
FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review


Abstract
Simple Summary
Abstract
1. Introduction
2. Small Molecule Anticancer Drugs
2.1. Various Protein Kinase Inhibitors as Anticancer Agents
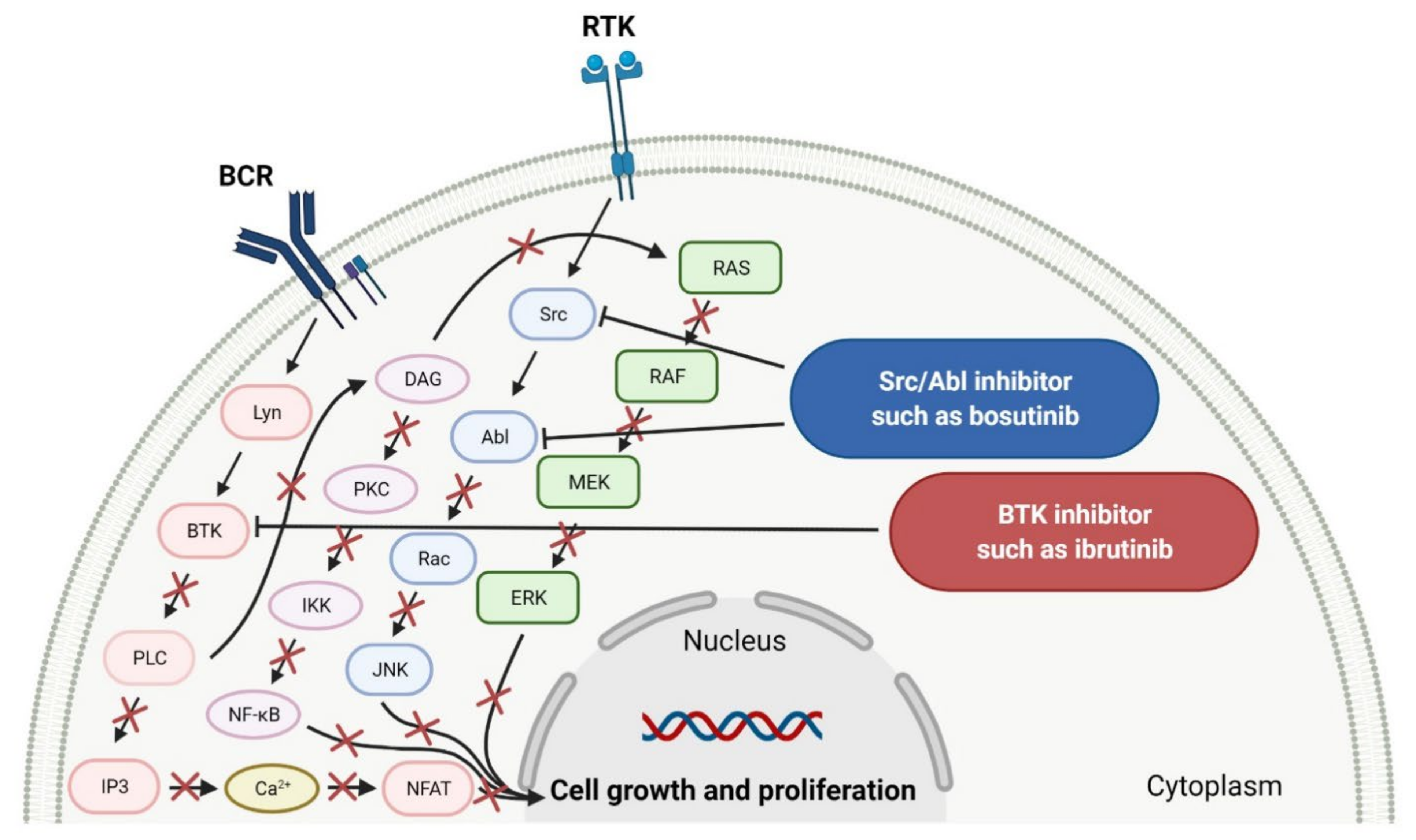
2.1.1. Tyrosine Kinase (TK) Inhibitors
2.1.2. Multi Kinase Inhibitors
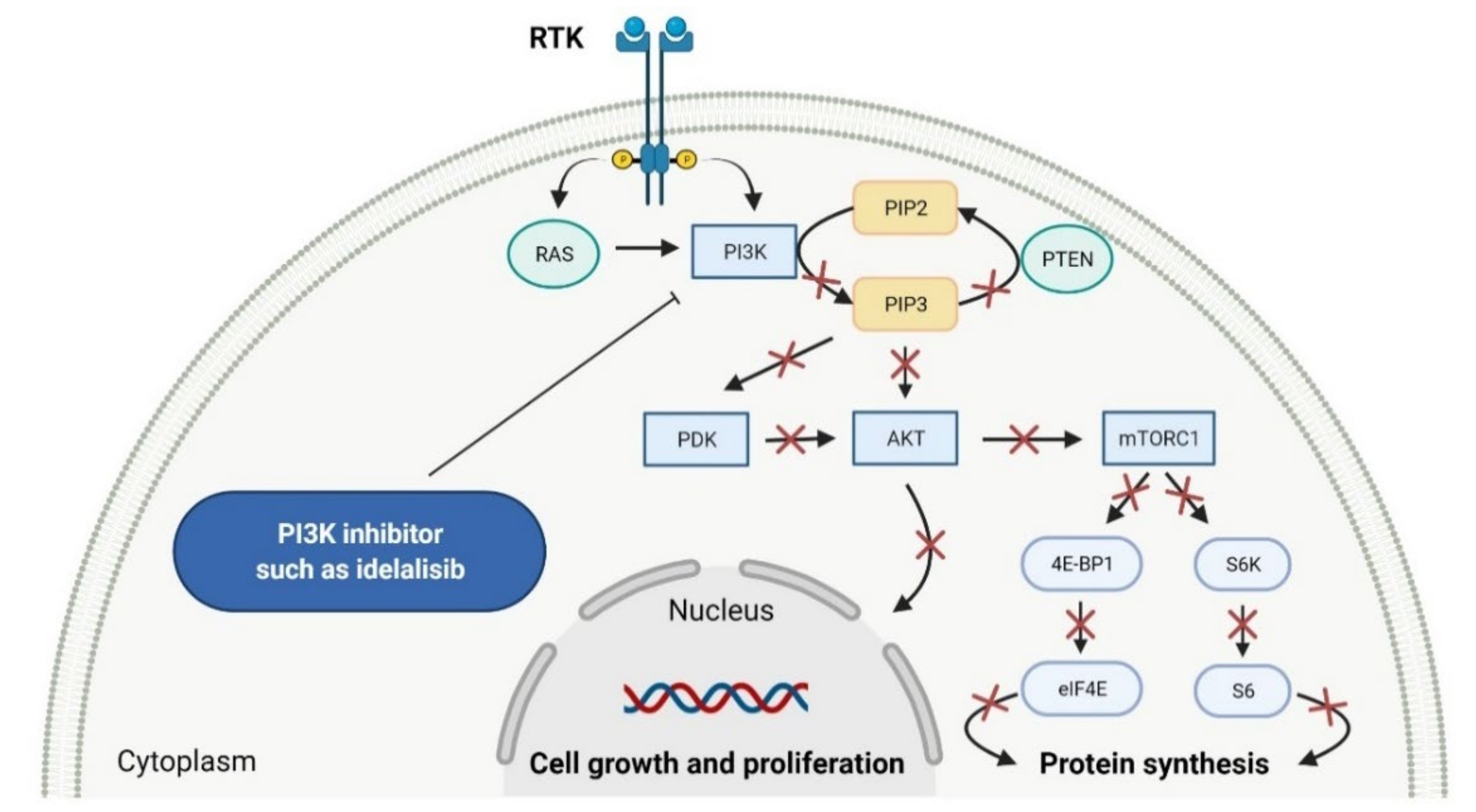
2.2. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors as Anticancer Agents
2.3. Various Enzymes Inhibitors as Anticancer Agents
2.3.1. Histone Deacetylase Inhibitors
2.3.2. Isocitrate Dehydrogenase Inhibitors
2.3.3. Enhancer of Zeste Homolog 2 Inhibitor
2.3.4. DNA and RNA Methyltransferases Inhibitor
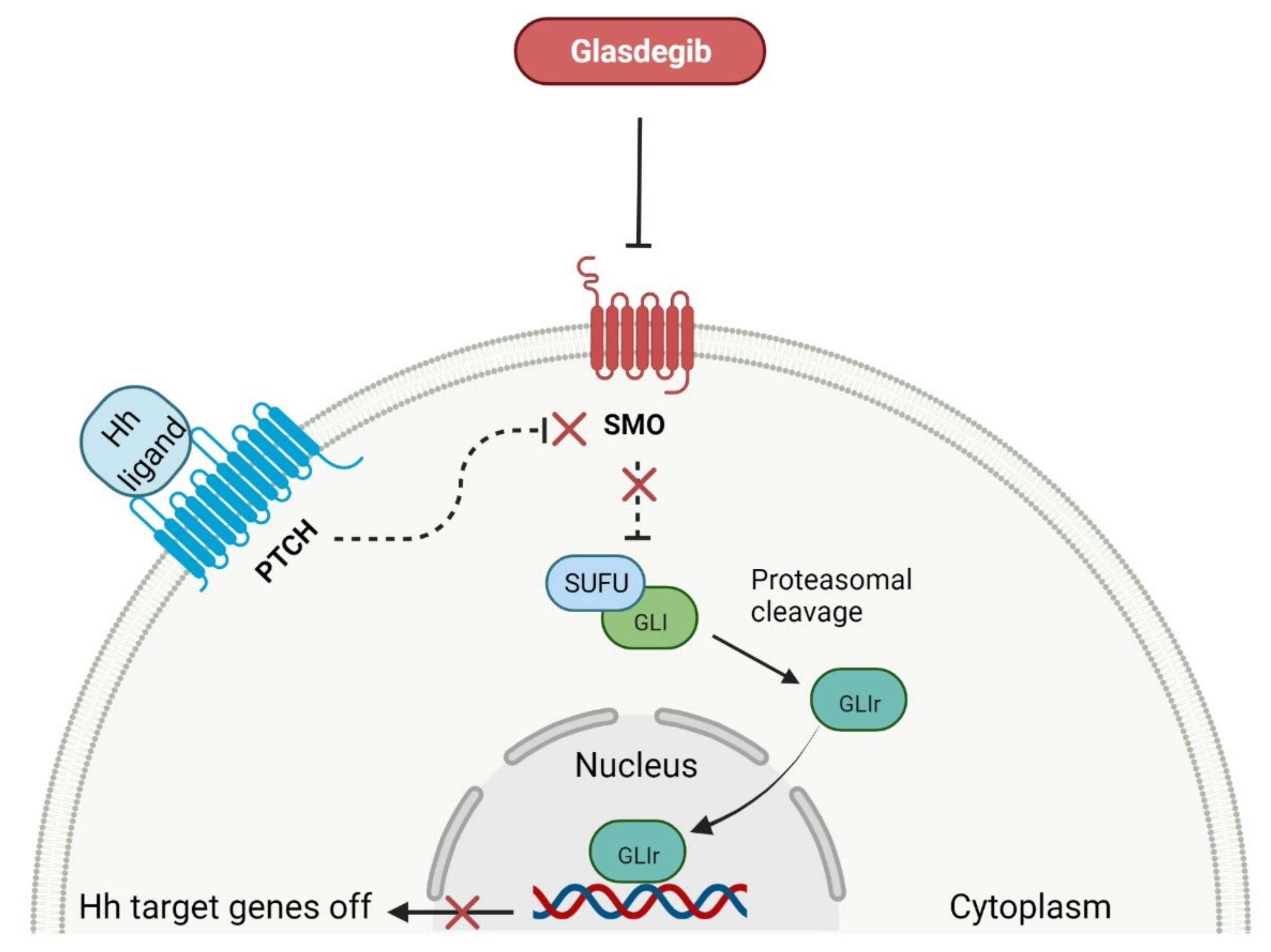
2.4. Smoothed Receptor Antagonists as Anticancer Agents
2.5. Various Proteins Inhibitors as Anticancer Agents
2.5.1. B-Cell Leukemia/Lymphoma-2 Proteins Inhibitor
2.5.2. Exportin-1 Inhibitors
2.5.3. Proteasome Inhibitors
2.5.4. Tubulin Inhibitor
2.6. Protein Translation Inhibitors as Anticancer Agents
2.7. Purine Antagonist as Anticancer Agent
2.8. Immunomodulators as Anticancer Agents
2.9. Fixed-Dose Combination Drugs as Anticancer Agents
3. Macromolecule Anticancer Drugs
3.1. Monoclonal Antibodies as Anticancer Agents
3.1.1. Anti-CD19 Monoclonal Antibody
3.1.2. Anti-CD20 Monoclonal Antibodies
3.1.3. Anti-CD38 Monoclonal Antibodies
3.1.4. Anti-SLAMF7 Monoclonal Antibody
3.1.5. Anti-PD-1 Monoclonal Antibodies
3.1.6. Anti-CCR4 Monoclonal Antibody
3.2. Bispecific Antibody as Anticancer Agent
3.3. Antibody-Drug Conjugates as Anticancer Agents
3.3.1. Anti-CD22 Antibody-Drug Conjugate
3.3.2. Anti-CD30 and Anti-CD79b Antibody-Drug Conjugates
3.3.3. Anti-BCMA Antibody-Drug Conjugate
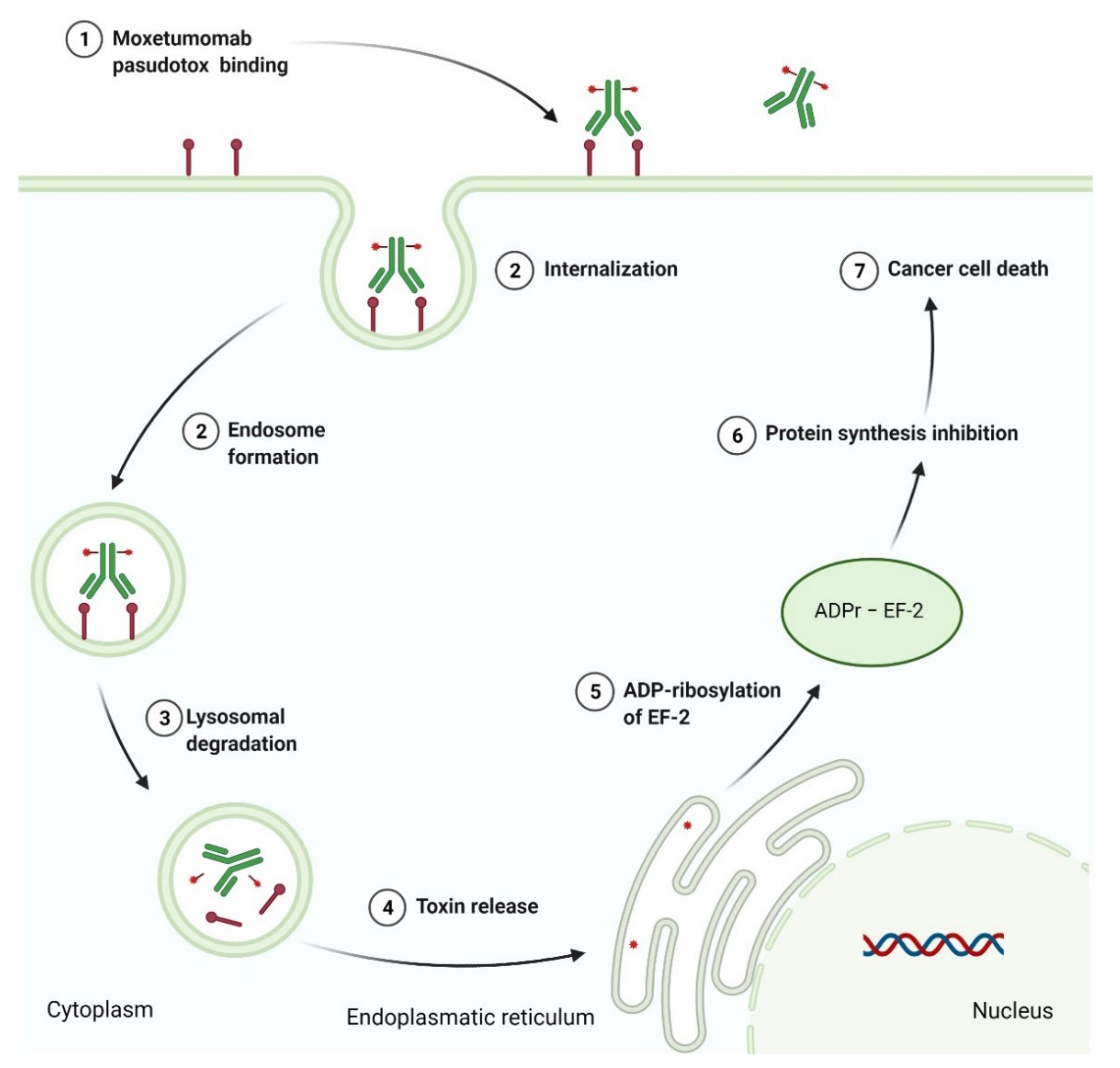
3.4. Recombinant Immunotoxin as Anticancer Agent
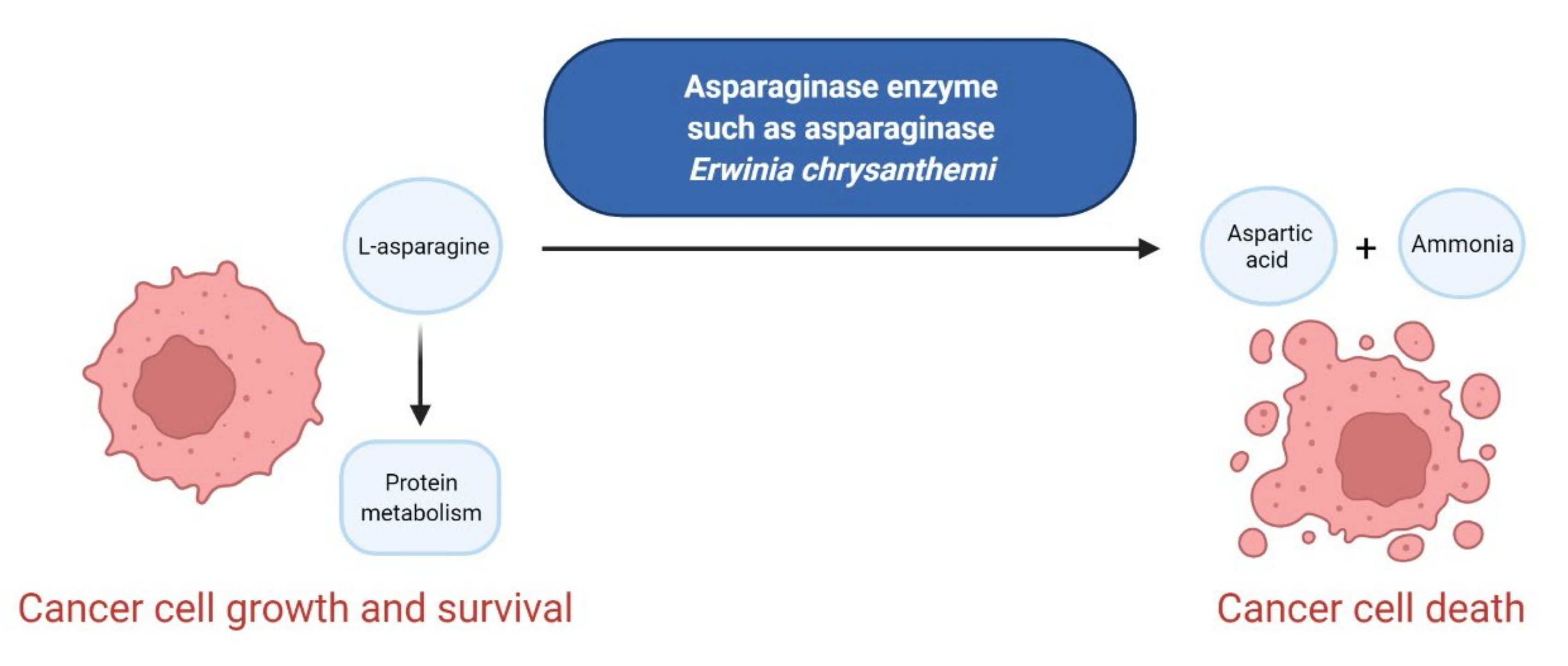
3.5. Enzymes as Anticancer Agents
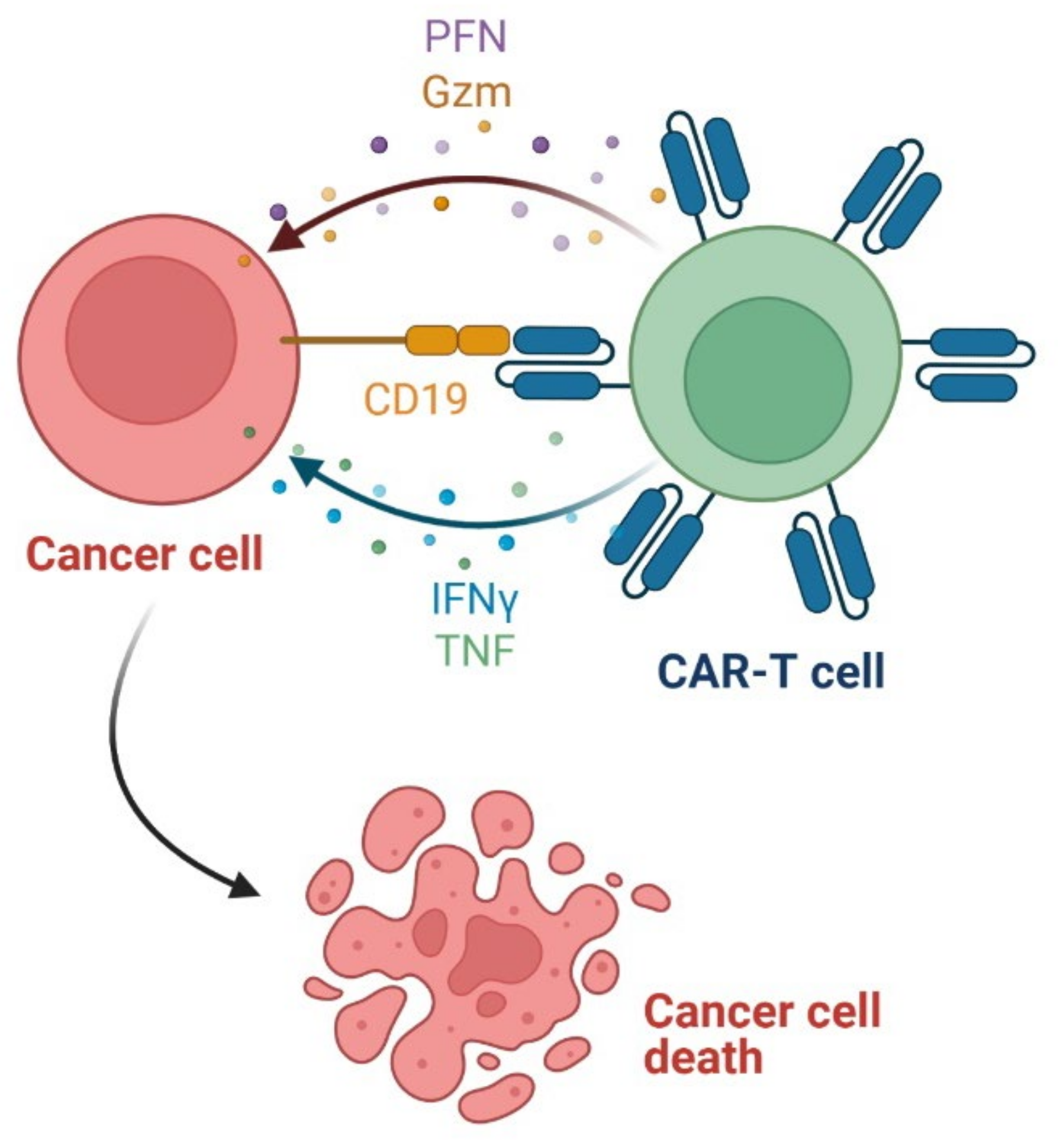
3.6. Chimeric Antigen Receptor T (CAR-T) Cells as Anticancer Agents
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Huang, L.; Huang, J.; Huang, J.; Xue, H.; Liang, Z.; Wu, J.; Chen, C. Nanomedicine—A promising therapy for hematological malignancies. Biomater Sci. 2020, 8, 2376–2393. [Google Scholar] [CrossRef]
- Nussbaumer, S.; Bonnabry, P.; Veuthey, J.-L.; Fleury-Souverain, S. Analysis of anticancer drugs: A review. Talanta 2011, 85, 2265–2289. [Google Scholar] [CrossRef]
- Goodman, L.S.; Wintrobe, M.M.; Huang, L.; Huang, J.; Huang, J.; Xue, H.; Liang, Z.; Wu, J.; Chen, C. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Mustargen NDA #006695—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=006695 (accessed on 23 August 2021).
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-Aminopteroyl-glutamic acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Methotrexate Sodium NDA #008085—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=008085 (accessed on 23 August 2021).
- Wright, J.C.; Prigot, A.; Wright, B.; Weintraub, S.; Wright, L.T. An evaluation of folic acid antagonists in adults with neoplastic diseases: A study of 93 patients with incurable neoplasms. J. Natl. Med. Assoc. 1951, 43, 211–240. [Google Scholar]
- Elion, G.B.; Burgi, E.; Hitchings, G.H. Studies on condensed pyrimidine systems. IX. The synthesis of some 6-substituted purines. J. Am. Chem. Soc. 1952, 74, 411–414. [Google Scholar] [CrossRef]
