
Inhibition of HDAC and Signal Transduction Pathways Induces Tight Junctions and Promotes Differentiation in p63-Positive Salivary Duct Adenocarcinoma

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cultures of Cell Lines and Treatments
2.3. Isolation and Culture of Human Salivary Gland Duct Epithelial Cells
2.4. GeneChip Analysis
2.5. RNA Interference and Transfection
2.6. Immunohistochemical Analysis
2.7. Transmission Electron Microscopy Analysis
2.8. Western Blot Analysis
2.9. Immunoprecipitation and Western Blot Analysis
2.10. Immunocytochemistry
2.11. Cell Cycle Assay
2.12. Migration Assay
2.13. Measurement of Transepithelial Electrical Resistance (TEER)
2.14. XF96 Extracellular Flux Measurements
2.15. Data Analysis
3. Results
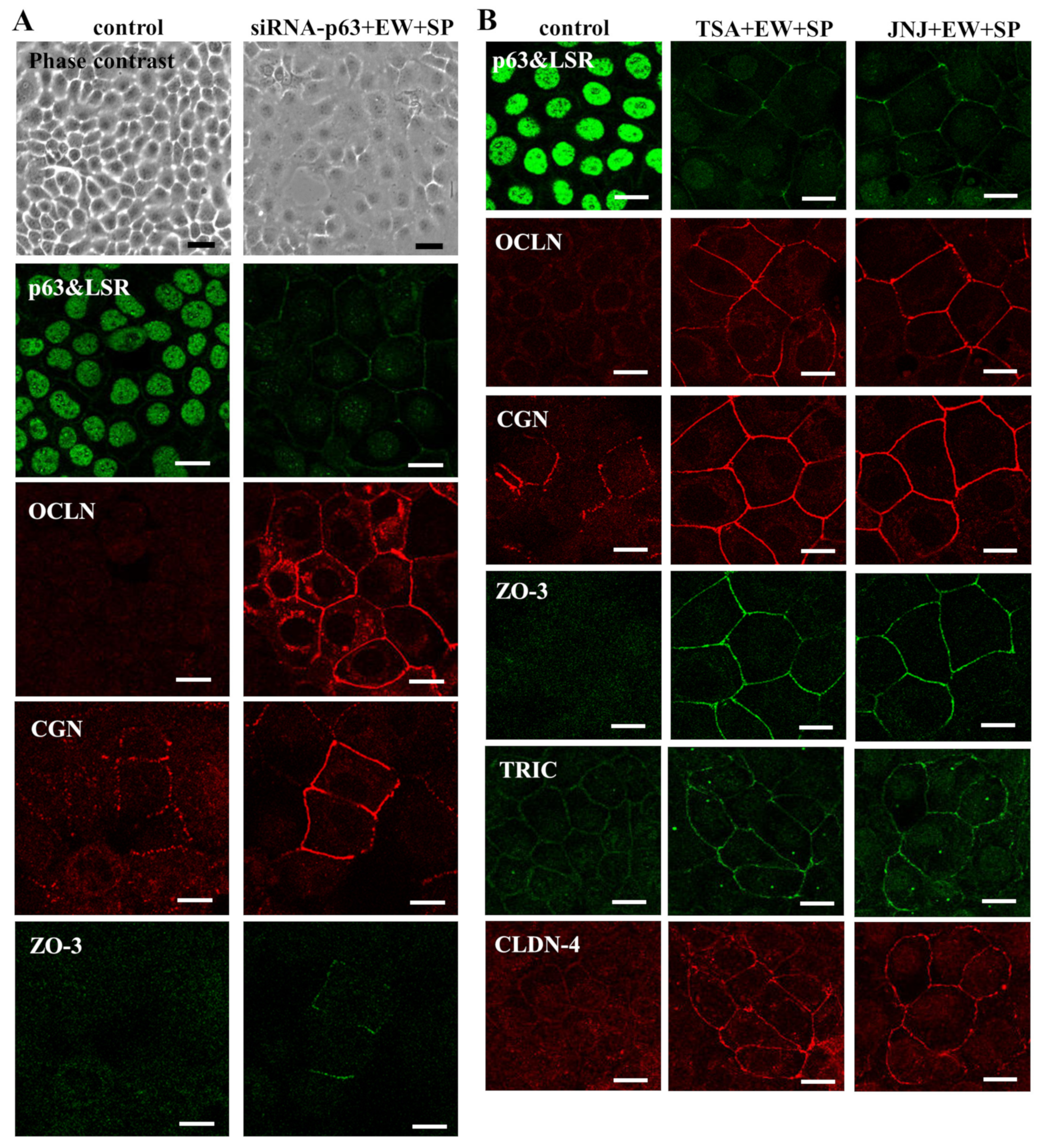
3.1. Knockdown of p63 Induced Tight Junction (TJ) Proteins CGN and ZO-3 and Differentiation Markers in Human Salivary Gland Duct Adenocarcinoma Cell Line A253
3.2. Knockdown of p63 Induced Tight Junction (TJ) Proteins at the Membranes in A253 Cells
3.3. HDAC Inhibitors Downregulate Expression of p63 and Upregulate Expression of TJ Proteins in A253 Cells
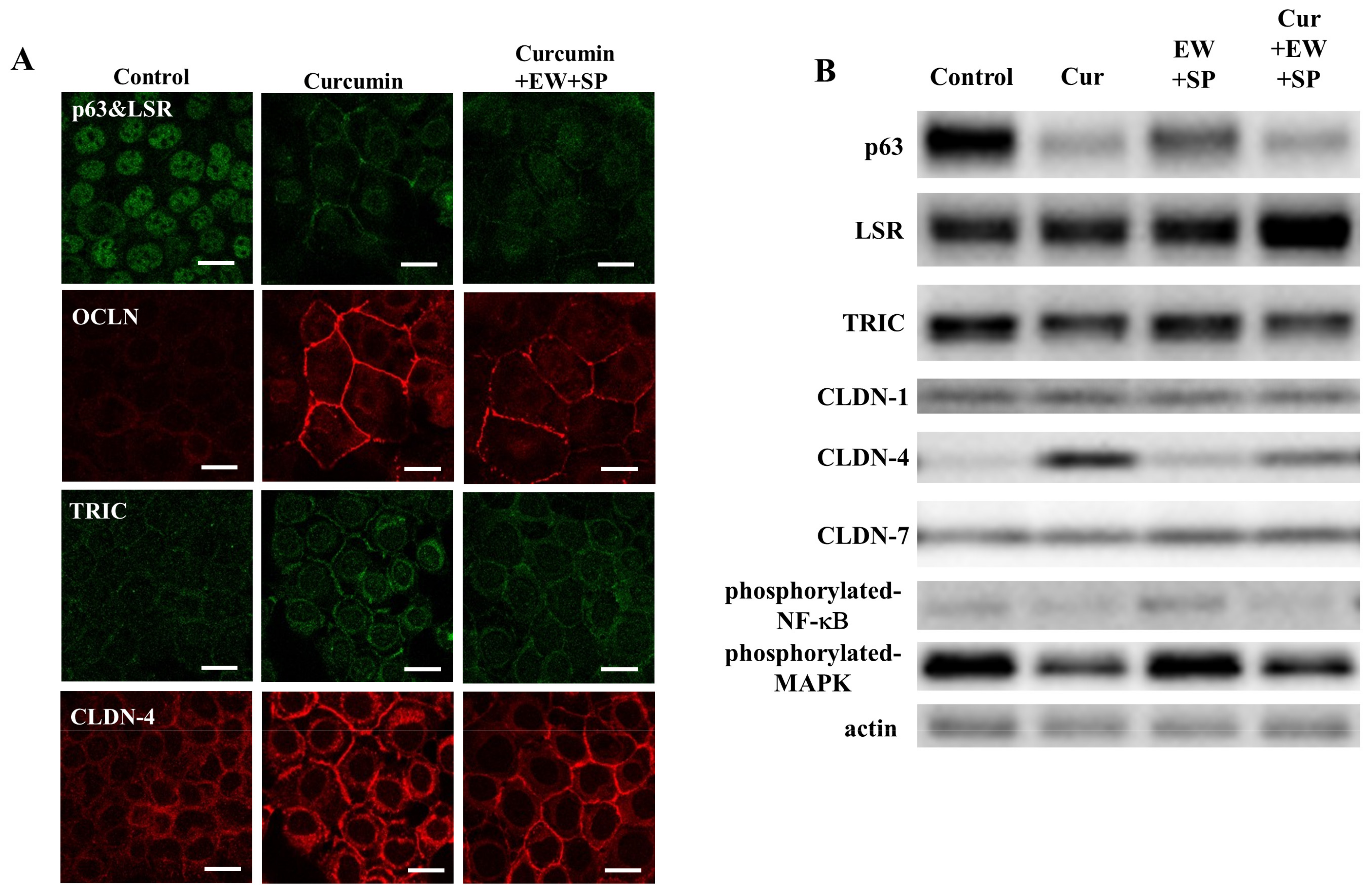
3.4. NF-κB Inhibitor Curcumin Downregulate Expression of p63 and Upregulate Expression of TJ Proteins in A253 Cells
3.5. CGN Induced by HDAC Inhibitors Binds Microtubules Formed Planar Apical Network in A253 Cells
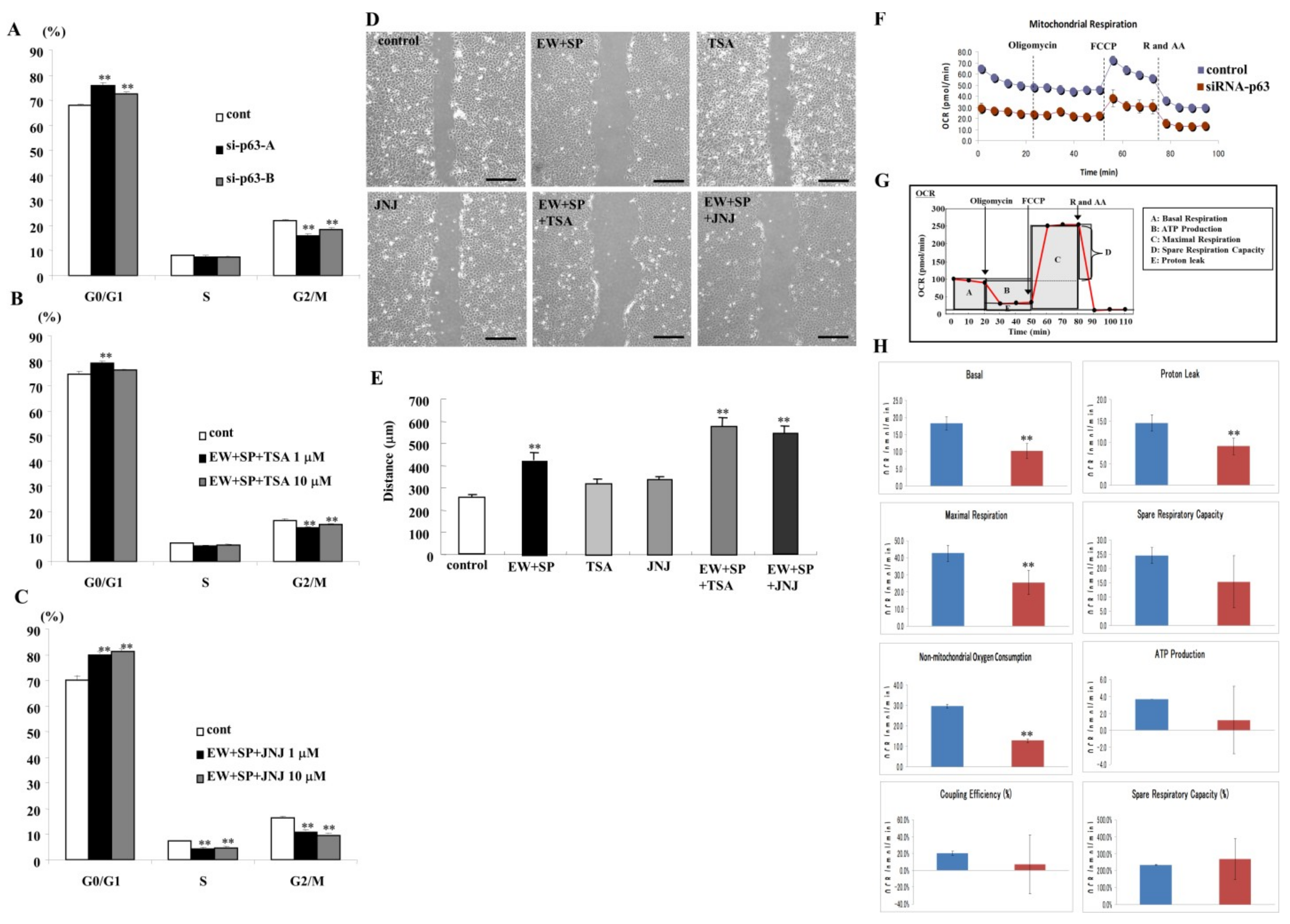
3.6. Knockdown of p63 and HDAC Inhibitors Prevent Cell Proliferation in A253 Cells
3.7. HDAC Inhibitors Prevent Cell Migration in A253 Cells
3.8. Knockdown of p63 Induced Aberrant Cell Metabolism in A253 Cells
3.9. Roles of p63, CGN and Angulin-1/LSR in Cultured Normal Human Salivary Gland Duct Epithelial (HSDE) Cells
3.10. Expression and Localization of p63, CGN, ZO-3 and OLFML3 in Human Salivary Duct Adenocarcinoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’heygere, E.; Meulemans, J.; Vander Poorten, V. Salivary duct carcinoma. Curr. Opin. Otolaryngol. Head Neck Surg. 2018, 26, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Freitag, V.; Lettmaier, S.; Semrau, S.; Hecht, M.; Mantsopoulos, K.; Müller, S.K.; Traxdorf, M.; Iro, H.; Agaimy, A.; Fietkau, R.; et al. High-grade salivary gland cancer: Is surgery followed by radiotherapy an adequate treatment to reach tumor control? Results from a tertiary referral centre focussing on incidence and management of distant metastases. Eur. Arch. Otorhinolaryngol. 