
A Histone Deacetylase Inhibitor Induces Acetyl-CoA Depletion Leading to Lethal Metabolic Stress in RAS-Pathway Activated Cells

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Treatment and Statistical Tests
2.2. Transcriptomic Analysis with R
2.3. Mouse Study
2.4. Flow Cytometry
2.5. Western Blot
2.6. Seahorse Experiment
2.7. Cell Lines, Treatment and Statistical Tests
2.8. Acetyl-CoA Quantification Analysis
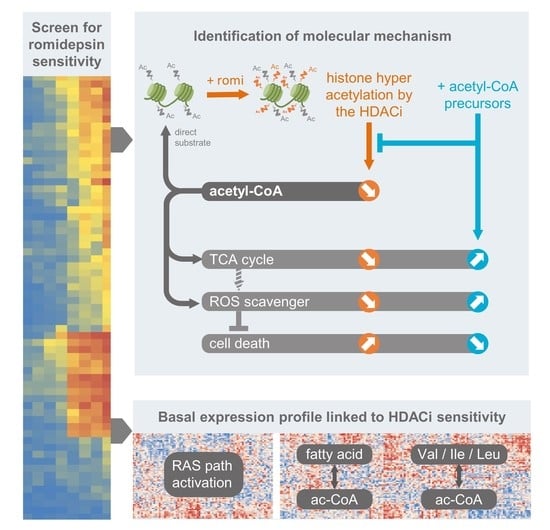
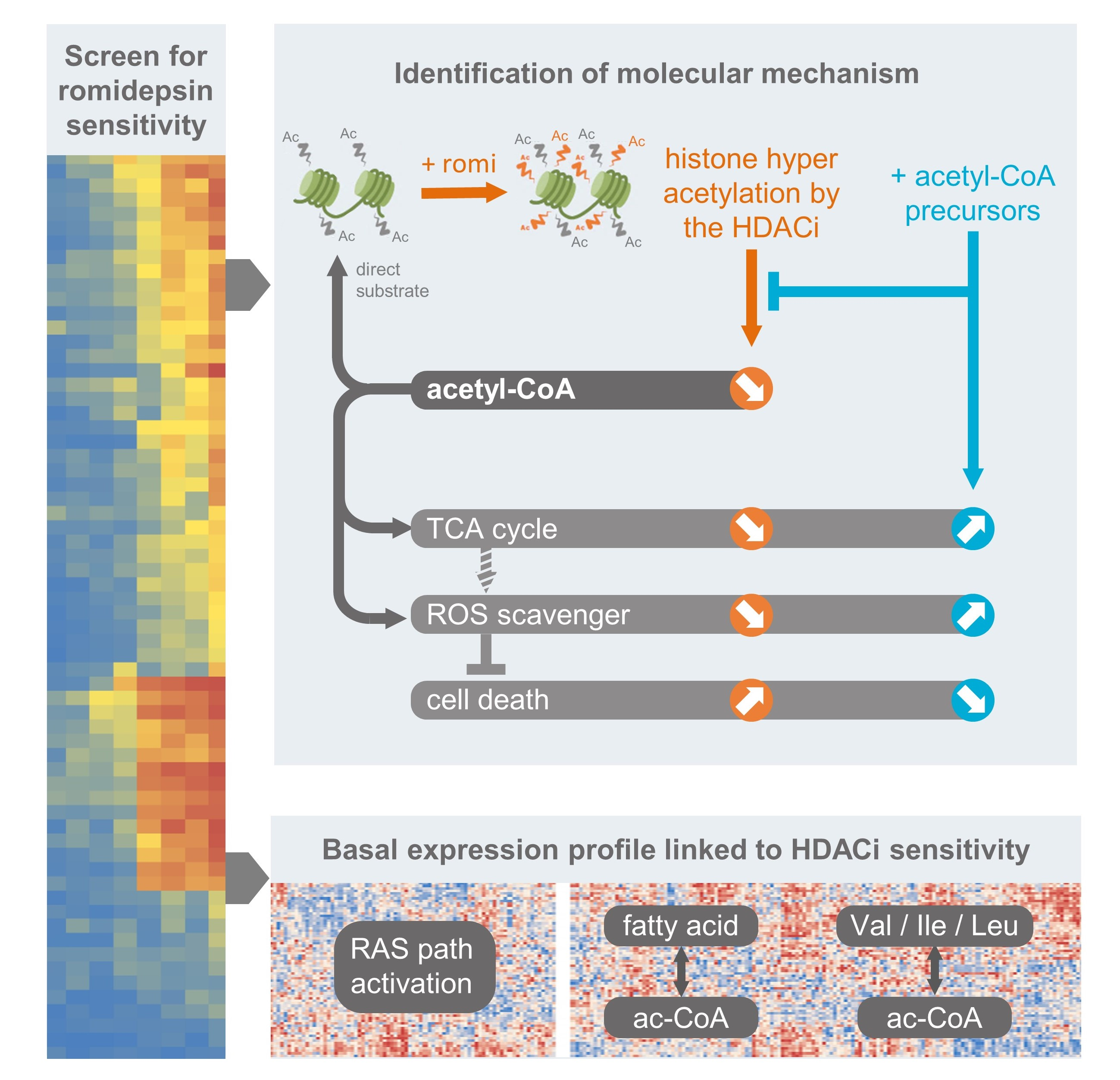
3. Results
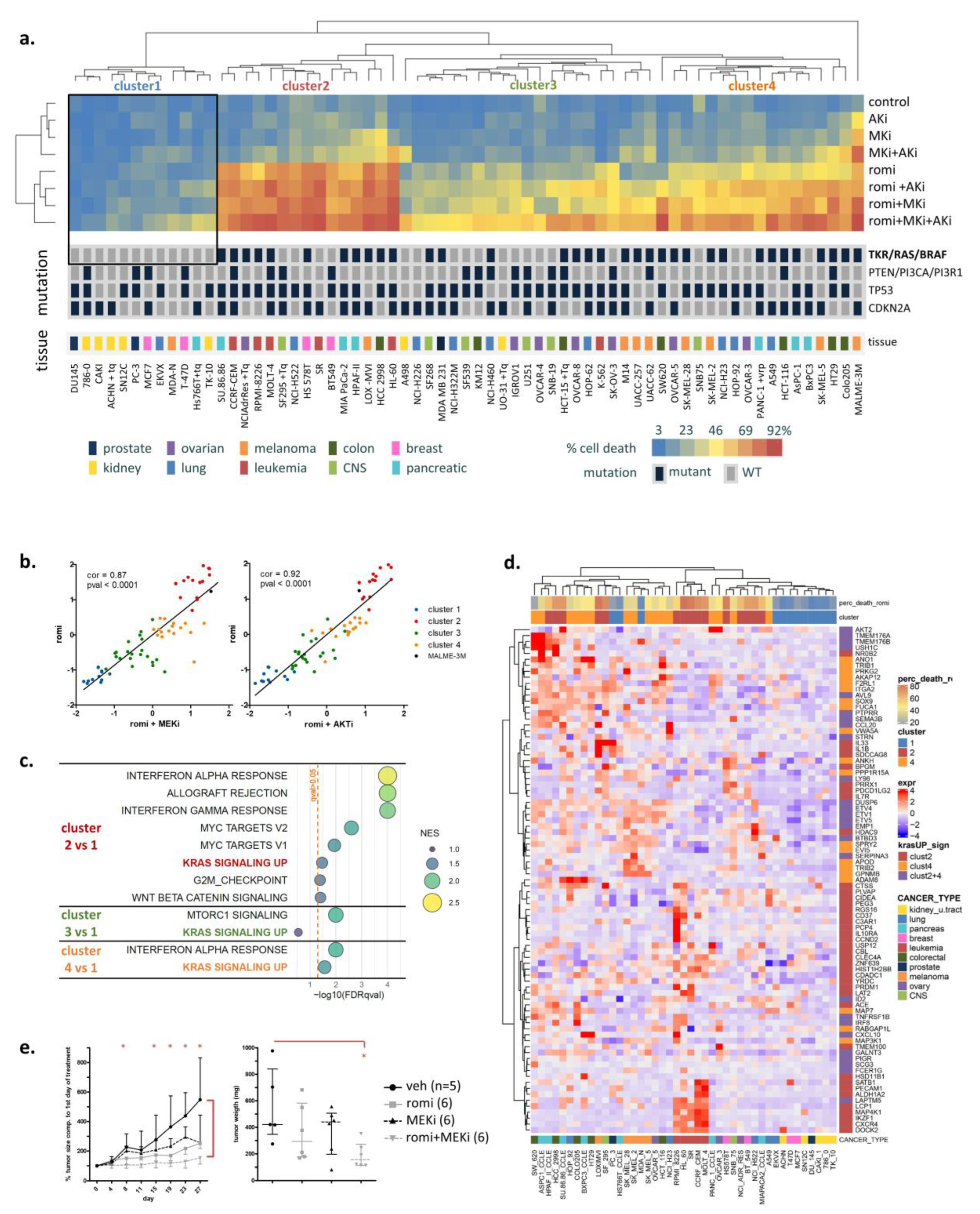
3.1. Activated RAS Pathway Is Linked to Romidepsin Sensitivity
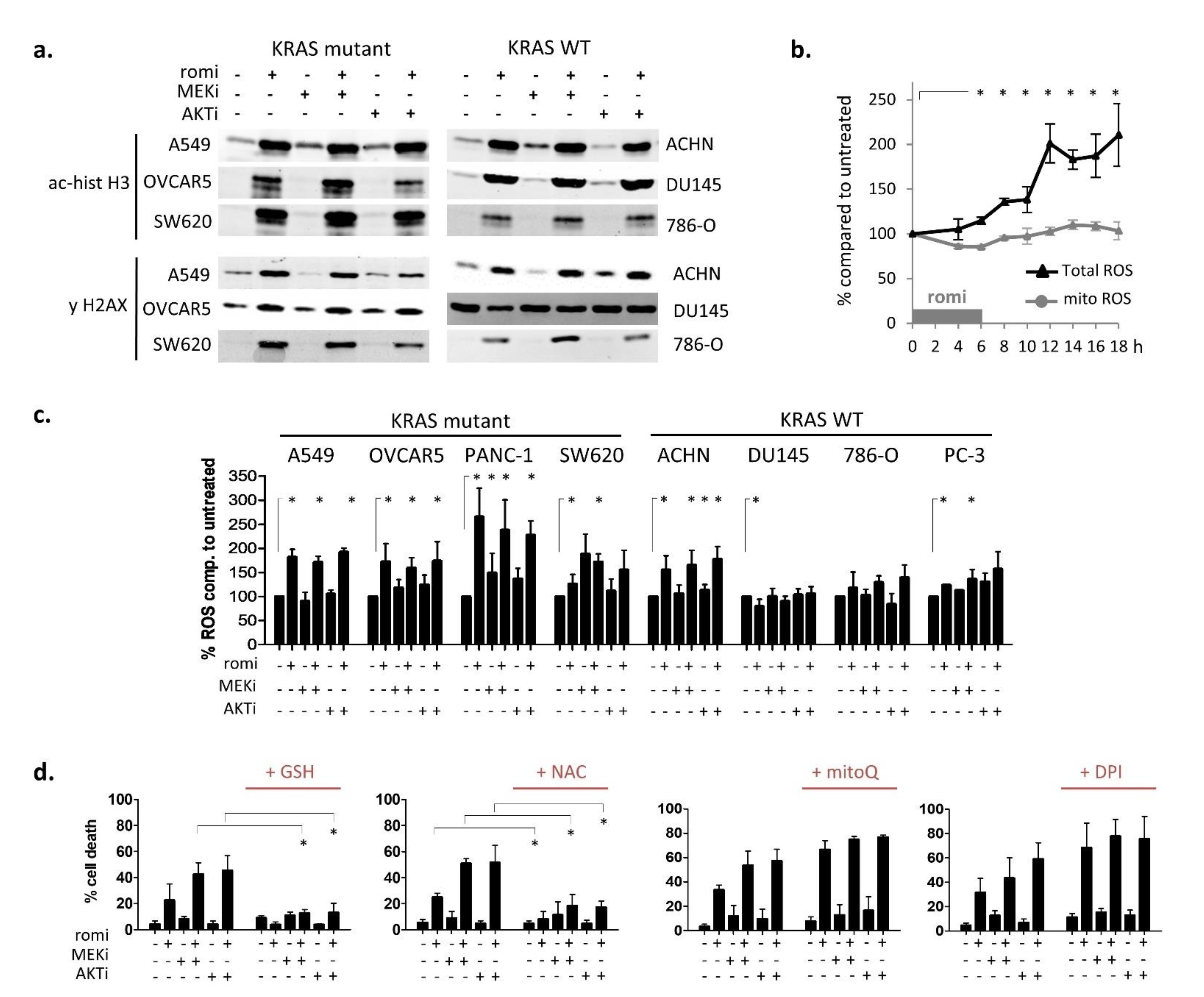
3.2. Early Cytoplasmic ROS Release Differentiates KRAS Sensitivity to Romidepsin, While Acetylation and DNA Damage Do Not
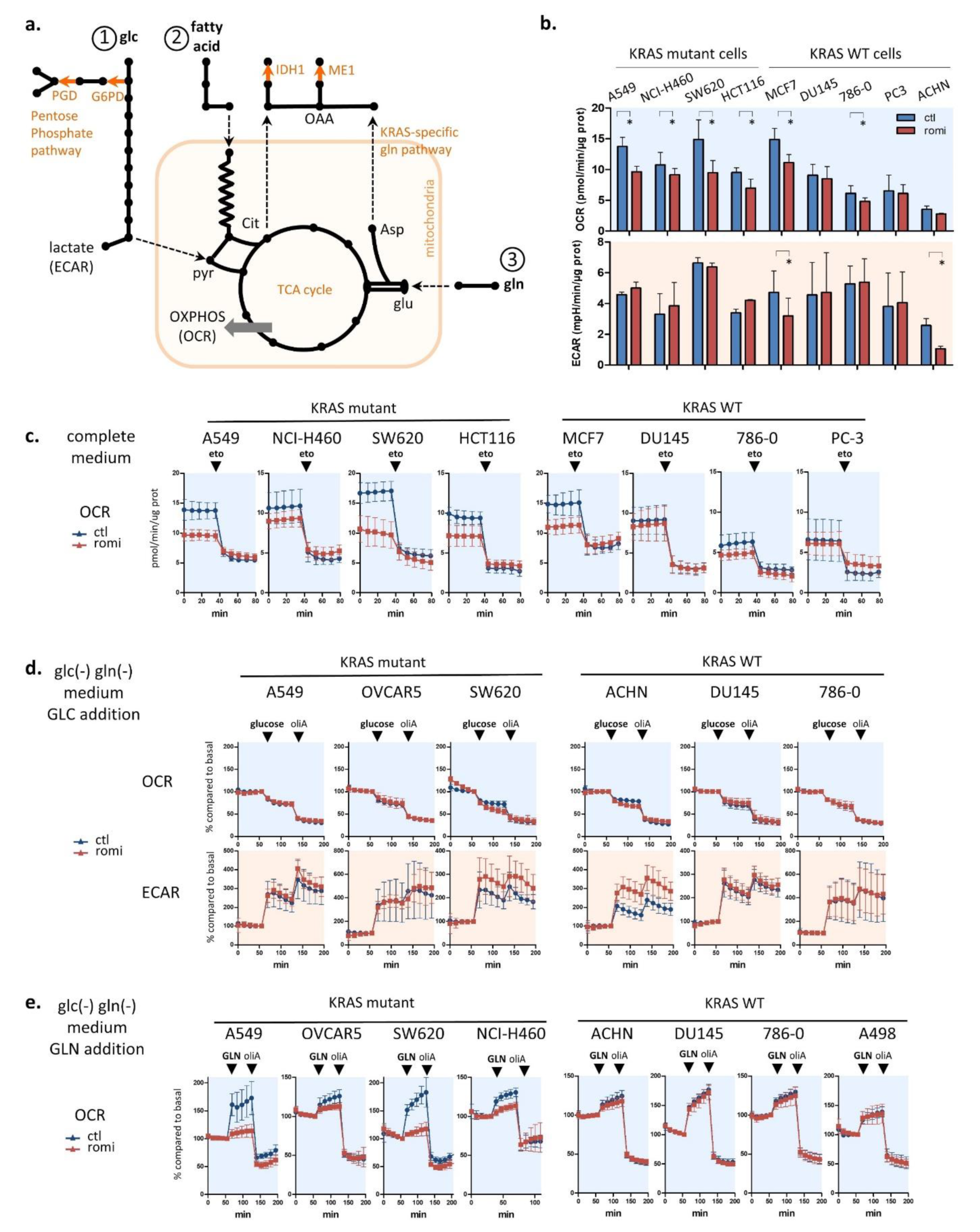
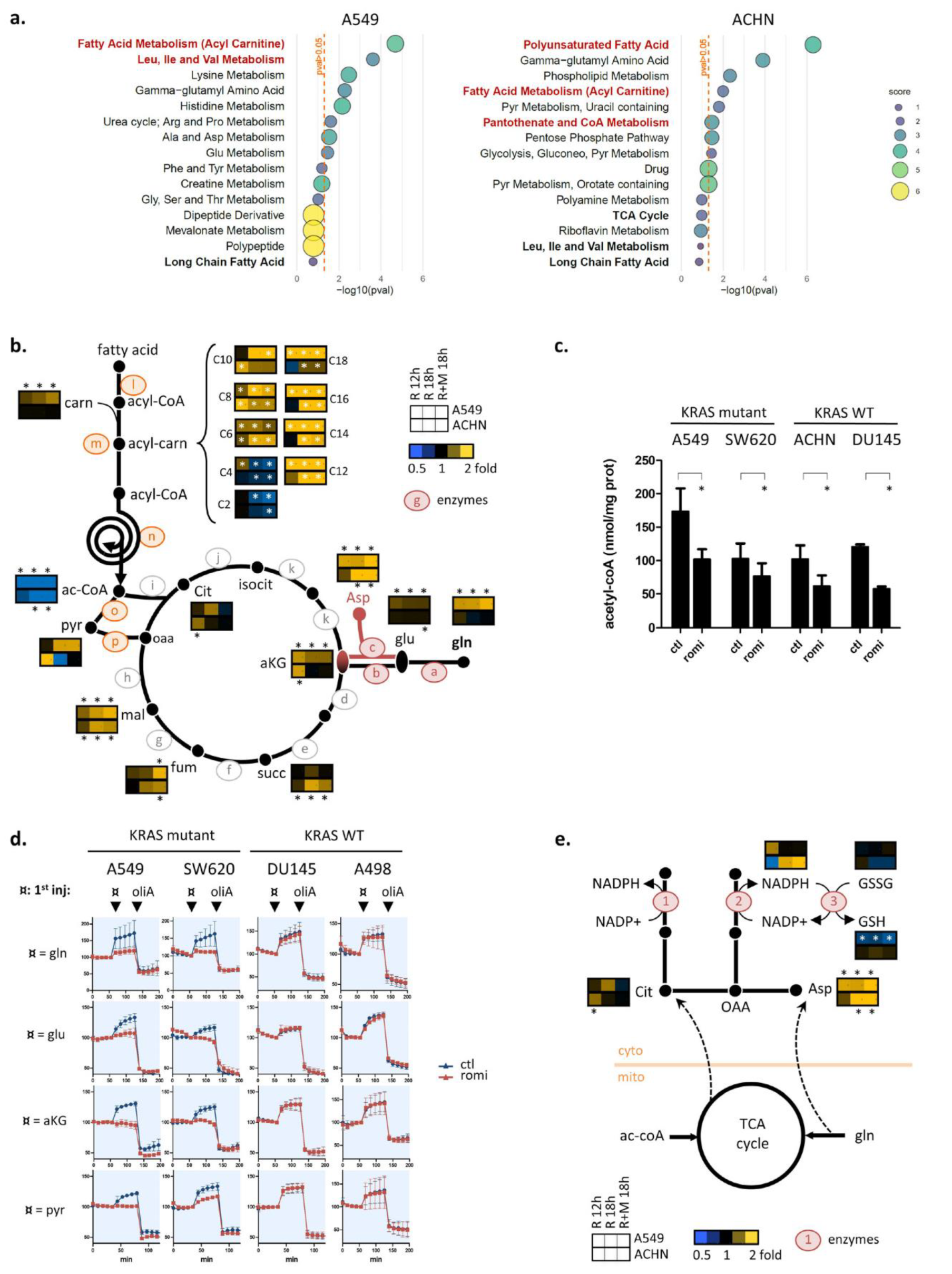
3.3. Romidepsin Affects Two TCA-Fueling Pathways: Fatty Acid Beta-Oxidation and Glutaminolysis