- Purinethol NDA #009053—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=009053 (accessed on 24 August 2021).
- Dushinsky, R.; Pleven, E.; Heidelberger, C. The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560. [Google Scholar] [CrossRef]
- Fluorouracil NDA 012209—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=012209 (accessed on 24 August 2021).
- Hurley, J.D. Chemotherapy of solid carcinoma. JAMA 1960, 174, 1696–1701. [Google Scholar] [CrossRef]
- Noble, R.L. The discovery of the vinca alkaloids—Chemotherapeutic agents against cancer. Biochem. Cell Biol. 1990, 68, 1344–1351. [Google Scholar] [CrossRef]
- Oncovin NDA #014103—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=014103 (accessed on 25 August 2021).
- Watson, R.A.; De La Peña, H.; Tsakok, M.T.; Joseph, J.; Stoneham, S.; Shamash, J.; Joffe, J.; Mazhar, D.; Traill, Z.; Ho, L.P.; et al. Development of a best-practice clinical guideline for the use of bleomycin in the treatment of germ cell tumours in the UK. Br. J. Cancer 2018, 119, 1044–1051. [Google Scholar] [CrossRef]
- Takeuchi, T. Antitumor antibiotics discovered and studied at the Institute of Microbial Chemistry. J. Cancer Res. Clin. Oncol. 1995, 121, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, G.; Monfardini, S.; De Lena, M.; Fossati-Bellani, F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br. Med. J. 1969, 3, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Doxorubicin Hydrochloride NDA #050467—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050467 (accessed on 25 August 2021).
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Peyrone, M. Ueber die einwirkung von ammoniak auf platinchlorü: Zweite abhandlung. Ann. Chem. Pharm. 1845, 55, 205–213. [Google Scholar] [CrossRef]
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Grillo-López, A.J.; White, C.A.; Dallaire, B.K.; Varns, C.L.; Shen, C.D.; Wei, A.; Leonard, J.E.; McClure, A.; Weaver, R.; Cairelli, S.; et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000, 1, 1–9. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, J.; Kim, S.K.; Wood, R.; et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2002, 8, 935–942. [Google Scholar]
- Nava-Parada, P.; Shelbaya, A.; Nabhan, C. Rituximab biosimilars in hematologic malignancies: The need for a real-world approach. Future Oncol. 2020, 16, 2017–2027. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [PubMed]
- IMBRUVICA (Ibrutinib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/205552s030,210563s006lblPI.pdf (accessed on 19 October 2021).
- CALQUENCE (Acalabrutinib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210259s006s007lbl.pdf (accessed on 19 October 2021).
- Wiestner, A. Targeting B-Cell receptor signaling for anticancer therapy: The Bruton’s tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J. Clin. Oncol. 2013, 31, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Bosutinib for Treatment of Newly-Diagnosed PH+ CML. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-bosutinib-treatment-newly-diagnosed-ph-cml (accessed on 22 December 2021).
- Cortes, J.E.; Kantarjian, H.M.; Brümmendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef]
