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Tsao, N.; Zhang, W.; Chang, Z.-F. NME3 Regulates Mitochondria to Reduce ROS-Mediated Genome Instability. Int. J. Mol. Sci. 2020, 21, 5048. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kunimoto, H.; Katayama, B.; Zhao, H.; Shiromizu, T.; Wang, L.; Ozawa, T.; Tomonaga, T.; Tsuruta, D.; Nakajima, K. Phospho-Ser727 Triggers a Multistep Inactivation of STAT3 by Rapid Dissociation of PY705–SH2 through C-Terminal Tail Modulation. Int. Immunol. 2020, 32, 73. [Google Scholar] [CrossRef] [PubMed]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative Metabolism in Cancer: A STAT Affair? JAKSTAT 2013, 2, e25764. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Guan, W.; Zhang, L.; Sun, W.; Zhou, D.; Lin, Q.; Ren, W.; Nadeem, L.; Xu, G. Physical Interaction of STAT1 Isoforms with TGF-β Receptors Leads to Functional Crosstalk between Two Signaling Pathways in Epithelial Ovarian Cancer. J. Exp. Clin. Cancer Res. 2018, 37, 103. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kakizuka, A.; Imamura, H. BTeam, a Novel BRET-Based Biosensor for the Accurate Quantification of ATP Concentration within Living Cells. Sci. Rep. 2016, 6, 39618. [Google Scholar] [CrossRef]
- Tolomeo, M.; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual Inhibition of STAT1 and STAT3 Activation Downregulates Expression of PD-L1 in Human Breast Cancer Cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. Correction: The Ratio of STAT1 to STAT3 Expression Is a Determinant of Colorectal Cancer Growth. Oncotarget 2018, 9, 33865. [Google Scholar] [CrossRef]
- Stephanou, A.; Latchman, D.S. Opposing Actions of STAT-1 and STAT-3. Growth Factors 2005, 23, 177–182. [Google Scholar] [CrossRef]
- Santoni, M.; Conti, A.; Piva, F.; Massari, F.; Ciccarese, C.; Burattini, L.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M.; Santini, D.; et al. Role of STAT3 Pathway in Genitourinary Tumors. Future Sci. OA 2015, 1, FSO15. [Google Scholar] [CrossRef]
- Chung, S.S.; Aroh, C.; Vadgama, J.V. Constitutive Activation of STAT3 Signaling Regulates HTERT and Promotes Stem Cell-like Traits in Human Breast Cancer Cells. PLoS ONE 2013, 8, e83971. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Fay, A.P.; Gray, K.P.; Callea, M.; Ho, T.H.; Albiges, L.; Bellmunt, J.; Song, J.; Carvo, I.; Lampron, M.; et al. PD-L1 Expression in Nonclear-Cell Renal Cell Carcinoma. Ann. Oncol. 2014, 25, 2178–2184. [Google Scholar] [CrossRef]
- Iacovelli, R.; Nolè, F.; Verri, E.; Renne, G.; Paglino, C.; Santoni, M.; Cossu Rocca, M.; Giglione, P.; Aurilio, G.; Cullurà, D.; et al. Prognostic Role of PD-L1 Expression in Renal Cell Carcinoma. A Systematic Review and Meta-Analysis. Target. Oncol. 2016, 11, 143–148. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Rossi de Vermandois, J.A.; Gubbiotti, M.; Giannantoni, A.; Mearini, E.; Maggi, F.; Nabissi, M.; Marinelli, O.; Santoni, M.; et al. Correlation between High PD-L1 and EMT/Invasive Genes Expression and Reduced Recurrence-Free Survival in Blood-Circulating Tumor Cells from Patients with Non-Muscle-Invasive Bladder Cancer. Cancers 2021, 13, 5989. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Yang, X.; Liu, Y.; Liu, Y.; Li, Y.; Sun, L.; Yang, X.; Niu, H. Bacillus Calmette-Guérin and Anti-PD-L1 Combination Therapy Boosts Immune Response against Bladder Cancer. Onco Targets Ther. 2018, 11, 2891–2899. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Jimeno, A. Atezolizumab: A Novel PD-L1 Inhibitor in Cancer Therapy with a Focus in Bladder and Non-Small Cell Lung Cancers. Drugs Today 2017, 53, 217–237. [Google Scholar] [CrossRef]
- Cai, S.; Chen, Z.; Wang, Y.; Wang, M.; Wu, J.; Tong, Y.; Chen, L.; Lu, C.; Yang, H. Reducing PD-L1 Expression with a Self-Assembled Nanodrug: An Alternative to PD-L1 Antibody for Enhanced Chemo-Immunotherapy. Theranostics 2021, 11, 1970–1981. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Crittenden, M.; Wargo, J. Combination Immunotherapy Development in Melanoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 197–207. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unigene | GenBank ID | Symbol | Cell Lines | |||||
|---|---|---|---|---|---|---|---|---|
| 5637 | T24 | A498 | ||||||
| 4 h | 12 h | 4 h | 12 h | 4 h | 12 h | |||
| NM_003998 | Nuclear factor of kappa light polypeptide gene enhancer in B cells-1 | NF-kB1 | 1.4 | −1.2 | 1.1 | −1.8 | 2.0 * | −1.2 |
| NM_020529 | Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha | NF-kBIA | −2.3 * | −2.7 * | −2.1 * | −1.7 | −1.2 | −1.7 |
| NM_005225 | E2F transcriptional factor 1 | E2F1 | N/A | N/A | N/A | N/A | IND * | 3.2 * |
| NM_005239 | V-ETs erythroblastosis virus E26 oncogene homolog 2 | ETS2 | −1.1 | −2.2 * | −1.0 | −2.8 * | 1.7 | −1.2 |
| NM_005252 | V-Fos FBJ murine osteosarcoma viral oncogene homolog | FOS | −1.4 | −3.0 * | −1.9 | −2.0 * | −2.3* | −7.7 * |
| NM_002228 | V-Jun sarcoma virus 17 oncogene homolog | JUN | −1.3 | −2.4 * | −1.1 | −2.8 * | −2.6* | −3.1 * |
| NM_002467 | V-mycmyelocitomatosis viral oncogene | MYC | 1.3 | −1.5 | 1.5 | −1.9 | 5.0 * | 2.0 * |
| NM−005163 | V-Akt murine thymoma viral oncogene homolog 1 | AKT | SIL * | −2.9 * | −2.3 * | −3.4 * | N/A | N/A |
| NM_004448 | V-Erb-b2 erythroblastic leukemia viral oncogene homolog 2 | ERBB2 | 1.1 | −2.4 * | 1.1 | −2.4 * | 3.2 * | 1.5 |
| NM_002880 | V-Raf-1 murine leukemia viral oncogene homolog 1 | RAF1 | 1.0 | −1.2 | 1.1 | −1.0 | 1.5 | 1.2 |
| NM_000245 | Met proto-oncogene | MET | N/A | N/A | N/A | N/A | IND * | 1.9 |
| NM-002755 | Mitogen-activated protein kinase kinase 1 | MAPKK1 | −2.6 * | −1.4 | −2.4 * | −1.0 | 2.7 * | 2.0 * |
| NM_181504 | Phosphoinositide-3-kinase, regulatory subunit polypeptide 1 | PI3K | 1.6 | 1.1 | 1.8 | 1.6 | 1.0 | −1.1 |
| NM_003177 | Spleen tyrosine kinase | SYK | N/A | N/A | N/A | N/A | IND * | 2.8 * |
| Unigene | GenBank ID | Symbol | Cell Lines | |||||
|---|---|---|---|---|---|---|---|---|