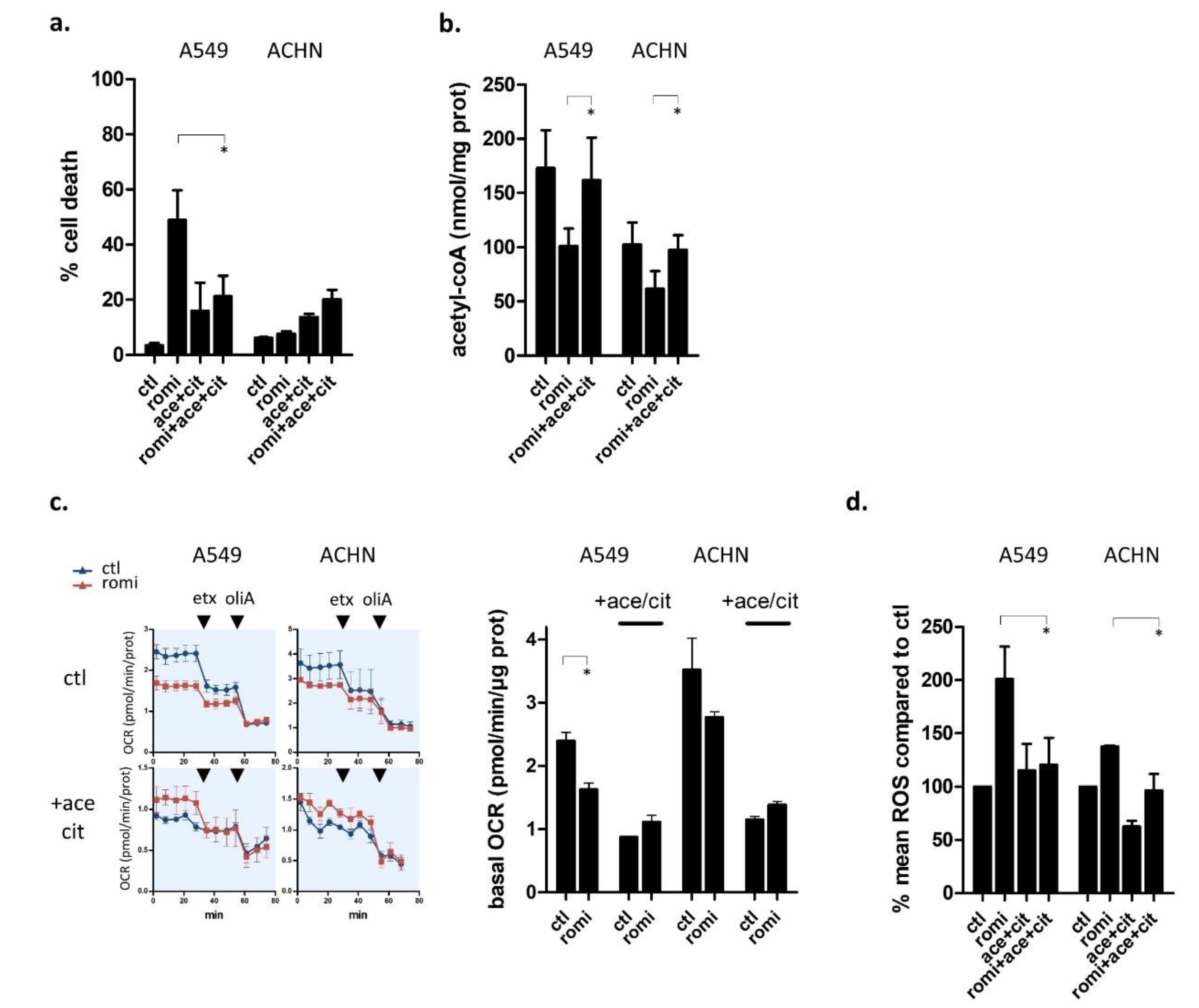
3.4. Romidepsin Affects Acetyl-CoA Availability
3.5. Acetyl-CoA Precursors Replenish Acetyl-CoA Stock and Rescue KRAS-Mutant Cells from Metabolic Stress
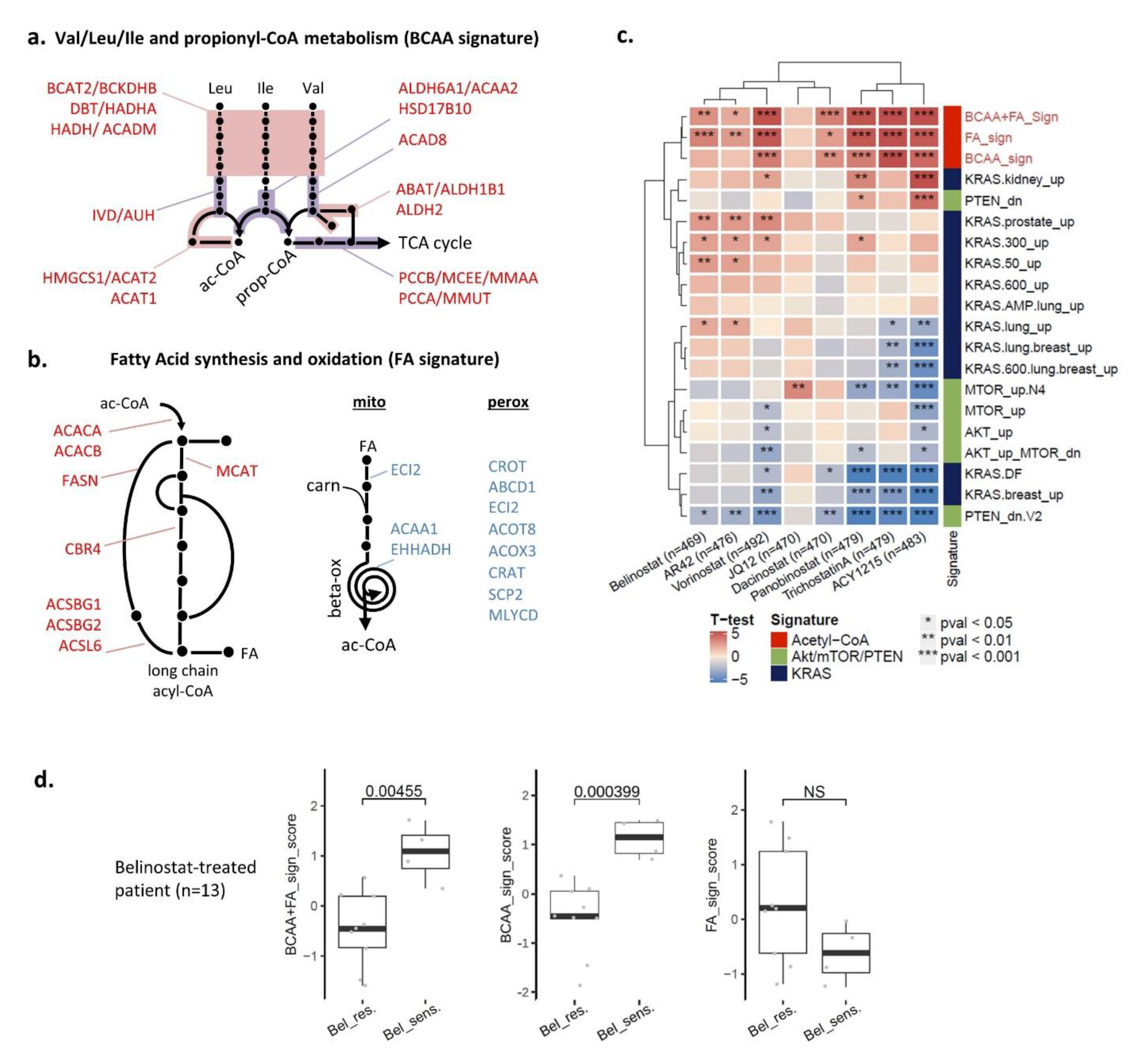
3.6. Two Acetyl-CoA Metabolism Pathways Are Markers of HDAC Inhibitor Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryan, M.B.; Corcoran, R.B. Therapeutic Strategies to Target RAS-Mutant Cancers. Nat. Rev. Clin. Oncol 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef]
- Bahr, J.C.; Robey, R.W.; Luchenko, V.; Basseville, A.; Chakraborty, A.R.; Kozlowski, H.; Pauly, G.T.; Patel, P.; Schneider, J.P.; Gottesman, M.M.; et al. Blocking Downstream Signaling Pathways in the Context of HDAC Inhibition Promotes Apoptosis Preferentially in Cells Harboring Mutant Ras. Oncotarget 2016, 7, 69804–69815. [Google Scholar] [CrossRef]