- Xue, C.; Wang, X.; Zhang, L.; Qu, Q.; Zhang, Q.; Jiang, Y. Ibrutinib in B-cell lymphoma: Single fighter might be enough? Cancer Cell Int. 2020, 20, 467. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- European Medicines Agency. BRUKINSA (Zanubrutinib). Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/19/2167-public-summary-opinion-orphan-designation-zanubrutinib-treatment-lymphoplasmatic-lymphoma_en.pdf (accessed on 1 December 2021).
- Markham, A.; Dhillon, S. Acalabrutinib: First global approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. CALQUENCE (Acalabrutinib). Available online: https://www.ema.europa.eu/en/documents/overview/calquence-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Cameron, F.; Sanford, M. Ibrutinib: First global approval. Drugs 2014, 74, 263–271. [Google Scholar] [CrossRef]
- European Medicines Agency. IMBRUVICA (Ibrutinib). Available online: https://www.ema.europa.eu/en/documents/overview/imbruvica-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Bosulif NDA #203341—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=203341 (accessed on 25 October 2021).
- BOSULIF (Bosutinib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203341s009lbl.pdf (accessed on 7 September 2021).
- European Medicines Agency. BOSULIF (Bosutinib). Available online: https://www.ema.europa.eu/en/documents/overview/bosulif-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef]
- Krug, M.; Hilgeroth, A. Recent advances in the development of multi-kinase inhibitors. Mini Rev. Med. Chem. 2008, 8, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.; Freeman, C.; Giles, F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia 2012, 26, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Aydin, E.; Faehling, S.; Saleh, M.; Llaó Cid, L.; Seiffert, M.; Roessner, P.M. Phosphoinositide 3-kinase signaling in the tumor microenvironment: What do we need to consider when treating chronic lymphocytic leukemia with PI3K inhibitors? Front. Immunol. 2021, 11, 595818. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Shah, N.P.; Bixby, D.; Mauro, M.J.; Flinn, I.; O’Hare, T.; Hu, S.; Narasimhan, N.I.; Rivera, V.M.; et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2012, 367, 2075–2088. [Google Scholar] [CrossRef]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. EPIC investigators. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Gainor, J.F.; Chabner, B.A. Ponatinib: Accelerated disapproval. Oncologist 2015, 20, 847–848. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Jaszczur, S.M.; Afifi, S.; Zhao, J.C.; Zeidan, A.M. Beyond Ruxolitinib: Fedratinib and other emergent treatment options for myelofibrosis. Cancer Manag. Res. 2019, 11, 10777–10790. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Tefferi, A.; Jamieson, C.; Gabrail, N.Y.; Lebedinsky, C.; Gao, G.; Liu, F.; Xu, C.; Cao, H.; Talpaz, M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015, 5, e335. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of Fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Blair, H.A. Fedratinib: First approval. Drugs 2019, 79, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- INREBIC (Fedratinib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212327s000lbl.pdf (accessed on 5 December 2021).