2022, 279, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.J.; Roh, J.L.; Cho, K.J.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Risk Factors and Survival Associated with Distant Metastasis in Patients with Carcinoma of the Salivary Gland. Ann. Surg. Oncol. 2016, 23, 4376–4383. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.D.; Perrone, F.; Losa, M.; Mela, M.; Casieri, P.; Orsenigo, M.; Cortelazzi, B.; Negri, T.; Tamborini, E.; Quattrone, P.; et al. Treatment relevant target immunophenotyp- ing of 139 salivary gland carcinomas (SGCs). Oral Oncol. 2009, 45, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.I.; Kim, S.; Mills, A.A.; DeMayo, F.J.; Roop, D.R. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004, 18, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCoste, R.C.; Carter, M.D.; Pasternak, S.; Fleming, K.E.; Gaston, D.; Legge, A.; Ly, T.Y.; Walsh, N.M. Relationship between p63 and p53 expression in Merkel cell carcinoma and corresponding abnormalities in TP63 and TP53: A study and a proposal. Hum. Pathol. 2021, 117, 31–41. [Google Scholar] [CrossRef]
- Steurer, S.; Riemann, C.; Büscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; Weidemann, S.; Fraune, C.; et al. p63 expression in human tumors and normal tissues: A tissue microarray study on 10,200 tumors. Biomark. Res. 2021, 9, 7. [Google Scholar] [CrossRef]
- Rocco, J.W.; Leong, C.-O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Woodstock, D.L.; Sammons, M.A.; Fischer, M. p63 and p53: Collaborative Partners or Dueling Rivals? Front. Cell Dev. Biol. 2021, 9, 701986. [Google Scholar] [CrossRef]
- Niu, M.; He, Y.; Xu, J.; Ding, L.; He, T.; Yi, Y.; Fu, M.; Guo, R.; Li, F.; Chen, H.; et al. Noncanonical TGF-β signaling leads to FBXO3-mediated degradation of ΔNp63α promoting breast cancer metastasis and poor clinical prognosis. PLoS Biol. 2021, 19, e3001113. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorná, Z.; Vysloužil, J.; Hrabal, V.; Vojtěšek, B.; Coates, P.J. The foggy world(s) of p63 isoform regulation in normal cells and cancer. J. Pathol. 2021, 254, 454–473. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, N.; Narwal, A.; Pandiar, D.; Devi, A.; Anand, R.; Bansal, D.; Kamboj, M. Diagnostic utility of p63/p40 in the histo- logic differentiation of salivary gland tumors: A systematic review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 133, 189–198. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Dong, L.; Wang, Y.X.; Li, S.L.; Yu, G.Y.; Gan, Y.H.; Li, D.; Wang, C.Y. TGF-beta1 promotes migration and invasion of salivary adenoid cystic carcinoma. J. Dent. Res. 2011, 90, 804–809. [Google Scholar] [CrossRef]
- Liang, Y.; Jiao, J.; Liang, L.; Zhang, J.; Lu, Y.; Xie, H.; Liang, Q.; Wan, D.; Duan, L.; Wu, Y.; et al. Tumor necrosis factor receptor-associated factor 6 mediated the promotion of salivary adenoid cystic carcinoma progression through Smad-p38- JNK signaling pathway induced by TGF-beta. J. Oral Pathol. Med. 2018, 47, 583–589. [Google Scholar] [CrossRef]
- Min, S.; Oyelakin, A.; Gluck, C.; Bard, J.E.; Song, E.-A.C.; Smalley, K.; Che, M.; Flores, E.; Sinha, S.; Romano, R.-A. p63 and Its Target Follistatin Maintain Salivary Gland Stem/Progenitor Cell Function through TGF-β/Activin Signaling. iScience 2020, 23, 101524. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Lee, H.J.; Park, S.J.; Park, H.J.; Lee, K.; Sheen, Y.Y.; et al. Discovery of N-((4-([1,2,4] triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2 -yl)methyl)-2-fluoroaniline (EW-7197): A highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immuno- therapeutic/antifibrotic agent. J. Med. Chem. 2014, 57, 4213–4238. [Google Scholar]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisagno, V.; Cadet, J.L. Histone Deacetylases and Immediate Early Genes: Key Players in Psychostimulant-Induced Neuronal Plasticity. Neurotox. Res. 2021, 39, 2134–2140. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Chen, K.; Cao, J.; Yu, H.; Tian, L.; Liu, M. A correlation analysis between HDAC1 over-expression and clinical features of laryngeal squamous cell carcinoma. Acta Oto-Laryngol. 2015, 136, 172–176. [Google Scholar] [CrossRef]