- Hanker, A.B.; Healy, K.D.; Nichols, J.; Der, C.J. Romidepsin Inhibits Ras-Dependent Growth Transformation of NIH 3T3 Fibroblasts and RIE-1 Epithelial Cells Independently of Ras Signaling Inhibition. J. Mol. Signal. 2009, 4, 5. [Google Scholar] [CrossRef][Green Version]
- Choudhary, S.; Wang, H.-C.R. Proapoptotic Ability of Oncogenic H-Ras to Facilitate Apoptosis Induced by Histone Deacetylase Inhibitors in Human Cancer Cells. Mol. Cancer Ther. 2007, 6, 1099–1111. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–683. [Google Scholar] [CrossRef]
- Safari, M.; Litman, T.; Robey, R.; Aguilera, A.; Chakraborty, A.R.; Reinhold, W.; Basseville, A.; Petrukhin, L.; Scotto, L.; O’Connor, O.A.; et al. R-Loop-Mediated SsDNA Breaks Accumulate Following Short-Term Exposure to the HDAC Inhibitor Romidepsin. Mol. Cancer Res. 2021, 19, 1361–1374. [Google Scholar] [CrossRef]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras Copy Number Defines Metabolic Reprogramming and Therapeutic Susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Review: Precision Medicine and Driver Mutations: Computational Methods, Functional Assays and Conformational Principles for Interpreting Cancer Drivers. PLoS Comput. Biol. 2019, 15, e1006658. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.-P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK Pathway Activation Leads to Bim Loss and Histone Deacetylase Inhibitor Resistance: Rationale to Combine Romidepsin with an MEK Inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Kurdistani, S.K. Chromatin: A Capacitor of Acetate for Integrated Regulation of Gene Expression and Cell Physiology. Curr. Opin. Genet. Dev. 2014, 26, 53–58. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The World of Protein Acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.-L.; Xiong, Y.; Lei, Q.-Y. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Gansemer, E.R.; McCommis, K.S.; Martino, M.; King-McAlpin, A.Q.; Potthoff, M.J.; Finck, B.N.; Taylor, E.B.; Rutkowski, D.T. NADPH and Glutathione Redox Link TCA Cycle Activity to Endoplasmic Reticulum Homeostasis. iScience 2020, 23, 101116. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Han, Y.; Liu, J.; Fang, Y.; Tian, Y.; Zhou, J.; Ma, D.; Wu, P. Trichostatin A Targets the Mitochondrial Respiratory Chain, Increasing Mitochondrial Reactive Oxygen Species Production to Trigger Apoptosis in Human Breast Cancer Cells. PLoS ONE 2014, 9, e91610. [Google Scholar] [CrossRef]
- Zhang, Y.; Ishida, C.T.; Ishida, W.; Lo, S.-F.L.; Zhao, J.; Shu, C.; Bianchetti, E.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; et al. Combined HDAC and Bromodomain Protein Inhibition Reprograms Tumor Cell Metabolism and Elicits Synthetic Lethality in Glioblastoma. Clin. Cancer Res. 2018, 24, 3941–3954. [Google Scholar] [CrossRef]
- Kaisar, M.M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via Both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front. Immunol. 2017, 8, 1429. [Google Scholar] [CrossRef]
- Silva, M.F.B.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J.A. Valproate Induces in Vitro Accumulation of Long-Chain Fatty Acylcarnitines. Mol. Genet. Metab. 2001, 73, 358–361. [Google Scholar] [CrossRef]
- Coudé, F.X.; Grimber, G.; Pelet, A.; Benoit, Y. Action of the Antiepileptic Drug, Valproic Acid, on Fatty Acid Oxidation in Isolated Rat Hepatocytes. Biochem. Biophys. Res. Commun. 1983, 115, 730–736. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose Metabolism as a Target of Histone Deacetylase Inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Lee, T.-I.; Kao, Y.-H.; Tsai, W.-C.; Chung, C.-C.; Chen, Y.-C.; Chen, Y.-J. HDAC Inhibition Modulates Cardiac PPARs and Fatty Acid Metabolism in Diabetic Cardiomyopathy. PPAR Res. 2016, 2016, 5938740. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting Histone Deacetylases Suppresses Glucose Metabolism and Hepatocellular Carcinoma Growth by Restoring FBP1 Expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef]