- European Medicines Agency. INREBIC (Fedratinib). Available online: https://www.ema.europa.eu/en/documents/overview/inrebic-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Dhillon, S. Gilteritinib: First global approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. XOSPATA (Gilteritinib). Available online: https://www.ema.europa.eu/en/documents/overview/xospata-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Kim, E.S. Midostaurin: First global approval. Drugs 2017, 77, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. RYDAPT (Midostaurin). Available online: https://www.ema.europa.eu/en/documents/overview/rydapt-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Jain, H.; Thorat, J.; Sengar, M.; Dubey, A. Ponatinib: A drug review. Cancer Res. Stat. Treat. 2019, 2, 190–196. [Google Scholar] [CrossRef]
- European Medicines Agency. ICLUSIG (Ponatinib). Available online: https://www.ema.europa.eu/en/documents/overview/iclusig-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Deisseroth, A.; Kaminskas, E.; Grillo, J.; Chen, W.; Saber, H.; Lu, H.L.; Rothmann, M.D.; Brar, S.; Wang, J.; Garnett, C.; et al. Food and Drug Administration approval: Ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin. Cancer Res. 2012, 18, 3212–3217. [Google Scholar] [CrossRef]
- European Medicines Agency. JAKAVI (Ruxolitinib). Available online: https://www.ema.europa.eu/en/documents/overview/jakavi-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Verheijen, J.C.; Zask, A. Phosphatidylinositol 3-kinase (PI3K) inhibitors as anticancer drugs. Drugs Future 2007, 32, 537–547. [Google Scholar] [CrossRef]
- Falasca, M. PI3K/Akt signalling pathway specific inhibitors: A novel strategy to sensitize cancer cells to anti-cancer drugs. Curr. Pharm. Des. 2010, 16, 1410–1416. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T. Phosphatidylinositol 3-kinase inhibitors: Promising drug candidates for cancer therapy. Cancer Sci. 2008, 99, 1734–1740. [Google Scholar] [CrossRef]
- Raedler, L.A. Zydelig (Idelalisib): First-in-class PI3 kinase inhibitor approved for the treatment of 3 hematologic malignancies. Am. Health Drug Benefits 2015, 8, 157–162. [Google Scholar]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 is a highly selective intravenous pI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N.; et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ, γ, is clinically active in advanced hematologic malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Hassenrück, F.; Hallek, M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des. Dev. Ther. 2018, 12, 2577–2590. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Duvelisib: First global approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef]
- European Medicines Agency. COPIKTRA (Duvelisib). Available online: https://www.ema.europa.eu/en/documents/overview/copiktra-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Markham, A. Copanlisib: First global approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef]
- European Medicines Agency. ALIQOPA (Copanlisib). Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/2064-public-summary-opinion-orphan-designation-copanlisib-treatment-marginal-zone-lymphoma_en.pdf (accessed on 2 December 2021).
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ZYDELIG (Idelalisib). Available online: https://www.ema.europa.eu/en/documents/overview/zydelig-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Roberts, S.; Gibb, A. Chapter 1–Introduction to enzymes, receptors and the action of small molecule drugs. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin, C., Roberts, S., Jefferis, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–55. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Yen, K.; Bittinger, M.; Su, S.; Fantin, V.R. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene 2010, 29, 6409–6417. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Abou Dalle, I.; DiNardo, C.D. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther. Adv. Hematol. 2018, 9, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA approval summary: Ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef]
- Reed, D.R.; Elsarrag, R.Z.; Morris, A.L.; Keng, M.K. Enasidenib in acute myeloid leukemia: Clinical development and perspectives on treatment. Cancer Manag. Res. 2019, 11, 8073–8080. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Ribrag, V.; Soria, J.-C.; Michot, J.-M.; Schmitt, A.; Postel-Vinay, S.; Bijou, F.; Thomson, B.; Keilhack, H.; Blakemore, S.J.; Reyderman, L.; et al. Phase 1 study of Tazemetostat (EPZ-6438), an inhibitor of enhancer of Zeste-homolog 2 (EZH2): Preliminary safety and activity in relapsed or refractory non-hodgkin lymphoma (NHL) patients. Blood 2015, 126, 473. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- TAZVERIK (Tazemetostat). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213400s000lbl.pdf (accessed on 14 September 2021).
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2017, 19, 81–92. [Google Scholar] [CrossRef]
- Schapira, M. Structural chemistry of human RNA methyltransferases. ACS Chem. Biol. 2016, 11, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Azacitidine. Drugs 2012, 72, 1111–1136. [Google Scholar] [CrossRef]
- VIDAZA (Azacitidine for Injection). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/050794s011lbl.pdf (accessed on 15 September 2021).
- ONUREG (Azacitidine). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214120s000lbl.pdf (accessed on 15 September 2021).
- Garcia-Manero, G.; Stoltz, M.L.; Ward, M.R.; Kantarjian, H.; Sharma, S. A pilot pharmacokinetic study of oral azacitidine. Leukemia 2008, 22, 1680–1684. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ONUREG (Azacitidine). Available online: https://www.ema.europa.eu/en/documents/overview/onureg-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- European Medicines Agency. TAZVERIK (Tazemetostat). Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/2004-public-summary-opinion-orphan-designation-tazemetostat-treatment-diffuse-large-b-cell-lymphoma_en.pdf (accessed on 2 December 2021).
- Dhillon, S. Ivosidenib: First global approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- European Medicines Agency. TIBSOVO (Ivosidenib). Available online: https://www.ema.europa.eu/en/documents/medicine-qa/questions-answers-withdrawal-marketing-authorisation-tibsovo-ivosidenib_en.pdf (accessed on 2 December 2021).
- Kim, E.S. Enasidenib: First global approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef]
- European Medicines Agency. IDHIFA (Enasidenib). Available online: https://www.ema.europa.eu/en/documents/medicine-qa/questions-answers-withdrawal-application-marketing-authorisation-idhifa-enasidenib_en.pdf (accessed on 2 December 2021).
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef]
- European Medicines Agency. FARYDAK (Panobinostat). Available online: https://www.ema.europa.eu/en/documents/overview/farydak-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- European Medicines Agency. BELEODAQ (Belinostat). Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/12/1055-public-summary-opinion-orphan-designation-belinostat-treatment-peripheral-t-cell-lymphoma-nodal/disseminated_en.pdf (accessed on 2 December 2021).
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural basis for smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 4355. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA approval summary: Glasdegib for newly diagnosed acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Thomas, X.; Heiblig, M. An evaluation of glasdegib for the treatment of acute myelogenous leukemia. Expert Opin. Pharmacother. 2020, 21, 523–530. [Google Scholar] [CrossRef]
- Hoy, S.M. Glasdegib: First global approval. Drugs 2019, 79, 207–213. [Google Scholar] [CrossRef]
- European Medicines Agency. DAURISMO (Glasdegib). Available online: https://www.ema.europa.eu/en/documents/overview/daurismo-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Kale, J.; Osterlund, E.; Andrews, D. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Besbes, S.; Mirshahi, M.; Pocard, M.; Billard, C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 2015, 6, 12862–12871. [Google Scholar] [CrossRef]
- Mihalyova, J.; Jelinek, T.; Growkova, K.; Hrdinka, M.; Simicek, M.; Hajek, R. Venetoclax: A new wave in hematooncology. Exp. Hematol. 2018, 61, 10–25. [Google Scholar] [CrossRef]
- VENCLEXTA (Venetoclax Tablets). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208573s009lbl.pdf (accessed on 17 September 2021).
- Abdul Razak, A.R.; Mau-Soerensen, M.; Gabrail, N.Y.; Gerecitano, J.F.; Shields, A.F.; Unger, T.J.; Saint-Martin, J.R.; Carlson, R.; Landesman, Y.; McCauley, D.; et al. First-in-class, first-in-human phase I study of Selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 4142–4150. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018, 36, 859–866. [Google Scholar] [CrossRef]
- XPOVIO (Selinexor). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212306s005lbl.pdf (accessed on 20 September 2021).
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Orlowski, M.; Wilk, S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch. Biochem. Biophys. 2000, 383, 1–16. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- NINLARO (Ixazomib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208462lbl.pdf (accessed on 21 September 2021).
- KYPROLIS (Carfilzomib). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202714s030lbl.pdf (accessed on 21 September 2021).
- Muz, B.; Ghazarian, R.N.; Ou, M.; Luderer, M.J.; Kusdono, H.D.; Azab, A.K. Spotlight on ixazomib: Potential in the treatment of multiple myeloma. Drug Des. Dev. Ther. 2016, 10, 217–226. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Offidani, M.; Vigna, E.; Corvatta, L.; Recchia, A.G.; Morabito, L.; Morabito, F.; Gentili, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2015, 24, 1287–1298. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef]
- Syed, Y.Y. Selinexor: First global approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- European Medicines Agency. NEXPOVIO (Selinexor). Available online: https://www.ema.europa.eu/en/documents/overview/nexpovio-epar-medicine-overview_en-1.pdf (accessed on 2 December 2021).
- Deeks, E.D. Venetoclax: First global approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. VENCLYXTO (Venetoclax). Available online: https://www.ema.europa.eu/en/documents/overview/venclyxto-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Shirley, M. Ixazomib: First global approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. NINLARO (Ixazomib). Available online: https://www.ema.europa.eu/en/documents/overview/ninlaro-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Waterhouse, D.N.; Madden, T.D.; Cullis, P.R.; Bally, M.B.; Mayer, L.D.; Webb, M.S. Preparation, characterization, and biological analysis of liposomal formulations of vincristine. Methods Enzymol. 2005, 391, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Farag, S.S. Treating relapsed or refractory Philadelphia chromosome-negative acute lymphoblastic leukemia: Liposome-encapsulated vincristine. Int. J. Nanomed. 2013, 8, 3479–3488. [Google Scholar] [CrossRef][Green Version]
- European Medicines Agency. MARQIBO (Vincristine Sulfate LIPOSOME Injection). Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/08/555-public-summary-positive-opinion-orphan-designation-vincristine-sulphate-liposomes-treatment_en.pdf (accessed on 2 December 2021).
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. Food and drug administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef]
- European Medicines Agency. KYPROLIS (Carfilzomib). Available online: https://www.ema.europa.eu/en/documents/overview/kyprolis-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Clark, D.; Pazdernik, N.; McGehee, M. Chapter 13—Protein synthesis. In Molecular Biology, 3rd ed.; Clark, D., Pazdernik, N., McGehee, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 397–444. [Google Scholar] [CrossRef]
- SYNRIBO (Omacetaxine Mepesuccinate). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203585s005lbl.pdf (accessed on 24 September 2021).
- Kim, T.D.; Frick, M.; le Coutre, P. Omacetaxine mepesuccinate for the treatment of leukemia. Expert Opin. Pharmacother. 2011, 12, 2381–2392. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. OncoTargets Ther. 2014, 7, 177–186. [Google Scholar] [CrossRef]
- Alvandi, F.; Kwitkowski, V.E.; Ko, C.W.; Rothmann, M.D.; Ricci, S.; Saber, H.; Ghosh, D.; Brown, J.; Pfeiler, E.; Chikhale, E.; et al. Food and Drug Administration approval summary: Omacetaxine mepesuccinate as treatment for chronic myeloid leukemia. Oncologist 2014, 19, 94–99. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. NDA 203585Orig1s000. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203585Orig1s000SumR.pdf (accessed on 2 December 2021).