- Luo, S.; Ma, K.; Zhu, H.; Wang, S.; Liu, M.; Zhang, W.; Liang, S.; Xu, N. Molecular, biological characterization and drug sensi- tivity of chidamide-resistant non-small cell lung cancer cells. Oncol. Lett. 2017, 14, 6869–6875. [Google Scholar]
- Pouloudi, D.; Manou, M.; Sarantis, P.; Tsoukalas, N.; Tsourouflis, G.; Dana, E.; Karamouzis, M.; Klijanienko, J.; Theocharis, S. Clinical Significance of Histone Deacetylase (HDAC)-1, -2, -4 and -6 Expression in Salivary Gland Tumors. Diagnostics 2021, 11, 517. [Google Scholar] [CrossRef]
- Sborov, D.W.; Canella, A.; Hade, E.M.; Mo, X.; Khountham, S.; Wang, J.; Ni, W.; Poi, M.; Coss, C.; Liu, Z.; et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 2017, 58, 2310–2318. [Google Scholar] [CrossRef]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- Liva, S.; Chen, M.; Mortazavi, A.; Walker, A.; Wang, J.; Dittmar, K.; Hofmeister, C.; Coss, C.C.; Phelps, M.A. Population Pharmacokinetic Analysis from First-in-Human Data for HDAC Inhibitor, REC-2282 (AR-42), in Patients with Solid Tumors and Hematologic Malignancies: A Case Study for Evaluating Flat vs. Body Size Normalized Dosing. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 807–816. [Google Scholar] [CrossRef]
- Parag-Sharma, K.; Tasoulas, J.; Musicant, A.M.; do Nascimento-Filho, C.H.V.; Zhu, Z.; Twomey, C.; Liu, P.; Castilho, R.M.; Amelio, A.L. Synergistic efficacy of combined EGFR and HDAC inhibitors overcomes tolerance to EGFR monotherapy in sali vary mucoepidermoid carcinoma. Oral Oncol. 2021, 115, 105166. [Google Scholar] [CrossRef]
- Chikamatsu, K.; Ishii, H.; Murata, T.; Sakakura, K.; Shino, M.; Toyoda, M.; Takahashi, K.; Masuyama, K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013, 104, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-C.; Liu, H.-T.; Wang, X.-Y.; Wu, S.-C.; Chen, L.-C.; Liu, Y.-W. Trichostatin A induces bladder cancer cell death via intrinsic apoptosis at the early phase and Sp1-survivin downregulation at the late phase of treatment. Oncol. Rep. 2017, 38, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wu, J.Q.; Yu, X.F.; Yang, X.S.; Yang, Y. Trichostatin A inhibits proliferation of triple negative breast cancer cells by inducing cell