- Davidson, S.M. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol Uptake Disruption, in Association with Chemotherapy, Is a Promising Combined Metabolic Therapy for Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, M.; Heuer, T.; Grimmer, K.; Crowley, R.; Waszczuk, J.; Fridlib, M.; Ventura, R.; Rubio, C.; Lai, J.; Buckley, D.; et al. Abstract LB-214: FASN Inhibitor TVB-2640 Shows Pharmacodynamic Effect and Evidence of Clinical Activity in KRAS-Mutant NSCLC Patients in a Phase I Study. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basseville, A.; Violet, P.-C.; Safari, M.; Sourbier, C.; Linehan, W.M.; Robey, R.W.; Levine, M.; Sackett, D.L.; Bates, S.E. A Histone Deacetylase Inhibitor Induces Acetyl-CoA Depletion Leading to Lethal Metabolic Stress in RAS-Pathway Activated Cells. Cancers 2022, 14, 2643. https://doi.org/10.3390/cancers14112643
Basseville A, Violet P-C, Safari M, Sourbier C, Linehan WM, Robey RW, Levine M, Sackett DL, Bates SE. A Histone Deacetylase Inhibitor Induces Acetyl-CoA Depletion Leading to Lethal Metabolic Stress in RAS-Pathway Activated Cells. Cancers. 2022; 14(11):2643. https://doi.org/10.3390/cancers14112643
Chicago/Turabian StyleBasseville, Agnes, Pierre-Christian Violet, Maryam Safari, Carole Sourbier, W. Marston Linehan, Robert W. Robey, Mark Levine, Dan L. Sackett, and Susan E. Bates. 2022. "A Histone Deacetylase Inhibitor Induces Acetyl-CoA Depletion Leading to Lethal Metabolic Stress in RAS-Pathway Activated Cells" Cancers 14, no. 11: 2643. https://doi.org/10.3390/cancers14112643
APA StyleBasseville, A., Violet, P.-C., Safari, M., Sourbier, C., Linehan, W. M., Robey, R. W., Levine, M., Sackett, D. L., & Bates, S. E. (2022). A Histone Deacetylase Inhibitor Induces Acetyl-CoA Depletion Leading to Lethal Metabolic Stress in RAS-Pathway Activated Cells. Cancers, 14(11), 2643. https://doi.org/10.3390/cancers14112643