- Liliemark, J.; Pettersson, B.; Engberg, B.; Lafolie, P.; Masquelier, M.; Peterson, C. On the paradoxically concentration-dependent metabolism of 6-mercaptopurine in WEHI-3b murine leukemia cells. Cancer Res. 1990, 50, 108–112. [Google Scholar]
- Gerbek, T.; Ebbesen, M.; Nersting, J.; Frandsen, T.L.; Appell, M.L.; Schmiegelow, K. Role of TPMT and ITPA variants in mercaptopurine disposition. Cancer Chemother. Pharmacol. 2018, 81, 579–586. [Google Scholar] [CrossRef]
- Bökkerink, J.P.; Stet, E.H.; De Abreu, R.A.; Damen, F.J.; Hulscher, T.W.; Bakker, M.A.; van Baal, J.A. 6-Mercaptopurine: Cytotoxicity and biochemical pharmacology in human malignant T-lymphoblasts. Biochem. Pharmacol. 1993, 45, 1455–1463. [Google Scholar] [CrossRef]
- Moon, W.; Loftus, E.V. Review article: Recent advances in pharmacogenetics and pharmacokinetics for safe and effective thiopurine therapy in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2016, 43, 863–883. [Google Scholar] [CrossRef]
- PURIXAN (Mercaptopurine). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205919s000lbl.pdf (accessed on 27 September 2021).
- Mulla, H.; Leary, A.; White, P.; Pandya, H. A step toward more accurate dosing for mercaptopurine in childhood acute lymphoblastic leukemia. J. Clin. Pharmacol. 2012, 52, 1610–1613. [Google Scholar] [CrossRef]
- Norman, P. Orphan drug approvals of 2014: Europe and the United States. Expert Opin. Orphan Drugs 2015, 3, 445–455. [Google Scholar] [CrossRef]
- European Medicines Agency. XALUPRINE (Mercaptopurine). Available online: https://www.ema.europa.eu/en/documents/overview/xaluprine-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Raje, N.; Anderson, K.C. Thalidomide and immunomodulatory drugs as cancer therapy. Curr. Opin. Oncol. 2002, 14, 635–640. [Google Scholar] [CrossRef]
- POMALYST (Pomalidomide). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/204026s024lbl.pdf (accessed on 28 September 2021).
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013, 3, e143. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335, Erratum in: Leukemia 2012, 26, 2445. [Google Scholar] [CrossRef] [PubMed]
- Elkinson, S.; McCormack, P.L. Pomalidomide: First global approval. Drugs 2013, 73, 595–604. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. IMNOVID (Pomalidomide). Available online: https://www.ema.europa.eu/en/documents/overview/imnovid-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Moon, C.; Oh, E. Rationale and strategies for formulation development of oral fixed dose combination drug products. J. Pharm. Investig. 2016, 46, 615–631. [Google Scholar] [CrossRef]
- Pillai, G. Chapter 9—Nanotechnology toward treating cancer: A comprehensive review. In Micro and Nano Technologies, Applications of Targeted Nano Drugs and Delivery Systems; Mohapatra, S., Ranjan, S., Dasgupta, N., Mishra, R., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 221–256. [Google Scholar] [CrossRef]
- El-Subbagh, H.; Al-Badr, A. Chapter 2—Cytarabine. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 34, pp. 37–113. [Google Scholar] [CrossRef]
- Cytarabine NDA #016793—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=016793 (accessed on 29 September 2021).
- Kwok, K.; Vincent, E.; Gibson, J. 36—Antineoplastic drugs. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Dowd, F., Johnson, B., Mariotti, A., Eds.; Mosby: Maryland, MI, USA, 2017; pp. 530–562. [Google Scholar] [CrossRef]
- Cerubidine NDA #050484—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050484 (accessed on 29 September 2021).
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Tzogani, K.; Penttilä, K.; Lapveteläinen, T.; Hemmings, R.; Koenig, J.; Freire, J.; Márcia, S.; Cole, S.; Coppola, P.; Flores, B.; et al. EMA review of daunorubicin and cytarabine encapsulated in liposomes (Vyxeos, CPX-351) for the treatment of adults with newly diagnosed, therapy-related acute myeloid leukemia or acute myeloid leukemia with myelodysplasia-related changes. Oncologist 2020, 25, e1414–e1420. [Google Scholar] [CrossRef] [PubMed]
- VYXEOS (Daunorubicin and Cytarabine). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf (accessed on 4 October 2021).
- Decitabine NDA #021790—Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021790 (accessed on 4 October 2021).
- Plimack, E.R.; Kantarjian, H.M.; Issa, J.P. Decitabine and its role in the treatment of hematopoietic malignancies. Leuk. Lymphoma 2007, 48, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- INQOVI (Decitabine and Cedazuridine). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212576s000lbl.pdf (accessed on 4 October 2021).
- Dhillon, S. Decitabine/Cedazuridine: First approval. Drugs 2020, 80, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA approval summary: (Daunorubicin and Cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. VYXEOS (Daunorubicin and Cytarabine). Available online: https://www.ema.europa.eu/en/documents/overview/vyxeos-liposomal-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Bayer, V. An overview of monoclonal antibodies. Semin. Oncol. Nurs. 2019, 35, 150927. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal antibodies in cancer therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Hoy, S.M. Tafasitamab: First approval. Drugs 2020, 80, 1731–1737. [Google Scholar] [CrossRef]
- MONJUVI (Tafasitamab-cxix). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761163s000lbl.pdf (accessed on 7 October 2021).
- Uchida, J.; Lee, Y.; Hasegawa, M.; Liang, Y.; Bradney, A.; Oliver, J.A.; Bowen, K.; Steeber, D.A.; Haas, K.M.; Poe, J.C.; et al. Mouse CD20 expression and function. Int. Immunol. 2004, 16, 119–129. [Google Scholar] [CrossRef]
- Niederfellner, G.; Lammens, A.; Mundigl, O.; Georges, G.J.; Schaefer, W.; Schwaiger, M.; Franke, A.; Wiechmann, K.; Jenewein, S.; Slootstra, J.W.; et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011, 118, 358–367. [Google Scholar] [CrossRef]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef]
- Herter, S.; Herting, F.; Mundigl, O.; Waldhauer, I.; Weinzierl, T.; Fauti, T.; Muth, G.; Ziegler-Landesberger, D.; Van Puijenbroek, E.; Lang, S.; et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol. Cancer Ther. 2013, 12, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- TRUXIMA (Rituximab-abbs). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761088s000lbl.pdf (accessed on 8 October 2021).
- RUXIENCE (Rituximab-pvvr). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761103s000lbl.pdf (accessed on 8 October 2021).
- RIABNI (Rituximab-arrx). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761140s000lbl.pdf (accessed on 8 October 2021).
- GAZYVA (Obinutuzumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125486s017s018lbl.pdf (accessed on 8 October 2021).
- Partida-Sánchez, S.; Cockayne, D.; Monard, S.; Jacobson, E.L.; Oppenheimer, N.; Garvy, B.; Kusser, K.; Goodrich, S.; Howard, M.; Harmsen, A.; et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001, 7, 1209–1216. [Google Scholar] [CrossRef]
- Martin, T.G.; Corzo, K.; Chiron, M.; van de Velde, H.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic opportunities with pharmacological inhibition of CD38 with Isatuximab. Cells 2019, 8, 1522. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Usmani, S.Z. CD38 antibodies in multiple myeloma: Mechanisms of action and modes of resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Richardson, P.G.; Beksaç, M.; Špička, I.; Mikhael, J. Isatuximab for the treatment of relapsed/refractory multiple myeloma. Expert Opin. Biol. Ther. 2020, 20, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- DARZALEX (Daratumumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761036s035lbl.pdf (accessed on 11 October 2021).
- SARCLISA (Isatuximab-irfc). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761113s003lbl.pdf (accessed on 11 October 2021).
- DARZALEX FASPRO (Daratumumab and Hyaluronidase-fihj). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761145s002lbl.pdf (accessed on 11 October 2021).
- Malaer, J.D.; Mathew, P.A. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am. J. Cancer Res. 2017, 7, 1637–1641. [Google Scholar] [PubMed]
- Magen, H.; Muchtar, E. Elotuzumab: The first approved monoclonal antibody for multiple myeloma treatment. Ther. Adv. Hematol. 2016, 7, 187–195. [Google Scholar] [CrossRef]
- EMPLICITI (Elotuzumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761035s008lbl.pdf (accessed on 12 October 2021).
- Guan, J.; Lim, K.S.; Mekhail, T.; Chang, C.C. Programmed death ligand-1 (PD-L1) expression in the programmed death receptor-1 (PD-1)/PD-L1 blockade: A key player against various cancers. Arch. Pathol. Lab. Med. 2017, 141, 851–861. [Google Scholar] [CrossRef]
- Prasad, V.; Kaestner, V. Nivolumab and pembrolizumab: Monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin. Oncol. 2017, 44, 132–135. [Google Scholar] [CrossRef] [PubMed]
- OPDIVO (Nivolumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf (accessed on 15 October 2021).
- KEYTRUDA (Pembrolizumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125514s096lbl.pdf (accessed on 15 October 2021).
- Wu, X.S.; Lonsdorf, A.S.; Hwang, S.T. Cutaneous T-cell lymphoma: Roles for chemokines and chemokine receptors. J. Investig. Dermatol. 2009, 129, 1115–1119. [Google Scholar] [CrossRef][Green Version]
- Watson, S.; Marx, J.B. Mogamulizumab-kpkc: A novel therapy for the treatment of cutaneous T-cell lymphoma. J. Adv. Pract. Oncol. 2019, 10, 883–888. [Google Scholar] [CrossRef]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/overview/minjuvi-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Cai, H.H. Therapeutic monoclonal antibodies approved by FDA in 2020. Clin. Res. Immunol. 2021, 4, 1–2. [Google Scholar]
- Dhillon, S. Isatuximab: First approval. Drugs 2020, 80, 905–912. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. SARCLISA (Isatuximab-irfc). Available online: https://www.ema.europa.eu/en/documents/overview/sarclisa-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- European Medicines Agency. RUXIENCE (Rituximab-pvvr). Available online: https://www.ema.europa.eu/en/documents/overview/ruxience-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- U.S. Food and Drug Administration. FDA Approves First Biosimilar for Treatment of Adult Patients with Non-Hodgkin’s Lymphoma. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-biosimilar-treatment-adult-patients-non-hodgkins-lymphoma (accessed on 10 September 2021).
- European Medicines Agency. TRUXIMA (Rituximab-Abbs). Available online: https://www.ema.europa.eu/en/documents/overview/truxima-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Nie, L.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA approval summary: Mogamulizumab-kpkc for mycosis fungoides and Sézary syndrome. Clin. Cancer Res. 2019, 25, 7275–7280. [Google Scholar] [CrossRef]
- European Medicines Agency. POTELIGEO (Mogamulizumab-Kpkc). Available online: https://www.ema.europa.eu/en/documents/overview/poteligeo-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Markham, A. Elotuzumab: First global approval. Drugs 2016, 76, 397–403. [Google Scholar] [CrossRef]
- European Medicines Agency. EMPLICITI (Elotuzumab). Available online: https://www.ema.europa.eu/en/documents/overview/empliciti-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- McKeage, K. Daratumumab: First global approval. Drugs 2016, 76, 275–281. [Google Scholar] [CrossRef]
- European Medicines Agency. DARZALEX (Daratumumab). Available online: https://www.ema.europa.eu/en/documents/overview/darzalex-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. FDA approval summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef]
- OPDIVO (Nivolumab). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125554s019lbl.pdf (accessed on 29 November 2021).