cycle arrest and apoptosis. Neoplasma 2018, 65, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xu, J.; Wang, H.; Wu, L.; Yuan, W.; Du, J.; Cai, S. Trichostatin A, a histone deacetylase inhibitor, reverses epithelial- mesenchymal transition in colorectal cancer SW480 and prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 2015, 456, 320–326. [Google Scholar] [CrossRef]
- Arts, J.; King, P.; Mariën, A.; Floren, W.; Beliën, A.; Janssen, L.; Pilatte, I.; Roux, B.; Decrane, L.; Gilissen, R.; et al. JNJ- 26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin. Cancer Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Diao, H.; Dong, N.; Su, X.; Wang, B.; Mo, Q.; Yu, H.; Wang, X.; Chen, C. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol. Toxicol. 2016, 32, 469–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Dai, L.; Zhang, X.; Chen, D.; Wu, J.; Feng, X.; Zhang, Y.; Xie, H.; Zhou, L.; Wu, J.; et al. The HDAC Inhibitor Quisino- stat (JNJ-26481585) Supresses Hepatocellular Carcinoma alone and Synergistically in Combination with Sorafenib by G0/G1 phase arrest and Apoptosis induction. Int. J. Biol. Sci. 2018, 14, 1845–1858. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Varadarajan, S.; Stephenson, R.E.; Miller, A.L. Multiscale dynamics of tight junction remodeling. J. Cell Sci. 2019, 132, jcs229286. [Google Scholar] [CrossRef]
- Al-Awqati, Q.; Vijayakumar, S.; Takito, J. Terminal Differentiation of Epithelia. Biol. Chem. 2003, 384, 1255–1258. [Google Scholar] [CrossRef]
- Furuse, M. Molecular Basis of the Core Structure of Tight Junctions. Cold Spring Harb. Perspect. Biol. 2009, 2, a002907. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porat-Shliom, N.; Tietgens, A.J.; Van Itallie, C.M.; Vitale-Cross, L.; Jarnik, M.; Harding, O.J.; Anderson, J.M.; Gutkind, J.S.; Wei- gert, R.; Arias, I.M. Liver kinase B1 regulates hepatocellular tight junction distribution and function in vivo. Hepatology 2016, 64, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Kojima, T.; Yamaguchi, H.; Ito, T.; Kyuno, D.; Kono, T.; Konno, T.; Sawada, N. Tight junctions in human pancreatic duct epithelial cells. Tissue Barriers 2013, 1, e24894. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Ninomiya, T.; Kohno, T.; Kikuchi, S.; Sawada, N.; Kojima, T. c-Jun N-terminal kinase inhibitor SP600125 enhances barrier function and elongation of human pancreatic cancer cell line HPAC in a Ca-switch model. Histochem. Cell Biol. 2014, 143, 471–479. [Google Scholar] [CrossRef]