| 5637 | T24 | A498 | ||||||
| 4 h | 12 h | 4 h | 12 h | 4 h | 12 h | |||
| NM_024013 | Interferon alpha 1 | IFN-α1 | 17.4 * | 12.2 * | 6.2 * | 4.9 * | 1.5 | 1.6 |
| NM_002176 | Interferon beta 1 fibroblast | IFN-β1 | 24.5 * | 19.8 * | 9.5 * | 5.3 * | 1.4 | 1.3 |
| NM_000618 | Insulin-like growth factor 1 | IGF-1 | N/A | N/A | N/A | N/A | N/A | N/A |
| NM_000584 | Interleukin-8 | IL-8 | 1.3 | 1.1 | 1.1 | −9.1 * | 1.8 | 1.1 |
| NM_002607 | Platelet-derived growth factor alpha polypeptide | PDGF-A | N/A | N/A | N/A | N/A | IND * | 3.0 * |
| NM_002607 | Platelet-derived growth factor beta polypeptide | PDGF-B | −1.1 | −1.5 | −1.7 | −1.9 | −3.7 * | −3.9 * |
| NM_000660 | Transforming growth factor beta-1 | TGF-β1 | 1.1 | 1.0 | 1.2 | 2.5 * | 1.5 | 4.0 * |
| NM_000594 | Tumor necrosis factor alpha | TNF-α | 1.9 | 1.7 | 1.4 | −1.6 | 2.9 * | 2.0 * |
| NM_000141 | Fibroblast growth factor receptor 2 | FGFR2 | 2.3 * | −1.3 | 2.3 * | −1.3 | 1.5 | 1.1 |
| NM_003246 | Transforming growth factor beta receptor 1 | TGFβR1 | 1.0 | 1.2 | 1.6 | 2.1 * | 2.0 * | 2.2 * |
| NM_003862 | Tumor necrosis factor receptor superfamily member 10b | TNFRSM10b | −1.4 | −2.4 * | −1.4 | −2.4 * | 2.8 * | 1.6 |
| NM_001065 | Tumor necrosis factor receptor superfamily member 1A | TNFRSM1A | −2.0 * | −3.6 * | −2.0 * | −3.6 * | 2.0 * | 1.1 |
| NM_003790 | Tumor necrosis factor receptor superfamily member 25 | TNFRSM25 | 1.7 | −1.6 | 1.7 | −1.6 | 2.8 * | 2.0 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, M.B.; Marinelli, O.; Aguzzi, C.; Zeppa, L.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Maggi, F.; Santoni, M.; Santoni, G. Unveiling the Molecular Mechanisms Driving the Capsaicin-Induced Immunomodulatory Effects on PD-L1 Expression in Bladder and Renal Cancer Cell Lines. Cancers 2022, 14, 2644. https://doi.org/10.3390/cancers14112644
Morelli MB, Marinelli O, Aguzzi C, Zeppa L, Nabissi M, Amantini C, Tomassoni D, Maggi F, Santoni M, Santoni G. Unveiling the Molecular Mechanisms Driving the Capsaicin-Induced Immunomodulatory Effects on PD-L1 Expression in Bladder and Renal Cancer Cell Lines. Cancers. 2022; 14(11):2644. https://doi.org/10.3390/cancers14112644
Chicago/Turabian StyleMorelli, Maria Beatrice, Oliviero Marinelli, Cristina Aguzzi, Laura Zeppa, Massimo Nabissi, Consuelo Amantini, Daniele Tomassoni, Federica Maggi, Matteo Santoni, and Giorgio Santoni. 2022. "Unveiling the Molecular Mechanisms Driving the Capsaicin-Induced Immunomodulatory Effects on PD-L1 Expression in Bladder and Renal Cancer Cell Lines" Cancers 14, no. 11: 2644. https://doi.org/10.3390/cancers14112644
APA StyleMorelli, M. B., Marinelli, O., Aguzzi, C., Zeppa, L., Nabissi, M., Amantini, C., Tomassoni, D., Maggi, F., Santoni, M., & Santoni, G. (2022). Unveiling the Molecular Mechanisms Driving the Capsaicin-Induced Immunomodulatory Effects on PD-L1 Expression in Bladder and Renal Cancer Cell Lines. Cancers, 14(11), 2644. https://doi.org/10.3390/cancers14112644