- European Medicines Agency. OPDIVO (Nivolumab). Available online: https://www.ema.europa.eu/en/documents/overview/opdivo-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Poole, R.M. Pembrolizumab: First global approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves Pembrolizumab for Treatment of Relapsed or Refractory PMBCL. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-treatment-relapsed-or-refractory-pmbcl (accessed on 10 September 2021).
- U.S. Food and Drug Administration. Pembrolizumab (KEYTRUDA) for Classical Hodgkin Lymphoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/pembrolizumab-keytruda-classical-hodgkin-lymphoma (accessed on 10 September 2021).
- European Medicines Agency. KEYTRUDA (Pembrolizumab). Available online: https://www.ema.europa.eu/en/documents/overview/keytruda-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Cameron, F.; McCormack, P.L. Obinutuzumab: First global approval. Drugs 2014, 74, 147–154. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. GAZYVARO (Obinutuzumab). Available online: https://www.ema.europa.eu/en/documents/overview/gazyvaro-epar-summary-public_en.pdf (accessed on 2 December 2021).
- Van Spriel, A.B.; van Ojik, H.H.; van de Winkel, J.G.J. Immunotherapeutic perspective for bispecific antibodies. Immunol. Today 2000, 21, 391–397. [Google Scholar] [CrossRef]
- Newman, M.J.; Benani, D.J. A review of blinatumomab, a novel immunotherapy. J. Oncol. Pharm. Pract. 2016, 22, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M. Blinatumomab: First global approval. Drugs 2015, 75, 321–327. [Google Scholar] [CrossRef]
- European Medicines Agency. BLINCYTO (Blinatumomab). Available online: https://www.ema.europa.eu/en/documents/overview/blincyto-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sineh Sepehr, K.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell Physiol. 2019, 235, 31–64. [Google Scholar] [CrossRef]
- Sato, S.; Miller, A.S.; Inaoki, M.; Bock, C.B.; Jansen, P.J.; Tang, M.L.; Tedder, T.F. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: Altered signaling in CD22-deficient mice. Immunity 1996, 5, 551–562. [Google Scholar] [CrossRef]
- Shor, B.; Gerber, H.P.; Sapra, P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015, 67 Pt A, 107–116. [Google Scholar] [CrossRef]
- Godwin, C.; Gale, R.; Walter, R. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- BESPONSA (Inotuzumab Ozogamicin). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 13 October 2021).
- Sabattini, E.; Pizzi, M.; Tabanelli, V.; Baldin, P.; Sacchetti, C.S.; Agostinelli, C.; Zinzani, P.L.; Pileri, S.A. CD30 expression in peripheral T-cell lymphomas. Haematologica 2013, 98, e81–e82. [Google Scholar] [CrossRef]
- Fichtner, M.; Dreyling, M.; Binder, M.; Trepel, M. The role of B cell antigen receptors in mantle cell lymphoma. J. Hematol. Oncol. 2017, 10, 164. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Diefenbach, C.S. Polatuzumab Vedotin: A new target for B cell malignancies. Curr. Hematol. Malig. Rep. 2020, 15, 125–129. [Google Scholar] [CrossRef] [PubMed]
- ADCETRIS (Brentuximab Vedotin). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s097lbl.pdf (accessed on 18 October 2021).
- Picardi, M.; Della Pepa, R.; Giordano, C.; Pugliese, N.; Mortaruolo, C.; Trastulli, F.; Rascato, M.G.; Cappuccio, I.; Raimondo, M.; Memoli, M.; et al. Brentuximab vedotin followed by bendamustine supercharge for refractory or relapsed Hodgkin lymphoma. Blood Adv. 2019, 3, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- POLIVY (Polatuzumab Vedotin-Piiq). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 18 October 2021).
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B cell maturation antigen (BCMA) in multiple myeloma: Potential uses of BCMA-based immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- BLENREP (Belantamab Mafodotin-Blmf). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 18 October 2021).
- Markham, A. Belantamab Mafodotin: First approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- European Medicines Agency. BLENREP (Belantamab Mafodotin-Blmf). Available online: https://www.ema.europa.eu/en/documents/overview/blenrep-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Deeks, E.D. Polatuzumab Vedotin: First global approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- European Medicines Agency. POLIVY (Polatuzumab Vedotin-piiq). Available online: https://www.ema.europa.eu/en/documents/overview/polivy-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Lamb, Y.N. Inotuzumab Ozogamicin: First global approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- European Medicines Agency. BESPONSA (Inotuzumab Ozogamicin). Available online: https://www.ema.europa.eu/en/documents/overview/besponsa-epar-summary-public_en.pdf (accessed on 2 December 2021).
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. Food and Drug Administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef]
- European Medicines Agency. ADCETRIS (Brentuximab Vedotin). Available online: https://www.ema.europa.eu/en/documents/overview/adcetris-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Kreitman, R.J. Immunotoxins in cancer therapy. Curr. Opin. Immunol. 1999, 11, 570–578. [Google Scholar] [CrossRef]
- LUMOXITI (Moxetumomab Pasudotox-Tdfk). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf (accessed on 19 October 2021).
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef]
- Dhillon, S. Moxetumomab Pasudotox: First global approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. LUMOXITI (Moxetumomab Pasudotox-tdfk). Available online: https://www.ema.europa.eu/en/documents/overview/lumoxiti-epar-medicine-overview_en.pdf (accessed on 2 December 2021).
- Wang, Z.; Xie, Q.; Zhou, H.; Zhang, M.; Shen, J.; Ju, D. Amino acid degrading enzymes and autophagy in cancer therapy. Front. Pharmacol. 2021, 11, 582587. [Google Scholar] [CrossRef] [PubMed]
- Salzer, W.L.; Asselin, B.L.; Plourde, P.V.; Corn, T.; Hunger, S.P. Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann. N. Y. Acad. Sci. 2014, 1329, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Jin, R.; Liu, C.; Cao, X.; Manning, M.L.; Di, X.M.; Przepiorka, D.; Namuswe, F.; Deisseroth, A.; Goldberg, K.B.; et al. FDA approval summary: Calaspargase Pegol-mknl For treatment of acute lymphoblastic leukemia in children and young adults. Clin. Cancer Res. 2020, 26, 328–331. [Google Scholar] [CrossRef] [PubMed]
- ERWINAZE (Asparaginase Erwinia Chrysanthemi). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125359lbl.pdf (accessed on 20 October 2021).
- ASPARLAS (Calaspargase Pegol—mknl). Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761102s000lbl.pdf (accessed on 20 October 2021).
- Keating, G.M. Asparaginase Erwinia chrysanthemi (Erwinaze®): A guide to its use in acute lymphoblastic leukemia in the USA. BioDrugs 2013, 27, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Abernethy, D.R.; Wang, Y.; Zhao, P.; Zineh, I. The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 2013, 102, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. List of Nationally Authorised Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/psusa/asparaginase-crisantaspase-pegaspargase-nationally-authorised-products-list-nationally-authorised/00003161/201808_en.pdf (accessed on 1 December 2021).
- Yang, X.; Wang, G.-X.; Zhou, J.-F. CAR T cell therapy for hematological malignancies. Curr. Med. Sci. 2019, 39, 874–882. [Google Scholar] [CrossRef]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current progress in CAR-T cell therapy for hematological malignancies. J. Cancer 2021, 12, 326–334. [Google Scholar] [CrossRef]
- Mueller, K.T.; Waldron, E.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Hamilton, J.; Awasthi, R.; et al. Clinical pharmacology of Tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2018, 24, 6175–6184. [Google Scholar] [CrossRef]
- KYMRIAH (Tisagenlecleucel). Prescribing Information. Available online: https://www.fda.gov/media/107296/download (accessed on 21 October 2021).
- Mian, A.; Hill, B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef]
- Riedell, P.A.; Bishop, M.R. Safety and efficacy of axicabtagene ciloleucel in refractory large B-cell lymphomas. Ther. Adv. Hematol. 2020, 11, 2040620720902899. [Google Scholar] [CrossRef] [PubMed]
- YESCARTA (Axicabtagene Ciloleucel). Prescribing Information. Available online: https://www.fda.gov/media/108377/download (accessed on 23 October 2021).
- TECARTUS (Brexucabtagene Autoleucel). Prescribing Information. Available online: https://www.fda.gov/media/140409/download (accessed on 23 October 2021).
- U.S. Food and Drug Administration. FDA Approves First Cell-Based Gene Therapy For Adult Patients with Relapsed or Refractory MCL. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-relapsed-or-refractory-mcl (accessed on 25 October 2021).
- European Medicines Agency. TECARTUS (Brexucabtagene Autoleucel). Available online: https://www.ema.europa.eu/en/documents/overview/tecartus-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA approval summary: Axicabtagene Ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.ema.europa.eu/en/documents/overview/yescarta-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA approval summary: Tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. KYMRIAH (Tisagenlecleucel). Available online: https://www.ema.europa.eu/en/documents/overview/kymriah-epar-medicine-overview_en.pdf (accessed on 1 December 2021).
