
BRG1 and NPM-ALK Are Co-Regulated in Anaplastic Large-Cell Lymphoma; BRG1 Is a Potential Therapeutic Target in ALCL

 , , , ,
, , , ,  ,
,  ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Inhibitors and Antibodies
2.3. Primary Patient Samples and Immunohistochemistry
2.4. Mice
2.5. SDS-PAGE and Western Blot
2.6. RNAseq
2.7. Isolation of RNA
2.8. Synthesis of cDNA
2.9. Quantitative Real-Time PCR Analysis
2.10. Lentiviral Transduction of shRNA
2.11. Viability and Caspase Assays
3. Results
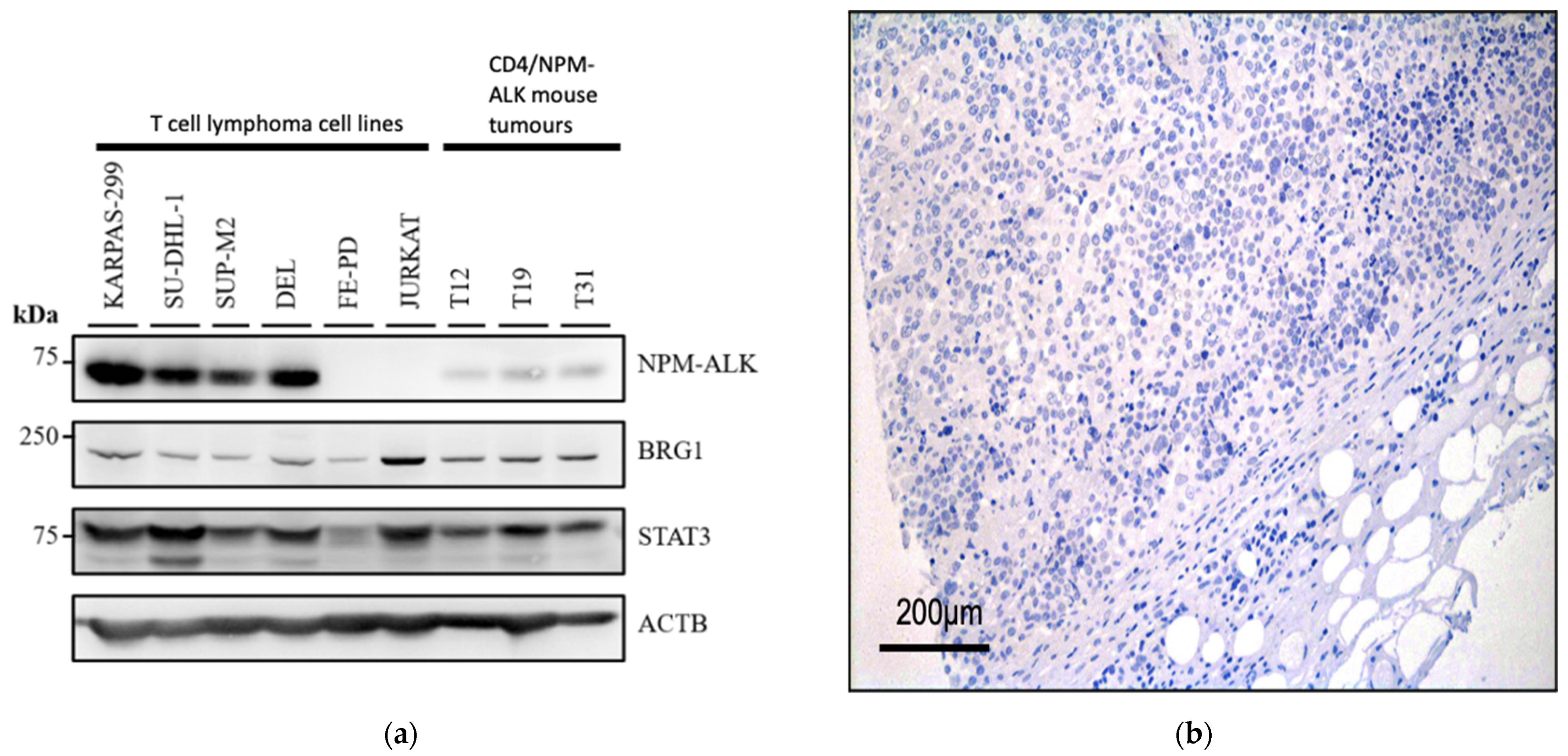
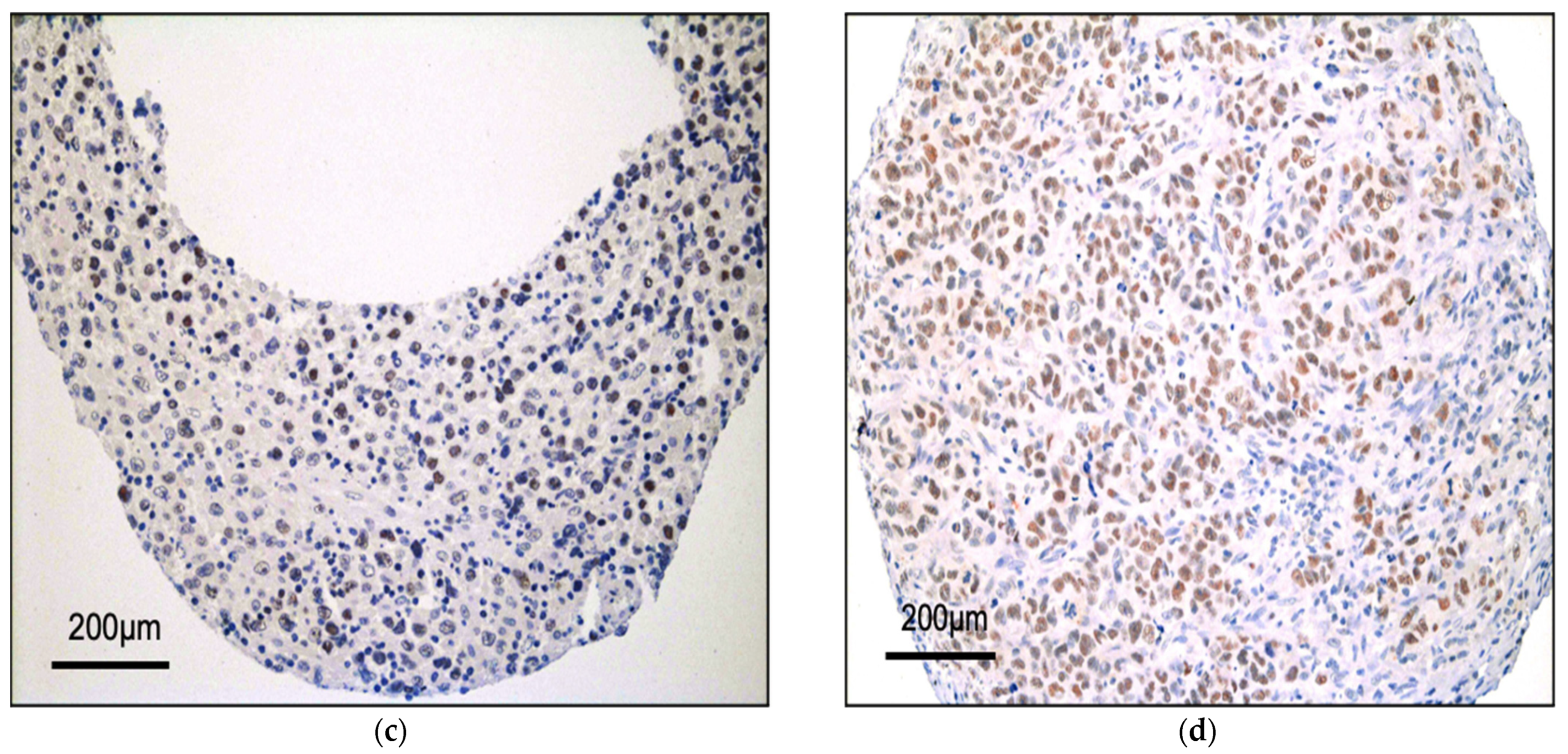
3.1. BRG1 Is Expressed in ALK+ ALCL