- Konno, T.; Kohno, T.; Kikuchi, S.; Shimada, H.; Satohisa, S.; Saito, T.; Kondoh, M.; Kojima, T. Epithelial barrier dysfunction and cell migration induction via JNK/cofilin/actin by angubindin-1. Tissue Barriers 2019, 8, 1695475. [Google Scholar] [CrossRef]
- Cordenonsi, M.; D’Atri, F.; Hammar, E.; Parry, D.A.; Kendrick-Jones, J.; Shore, D.; Citi, S. Cingulin contains globular and coiled- coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J. Cell Biol. 1999, 147, 1569–1582. [Google Scholar] [CrossRef]
- Yano, T.; Matsui, T.; Tamura, A.; Uji, M.; Tsukita, S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 2013, 203, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, T.; Kohno, T.; Kikuchi, S.; Shimada, H.; Satohisa, S.; Takano, K.; Saito, T.; Kojima, T. Localization of Tricellular Tight Junction Molecule LSR at Midbody and Centrosome During Cytokinesis in Human Epithelial Cells. J. Histochem. Cytochem. 2020, 68, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Bordin, M.; D’Atri, F.; Guillemot, L.; Citi, S. Histone deacetylase inhibitors up-regulate the expression of tight junction proteins. Mol. Cancer Res. 2004, 2, 692–701. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, T.; Takano, K.; Kaneko, Y.; Kakuki, T.; Nomura, K.; Yajima, R.; Kakiuchi, A.; Kohno, T.; Himi, T.; Kojima, T. Guanylate binding protein-1-mediated epithelial barrier in human salivary gland duct epithelium. Exp. Cell Res. 2018, 371, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lopardo, T.; Iacono, N.L.; Marinari, B.; Giustizieri, M.L.; Cyr, D.G.; Merlo, G.; Crosti, F.; Costanzo, A.; Guerrini, L. Claudin-1 Is a p63 Target Gene with a Crucial Role in Epithelial Development. PLoS ONE 2008, 3, e2715. [Google Scholar] [CrossRef] [PubMed]