- Plescia, J.; Moitessier, N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 2020, 195, 112270. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide bond bioisosteres: Strategies, synthesis, and successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef]
- Liu, W.; Liang, Y.; Si, X. Hydroxamic acid hybrids as the potential anticancer agents: An overview. Eur. J. Med. Chem. 2020, 205, 112679. [Google Scholar] [CrossRef]
- Tanii, H.; Hashimoto, K. Studies on the mechanism of acute toxicity of nitriles in mice. Arch. Toxicol. 1984, 55, 47–54. [Google Scholar] [CrossRef]
- Rosenkranz, H.S.; Mermelstein, R. Mutagenicity and genotoxicity of nitroarenes. All nitro-containing chemicals were not created equal. Mutat. Res. 1983, 114, 217–267. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Zanubrutinib | BRUKINSA BeiGene, Ltd., Beijing, China | FDA: 14 November 2019 EMA: 29 May 2019 |  | BTK 1 | Oral | Mantle Cell Lymphoma | Decreased neutrophil count, anemia, neutropenia, pneumonia, decreased platelet count, upper respiratory tract infection, rash, bruising, diarrhea, cough | [39,40] |
| 2 | Acalabrutinib | CALQUENCE AstraZeneca, Cambridge, UK | FDA: 31 October 2017 EMA: 5 November 2020 |  | BTK 1 | Oral | Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, Small Lymphocytic Lymphoma | Headache, diarrhea, fatigue, nausea, contusion, neutropenia, anemia, pneumonia, thrombocytopenia | [31,41,42] |
| 3 | Ibrutinib | IMBRUVICA AbbVie Inc., Lake Bluff, IL, USA | FDA: 13 November 2013 EMA: 21 October 2014 |  | BTK 1 | Oral | Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, Waldenström’s Macroglobulinemia, Small Lymphocytic Lymphoma, Marginal Zone Lymphoma | Diarrhea, fatigue, nausea, dyspnea, constipation, peripheral edema, upper respiratory tract infection, rash, cough, arthralgia, vomiting, decreased appetite, thrombocytopenia, neutropenia, anemia, pneumonia, dehydratation | [30,43,44] |
| 4 | Bosutinib | BOSULIF Pfizer Inc., New York, NY, USA | FDA: 4 September 2012 EMA: 27 March 2013 |  | Src 2, Abl 3 | Oral | Chronic Myelogenous Leukemia | Diarrhea, nausea, abdominal pain, vomiting, thrombocytopenia, anemia, neutropenia | [45,46,47] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Fedratinib | INREBIC Celgene Corporation, Summit, NJ, USA | FDA: 16 August 2019 EMA: 8 February 2021 |  | JAK2 2 | Oral | Myelofibrosis | Diarrhea, nausea, vomiting, constipation, anemia, thrombocytopenia | [66] 1, [67,68] |
| 2 | Gilteritinib | XOSPATA Astellas Pharma US, Inc., Northbrook, IL, USA | FDA: 28 November 2018 EMA: 24 October 2019 |  | FLT3 3, AXL 4, ALK 5 | Oral | Acute Myeloid Leukemia | Myalgia, arthralgia, increased levels of transaminases, fatigue, malaise, fever, diarrhea, dyspnea, edema, rash, pneumonia, sepsis, renal impairment | [69,70] |
| 3 | Midostaurin | RYDAPT Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA | FDA: 28 April 2017 EMA: 18 September 2017 |  | c-Kit 6, PDGFRA 7, PDGFRB 8, FLT3 3, PKC 9, CDK1 10, SYK 11, VEGFR-2 12 | Oral | Acute Myeloid Leukemia, Cutaneous Mastocytosis | Febrile neutropenia, nausea, vomiting, diarrhea, edema, mucositis, headache, device-related infection, abdominal pain, fatigue, pyrexia, dyspnea, musculoskeletal pain, constipation, epistaxis, upper respiratory tract infection, petechial, hyperglycemia, | [71,72] |
| 4 | Ponatinib | ICLUSIG Ariad Pharmaceuticals, Inc., Cambridge, MA, USA | FDA: 14 December 2012 EMA: 1 July 2013 |  | BCR-ABL 13, VEGFRs 14, FGFRs 15, PDGFRs 16, RET 17, c-Kit 6, TIE2 18, FLT3 3 | Oral | Chronic Myelogenous Leukemia, Acute Lymphoblastic Leukemia | Hypertension, cardiac failure, abdominal pain, constipation, diarrhea, oral mucositis, febrile neutropenia, fatigue, pneumonia, headache, peripheral neuropathy, dizziness, pleural effusion, cough, dyspnea, rush, dry skin, arthralgia, myalgia, spasms, decreased appetite, edema, weight loss, insomnia | [73,74] |
| 5 | Ruxolitinib | JAKAFI Incyte Corporation, Wilmington, DE, USA | FDA: 16 November 2011 EMA: 23 August 2012 |  | JAK1 19, JAK2 2 | Oral | Myelofibrosis, Polycythemia Vera, Graft-versus-host disease | Anemia, thrombocytopenia, neutropenia | [75,76] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Duvelisib | COPIKTRA Verastem, Inc. Needham, MA, USA | FDA: 24 September 2018 EMA: 19 May 2021 |  | PI3K-δ 1, PI3K-γ 2 | Oral | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Neutropenia, thrombocytopenia, anemia, diarrhea, pyrexia, nausea, vomiting, anorexia | [84,85] |
| 2 | Copanlisib | ALIQOPA Bayer HealthCare Pharmaceuticals Inc., HanoverWhippany, NJ, USA | FDA: 14 September 2017 EMA: Not approved |  | PI3K-α 3, PI3K-δ 1 | Intravenous infusion | Follicular Lymphoma | Hyperglycemia, hypertension, infections, neutropenia | [86,87] |
| 3 | Idelalisib | ZYDELIG Gilead Sciences, Inc., Foster City, CA, USA | FDA: 23 July 2014 EMA: 18 September 2014 |  | PI3K-δ 1 | Oral | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Diarrhea, nausea, vomiting, fatigue, headache, pneumonia, chill, dyspnea, rash, neutropenia, pyrexia, sepsis, decreased neutrophil count, hypertriglyceridemia, hyperglycemia, elevated alanine and aspartate transaminases | [88,89] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Azacitidine | ONUREG Bristol-Myers Squibb Company, New York, NY, USA | FDA: 1 September 2020 EMA: 17 June 2021 |  | DNMTs 1, RNMTs 2 | Oral | Acute Myeloid Leukemia | Nausea, vomiting, diarrhea, fatigue, constipation, pneumonia, arthralgia, decreased appetite, febrile neutropenia, dizziness, abdominal pain. | [112,114] |
| 2 | Tazemetostat | TAZVERIK Epizyme, Inc., Cambridge, MA, USA | FDA: 23 January 2020 EMA: Not approved |  | EZH2 3 | Oral | Epithelioid Sarcoma, Follicular Lymphoma | Fatigue, nausea, decreased appetite, vomiting, constipation, upper respiratory tract infection, abdominal pain, musculoskeletal pain | [106,115] |
| 3 | Ivosidenib | TIBSOVO Agios Pharmaceuticals, Inc., Cambridge, MA, USA | FDA: 20 July 2018 EMA: Not approved |  | IDH1 4 | Oral | Acute Myeloid Leukemia | Diarrhea, leukocytosis, nausea, fatigue, dyspnea, electrocardiogram QT prolonged, edema, anemia, pyrexia, cough, febrile neutropenia, isocitrate dehydrogenase differentiation syndrome | [116,117] |
| 4 | Enasidenib | IDHIFA Celgene Corporation, Summit, MA, USA | FDA: 1 August 2017 EMA: Not approved |  | IDH2 5 | Oral | Acute Myeloid Leukemia | Elevated bilirubin, nausea, diarrhea, decreased appetite, vomiting | [118,119] |
| 5 | Panobinostat | FARYDAK, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA | FDA: 23 February 2015 EMA: 28 August 2015 |  | HDAC 6 | Oral | Multiple Myeloma | Diarrhea, fatigue, nausea, peripheral edema, decreased appetite, pyrexia, vomiting, thrombocytopenia, lymphopenia, leukopenia, neutropenia, anemia, hypophosphatemia, hypokalemia, hyponatremia, increased creatinine | [120,121] |
| 6 | Belinostat | BELEODAQ, Spectrum Pharmaceuticals, Inc., Henderson, NV, USA | FDA: 3 July 2014 EMA: Not approved |  | HDAC 6 | Intravenous infusion | Peripheral T-cell Lymphoma | Nausea, vomiting, diarrhea, fatigue, pyrexia, anemia, constipation, dyspnea, rash, peripheral edema | [122,123] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Glasdegib | DAURISMO Pfizer Inc., New York, NY, USA | FDA: 21 November 2018 EMA: 26 June 2020 |  | SMO receptor 1 | Oral | Acute Myeloid Leukemia | Anemia, febrile neutropenia, thrombocytopenia | [129,130] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Selinexor | XPOVIO Karyopharm Therapeutics Inc., Newton Centre, MA, USA | FDA: 3 July 2019 EMA: 26 March 2021 |  | XPO1 1 | Oral | Multiple Myeloma, Diffuse Large B-cell Lymphoma | Thrombocytopenia, fatigue, nausea, anemia, decreased appetite, decreased weight, diarrhea, vomiting, hyponatremia, neutropenia, leukopenia, constipation, dyspnea and upper respiratory tract infection | [149,150] |
| 2 | Venetoclax | VENCLEXTA AbbVie Inc., Lake Bluff, IL, USA | FDA: 11 April 2016 EMA: 5 December 2016 |  | BCL-2 2 | Oral | Chronic Lymphocytic Leukemia, Acute Myeloid Leukemia | Neutropenia, diarrhea, nausea, anemia, thrombocytopenia, upper respiratory tract infection, fatigue | [151,152] |
| 3 | Ixazomib citrate | NINLARO Takeda Pharmaceuticals U.S.A., Inc., Deerfield, IL, usa | FDA: 20 November 2015 EMA: 21 November 2016 |  | Proteasome | Oral | Multiple Myeloma | Diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting, back pain, rash | [153,154] |
| 4 | Vincristine sulfate | MARQIBO Talon Therapeutics Inc., Irvine, CA, USA | FDA: 9 August 2012 EMA: Not approved |  | Tubulin | Intravenous | Acute Lymphoblastic Leukemia | Constipation, nausea, pyrexia, fatigue, peripheral neuropathy, febrile neutropenia, diarrhea, anemia, decreased appetite, insomnia | [155,156,157] |
| 5 | Carfilzomib | KYPROLIS Amgen Inc., Sauzend Oaks, CA, USA | FDA: 20 July 2012 EMA: 19 November 2015 |  | Proteasome | Intravenous | Multiple Myeloma | Fatigue, anemia, nausea, thrombocytopenia, dyspnea, diarrhea, pyrexia | [158,159] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Omacetaxine mepesuccinate | SYNRIBO Teva Pharmaceuticals, Tel Aviv, Israel | FDA: 26 October 2012 EMA: Not approved |  | Protein level | Subcutaneous injection | Chronic Myelogenous Leukemia | Thrombocytopenia, anemia, diarrhea, pyrexia, fatigue, nausea, neutropenia, injection site reaction, infection, lymphopenia | [164,165] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Mercaptopurine | PURIXAN Nova Laboratories, Ltd., Leicester, UK | FDA: 28 April 2014 EMA: 9 March 2012 |  | DNA 1 | Oral | Acute Lymphoblastic Leukemia | Anemia, neutropenia, thrombocytopenia | [170,172,173] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pomalidomide | POMALYST Bristol-Myers Squibb Company, New York, NY, USA | FDA: 8 February 2013 EMA: 5 August 2013 |  | CRBN 1 | Oral | Multiple Myeloma, Kaposi’s Sarcoma | Neutropenia, thrombocytopenia, anemia, fatigue | [179,180] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Decitabine | INQOVI Astex Pharmaceuticals, Taiho Oncology, and Otsuka Pharmaceutical, Tokyo, Japan | FDA: 7 July 2020 EMA: Not approved |  | DNMTs 1 | Oral | Myelodysplastic Syndrome | Fatigue, constipation, hemorrhage, myalgia, nausea, arthralgia, pneumonia, sepsis, decreased leukocytes, decreased platelet count, decreased neutrophil count | [193] |
| Cedazuridine |  | CDA 2 | |||||||
| 2 | Cytarabine | VYXEOS Jazz Pharmaceuticals plc, Dublin, Ireland | FDA: 3 August 2017 EMA: 23 August 2018 |  | DNA 3 | Intravenous | Acute Myeloid Leukemia | Hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, vomiting | [194,195] |
| Daunorubicin |  | TOP2 4 |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Rituximab-arrx | RIABNI Amgen Inc., Sauzend Oaks, CA, USA | FDA: 17 December 2020 EMA: Not approved | Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma, Chronic Lymphocytic Leukemia | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia, neutropenia | [207] |
| 2 | Tafasitamab-cxix | MONJUVI MorphoSys AG, Planegg, Germany | FDA: 31 July 2020 EMA: 26 August 2021 | Humanized immunoglobulin G1/2 (IgG1/2) hybrid monoclonal antibody | CD19 2 | Intravenous | Diffuse Large B-Cell Lymphoma | Neutropenia, fatigue, anemia, diarrhea, thrombocytopenia, cough, pyrexia, peripheral edema, respiratory tract infection, decreased appetite | [199,225] |
| 3 | Daratumumab and hyaluronidase-fihj | DARZALEX FASPRO Janssen Pharmaceuticals, Inc., Belce, Belgium | FDA: 1 May 2020 EMA:Not approved | Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody and an endoglycosidase | CD38 3 | Intravenous | Multiple Myeloma | Upper respiratory tracts infection, constipation, nausea, fatigue, pyrexia, peripheral sensory neuropathy, diarrhea, cough, insomnia, vomiting, back pain, muscle spasms, pneumonia, dyspnea | [215,226] |
| 4 | Isatuximab | SARCLISA Sanofi, Paris, France | FDA: 2 March 2020 EMA: 30 May 2020 | Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD38 3 | Intravenous | Multiple Myeloma | Infusion reactions, upper respiratory tract infections, bronchitis, pneumonia | [227,228] |
| 5 | Rituximab-pvvr | RUXIENCE Pfizer Inc., New York, NY, USA | FDA: 23 July 2019 EMA: 1 April 2020 | Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma, Chronic Lymphocytic Leukemia | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia, neutropenia | [206,229] |
| 6 | Rituximab-abbs | TRUXIMA Celltrion, Inc., Incheon, Korea | FDA: 28 November 2018 EMA: 17 February 2017 | Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia | [230,231] |
| 7 | Mogamulizumab-kpkc | POTELIGEO Kyowa Kirin, Inc., Bedminster, NJ, USA | FDA: 8 August 2018 EMA: 22 November 2018 | Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CCR4 4 | Intravenous | Mycosis Fungoides, Sézary syndrome | Rash, infusion related reactions, fatigue, diarrhea, musculoskeletal pain, and upper respiratory tract infection | [232,233] |
| 8 | Elotuzumab | EMPLICITI Bristol-Myers Squibb Company and AbbVie, Lake Bluff, IL, USA | FDA: 30 November 2015 EMA: 11 May 2016 | Humanized immunoglobulin G1 (IgG1) monoclonal antibody | SLAMF7 5 | Intravenous | Multiple Myeloma | Fatigue, diarrhea, pyrexia, constipation, cough, peripheral neuropathy, nasopharyngitis, upper respiratory tract infection, decreased appetite, pneumonia | [234,235] |
| 9 | Daratumumab | DARZALEX Janssen Biotech, Inc., Horsham, PA, USA | FDA: 16 November 2015 EMA: 20 May 2016 | Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD38 3 | Intravenous | Multiple Myeloma | Infusion-related reactions, lymphopenia, neutropenia, thrombocytopenia, anemia | [236,237] |
| 10 | Nivolumab | OPDIVO Bristol-Myers Squibb Company, New York, NY, USA | FDA: 22 December 2014 EMA: 19 June 2015 | Human immunoglobulin G4 kappa (IgG4 κ) monoclonal antibody | PD-1 6 | Intravenous | Hodgkin’s Lymphoma | Fatigue, upper respiratory tract infection, pyrexia, diarrhea, cough | [238,239,240] |
| 11 | Pembrolizumab | KEYTRUDA Merck, Kenilworth, NJ, USA | FDA: 4 September 2014 EMA: 17 July 2015 | Humanized immunoglobulin G4 (IgG4) monoclonal antibody | PD-1 6 | Intravenous | Primary Mediastinal Large B-Cell Lymphoma, Hodgkin’s Lymphoma | Fatigue, cough, pruritus, nausea, rash, decreased appetite, constipation, arthralgia, diarrhea, anemia, hyperglycemia, hyponatremia, hypoalbuminemia, hypertriglyceridemia,, hypocalcaemia, elevated aspartate transaminase | [241,242,243,244] |
| 12 | Obinutuzumab | GAZYVA Genentech, Inc., South San Francisco, CA, USA | FDA: 1 November 2013 EMA: 23 July 2014. | Humanized immunoglobulin G1 (IgG1) monoclonal antibody | CD20 1 | Intravenous | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Infusion reactions, neutropenia | [245,246] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Blinatumomab | BLINCYTO Amgen Inc., Thousand Oaks, CA, USA | FDA: 3 December 2014 EMA: 23 November 2015 | Bispecific T-cell engaging (BiTE) antibody | CD19 1, CD3 2 | Intravenous | Acute Lymphoblastic Leukemia | Pyrexia, headache, peripheral edema, nausea, febrile neutropenia, hypokalemia, constipation, febrile neutropenia, pneumonia, device-related infection, tremor, encephalopathy, infection, overdose, confusion, Staphylococcal bacteremia, headache | [249,250] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Belantamab mafodotin-blmf | BLENREP GlaxoSmithKline, Brentford, England | FDA: 5 August 2020 EMA: 25 August 2020 |  | BCMA 1 | Intravenous | Multiple Myeloma | Ocular toxicity, thrombocytopenia, infusion-related reactions, gastrointestinal disorders, pyrexia, fatigue | [265,266] |
| 2 | Polatuzumab vedotin-piiq | POLIVY Genentech, Inc., South San Francisco, CA, USA | FDA: 10 June 2019 EMA: 16 January 2020 |  | CD79b 2 | Intravenous | Diffuse Large B-Cell Lymphoma | Cytopenias | [262,267,268] |
| 3 | Inotuzumab ozogamicin | BESPONSA Pfizer Inc., New York, NY, USA | FDA: 17 August 2017 EMA: 29 June 2017 |  | CD22 3 | Intravenous | Acute Lymphoblastic Leukemia | Cytopenias (including febrile neutropenia), infections, nausea, pyrexia, abnormal liver function and venoocclusive liver disease | [269,270] |
| 4 | Brentuximab vedotin | ADCETRIS Seattle Genetics, Inc., Bothell, WA, USA | FDA: 19 August 2011 EMA: 25 October 2012 |  | CD30 4 | Intravenous | Lymphoma, Hodgkin’s Lymphoma, Mycosis Fungoides | Neutropenia, anemia, peripheral sensory neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia | [260,271,272] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Moxetumomab pasudotox-tdfk | LUMOXITI AstraZeneca, Cambridge, England | FDA: 13 September 2018 EMA: Not approved | Murine immunoglobulin and PE38 conjugate | CD22 1 | Intravenous | Hairy Cell Leukemia | Peripheral edema, nausea, fatigue, headache, pyrexia, decreased lymphocyte count, hemolytic uremic syndrome, | [276,277] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Calaspargase pegol-mknl | ASPARLAS Servier Pharmaceuticals, Boston, MA, USA | FDA: 20 December 2018 EMA: Not approved | Asparagine-specific enzyme | Asparagine level | Intravenous | Acute Lymphoblastic Leukemia | Elevated transaminase, bilirubin increased, pancreatitis, abnormal clotting studies | [280] |
| 2 | Asparaginase Erwinia chrysanthemi | ERWINAZE Jazz Pharmaceuticals plc, Dublin, Ireland | FDA: 18 November 2011 EMA: nationally authorized | Asparagine-specific enzyme | Asparagine level | Intravenous | Acute Lymphoblastic Leukemia | Anaphylaxis, pancreatitis, abnormal transaminases, thrombosis, hemorrhage, nausea, vomiting, hyperglycemia. | [283,284,285] |
| No. | Generic Name of Drug | Brand Name and Company | First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Brexucabtagene autoleucel | TECARTUS Kite, a Gilead Company, Foster City, CA, USA | FDA: 24 July 2020 EMA: 14 December 2020 | Genetically modified autologous T cells | CD19 1 | Intravenous | Mantle Cell Lymphoma | Cytokine release syndrome, cytopenias, hypotension, encephalopathy, fever, fatigue, tachycardia, arrhythmia, infection with pathogen unspecified, chills, hypoxia, cough, tremor, musculoskeletal pain, headache, nausea, edema, motor dysfunction, constipation, diarrhea, decreased appetite, dyspnea, rash, insomnia, pleural effusion, aphasia | [290,294,295] |
| 2 | Axicabtagene ciloleucel | YESCARTA Kite Pharma, Inc., Los Angeles, CA, USA | FDA: 18 October 2017 EMA: 23 August 2018 | Genetically modified autologous T cells | CD19 1 | Intravenous | Large B-Cell Lymphoma, Follicular Lymphoma | Cytokine release syndrome, fever, hypotension, encephalopathy, tachycardia, fatigue, headache, febrile neutropenia, nausea, infections with pathogen unspecified, decreased appetite, chills, diarrhea, tremor, musculoskeletal pain, cough, hypoxia, constipation, vomiting, arrhythmias, dizziness | [296,297] |
| 3 | Tisagenlecleucel | KYMRIAH Novartis Pharmaceuticals Corporation, Basel, Switzerland | FDA: 30 August 2017 EMA: 23 August 2018 | Genetically modified autologous T cells | CD19 1 | Intravenous | Acute Lymphoblastic Leukemia, Large B-Cell Lymphoma | Cytokine release syndrome, infections-pathogen unspecified, pyrexia, decreased appetite, hypogammaglobinemia, headache, encephalopathy, hypotension, bleeding, episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, edema, cough, delirium | [298,299] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review. Cancers 2022, 14, 87. https://doi.org/10.3390/cancers14010087
Sochacka-Ćwikła A, Mączyński M, Regiec A. FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review. Cancers. 2022; 14(1):87. https://doi.org/10.3390/cancers14010087
Chicago/Turabian StyleSochacka-Ćwikła, Aleksandra, Marcin Mączyński, and Andrzej Regiec. 2022. "FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review" Cancers 14, no. 1: 87. https://doi.org/10.3390/cancers14010087
APA StyleSochacka-Ćwikła, A., Mączyński, M., & Regiec, A. (2022). FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review. Cancers, 14(1), 87. https://doi.org/10.3390/cancers14010087