- Kakuki, T.; Kurose, M.; Takano, K.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; Takahashi, S.; et al. Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 33887–33900. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Bellmann, U.; Bootz, F.; Wittekind, C.; Tannapfel, A. Expression of p53 and its homologues in primary and recurrent squamous cell carcinomas of the head and neck. Int. J. Cancer 2002, 99, 22–28. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, W.; Huang, B.; Liu, Z.; Sun, L.; Zhang, Q.; Qiu, X.; Xu, K.; Wang, E. Overexpression of JAM-A in Non-Small Cell Lung Cancer Correlates with Tumor Progression. PLoS ONE 2013, 8, e79173. [Google Scholar] [CrossRef] [Green Version]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef]
- Thépot, A.; Hautefeuille, A.; Cros, M.-P.; Abedi-Ardekani, B.; Pétré, A.; Damour, O.; Krutovskikh, V.; Hainaut, P. Intraepithelial p63-dependent expression of distinct components of cell adhesion complexes in normal esophageal mucosa and squamous cell carcinoma. Int. J. Cancer 2010, 127, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, A.; Kakuki, T.; Ohwada, K.; Kurose, M.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; et al. HDAC inhibitors suppress the proliferation, migration and invasiveness of human head and neck squamous cell carcinoma cells via p63-mediated tight junction molecules and p21-mediated growth arrest. Oncol. Rep. 2021, 45, 46. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; Kojima, T. Effects of histone deacetylase inhibitors Tricostatin A and Quisinostat on tight junction proteins of human lung adenocarcinoma A549 cells and normal lung epithelial cells. Histochem. Cell Biol. 2021, 155, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T. From Warburg effect to Reverse Warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef] [PubMed]
- Nantasupha, C.; Thonusin, C.; Charoenkwan, K.; Chattipakorn, S.; Chattipakorn, N. Metabolic reprogramming in epithelial ovarian cancer. Am. J. Transl. Res. 2021, 13, 9950–9973. [Google Scholar]
- Ciuffoli, V.; Lena, A.M.; Gambacurta, A.; Melino, G.; Candi, E. Myoblasts rely on TAp63 to control basal mitochondria respiration. Aging 2018, 10, 3558–3573. [Google Scholar] [CrossRef]
- Ogawa, E.; Okuyama, R.; Egawa, T.; Nagoshi, H.; Obinata, M.; Tagami, H.; Ikawa, S.; Aiba, S. p63/p51-induced onset of keratinocyte differentiation via the c-Jun N-terminal kinase pathway is counteracted by keratinocyte growth factor. J. Biol. Chem. 2008, 283, 34241–34249. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ID | Gene Bank ID | Fold-Change Control vs. Treatment |
|---|---|---|---|
| TP63 | H300019931 | NM_001114982.1 | 0.34 |
| CGN | H30009163 | NM_020770.2 | 2.77 |
| TJP3 (ZO-3) | H200003536 | NM_001267561.1 | 12.47 |
| OLFL3 | AHsV10002740 | NM_020190.3 | 14.56 |
| CY1A1 | AHsV10002241 | NM_001319217.1 | 66.44 |
| AQP7 | H300004487 | XM_011517868.1 | 2.26 |
| TLR1 | CHsGV10003913 | NM_003263.3 | 2.38 |
| TLR5 | H200011920 | NM_003268.5 | 2.28 |
| ELF3 | H200007656 | NM_001114309.1 | 2.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, M.; Ohwada, K.; Shindo, Y.; Konno, T.; Kohno, T.; Kikuchi, S.; Tsujiwaki, M.; Ishii, D.; Nishida, S.; Kakuki, T.; et al. Inhibition of HDAC and Signal Transduction Pathways Induces Tight Junctions and Promotes Differentiation in p63-Positive Salivary Duct Adenocarcinoma. Cancers 2022, 14, 2584. https://doi.org/10.3390/cancers14112584
Nakano M, Ohwada K, Shindo Y, Konno T, Kohno T, Kikuchi S, Tsujiwaki M, Ishii D, Nishida S, Kakuki T, et al. Inhibition of HDAC and Signal Transduction Pathways Induces Tight Junctions and Promotes Differentiation in p63-Positive Salivary Duct Adenocarcinoma. Cancers. 2022; 14(11):2584. https://doi.org/10.3390/cancers14112584
Chicago/Turabian StyleNakano, Masaya, Kizuku Ohwada, Yuma Shindo, Takumi Konno, Takayuki Kohno, Shin Kikuchi, Mitsuhiro Tsujiwaki, Daichi Ishii, Soshi Nishida, Takuya Kakuki, and et al. 2022. "Inhibition of HDAC and Signal Transduction Pathways Induces Tight Junctions and Promotes Differentiation in p63-Positive Salivary Duct Adenocarcinoma" Cancers 14, no. 11: 2584. https://doi.org/10.3390/cancers14112584
APA StyleNakano, M., Ohwada, K., Shindo, Y., Konno, T., Kohno, T., Kikuchi, S., Tsujiwaki, M., Ishii, D., Nishida, S., Kakuki, T., Obata, K., Miyata, R., Kurose, M., Kondoh, A., Takano, K., & Kojima, T. (2022). Inhibition of HDAC and Signal Transduction Pathways Induces Tight Junctions and Promotes Differentiation in p63-Positive Salivary Duct Adenocarcinoma. Cancers, 14(11), 2584. https://doi.org/10.3390/cancers14112584