and Other Peripheral T-Cell Lymphomas
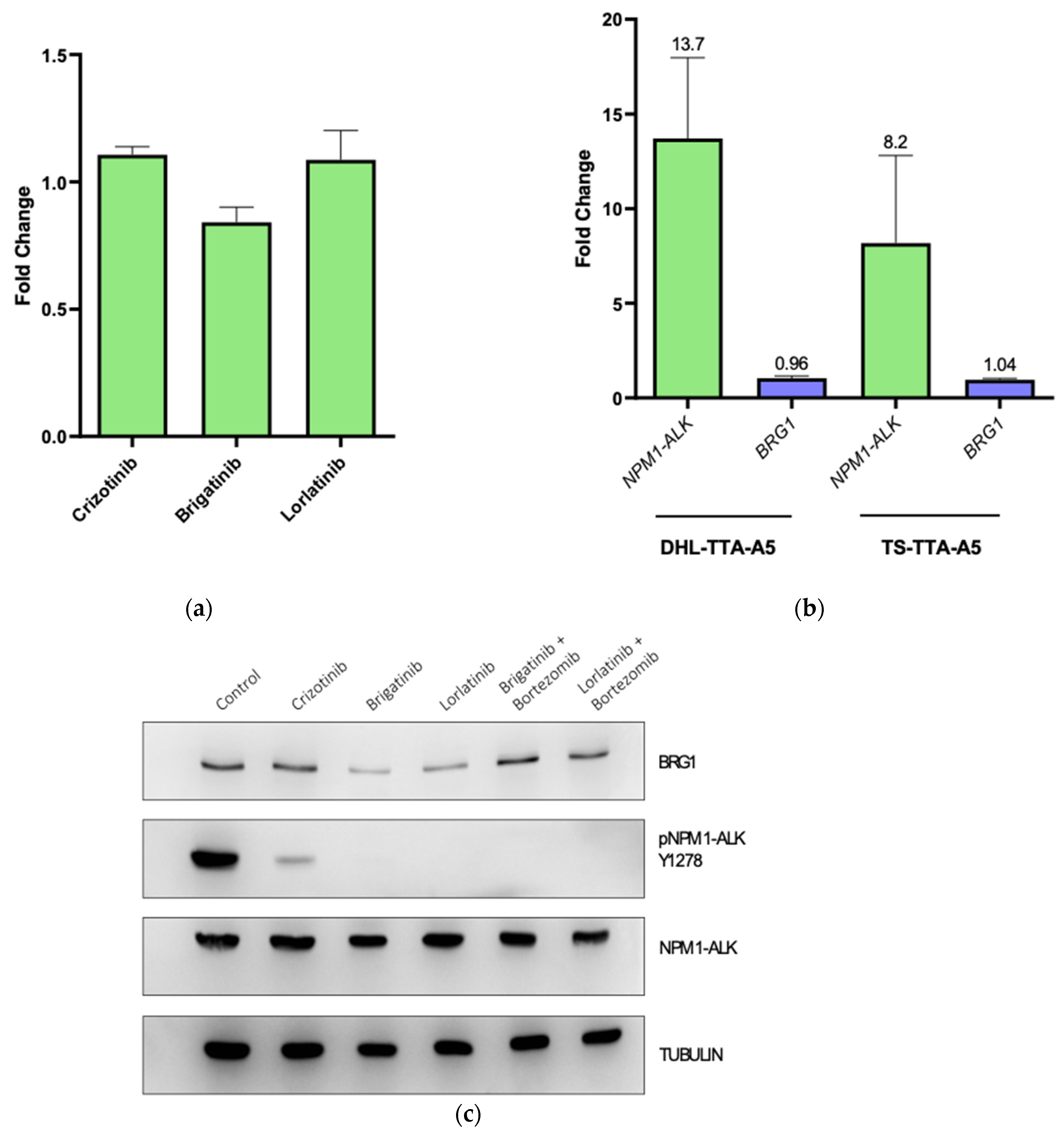
3.2. NPM-ALK Regulates Expression of BRG1 by Protecting It from Proteasomal Degradation
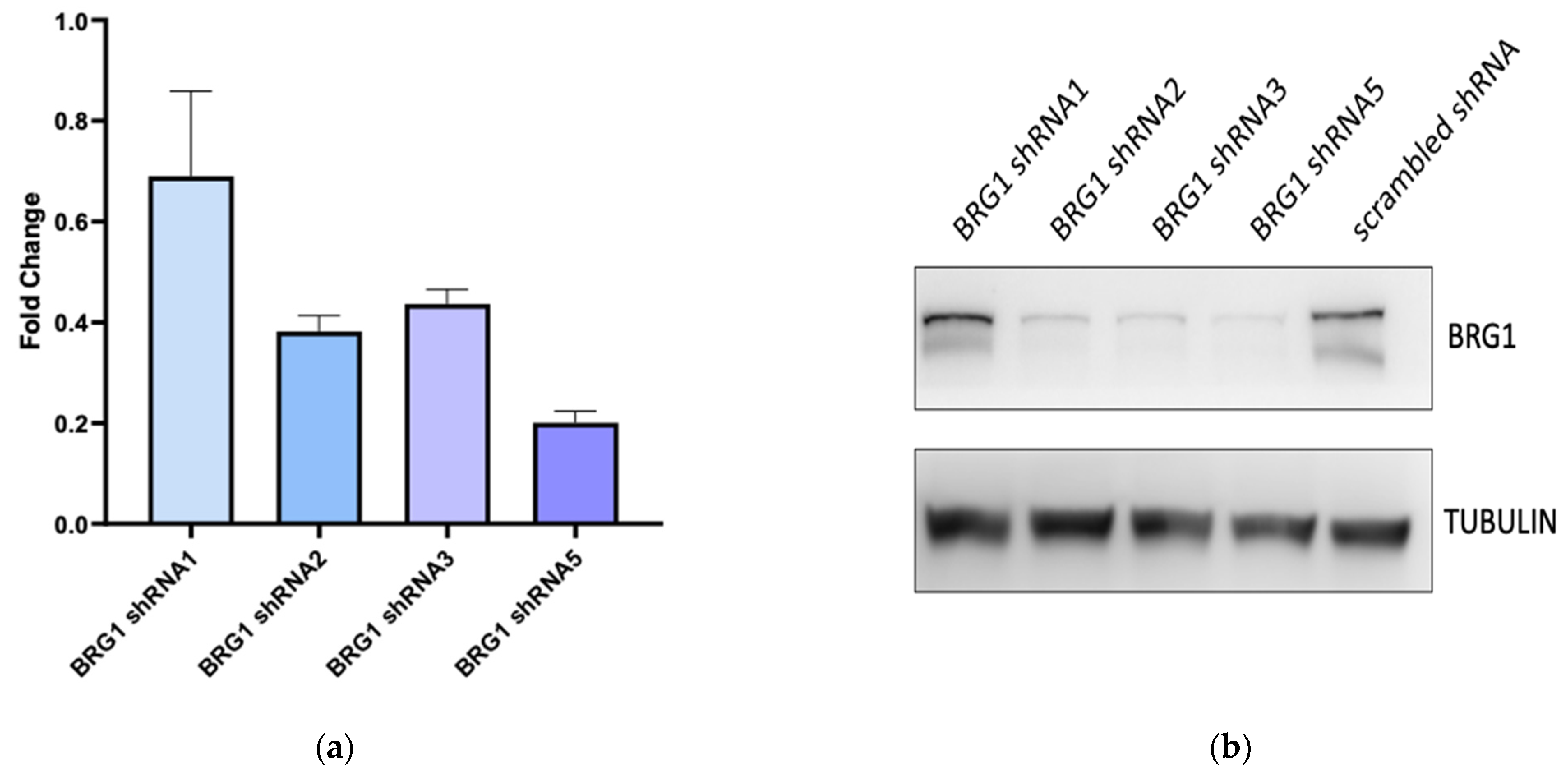
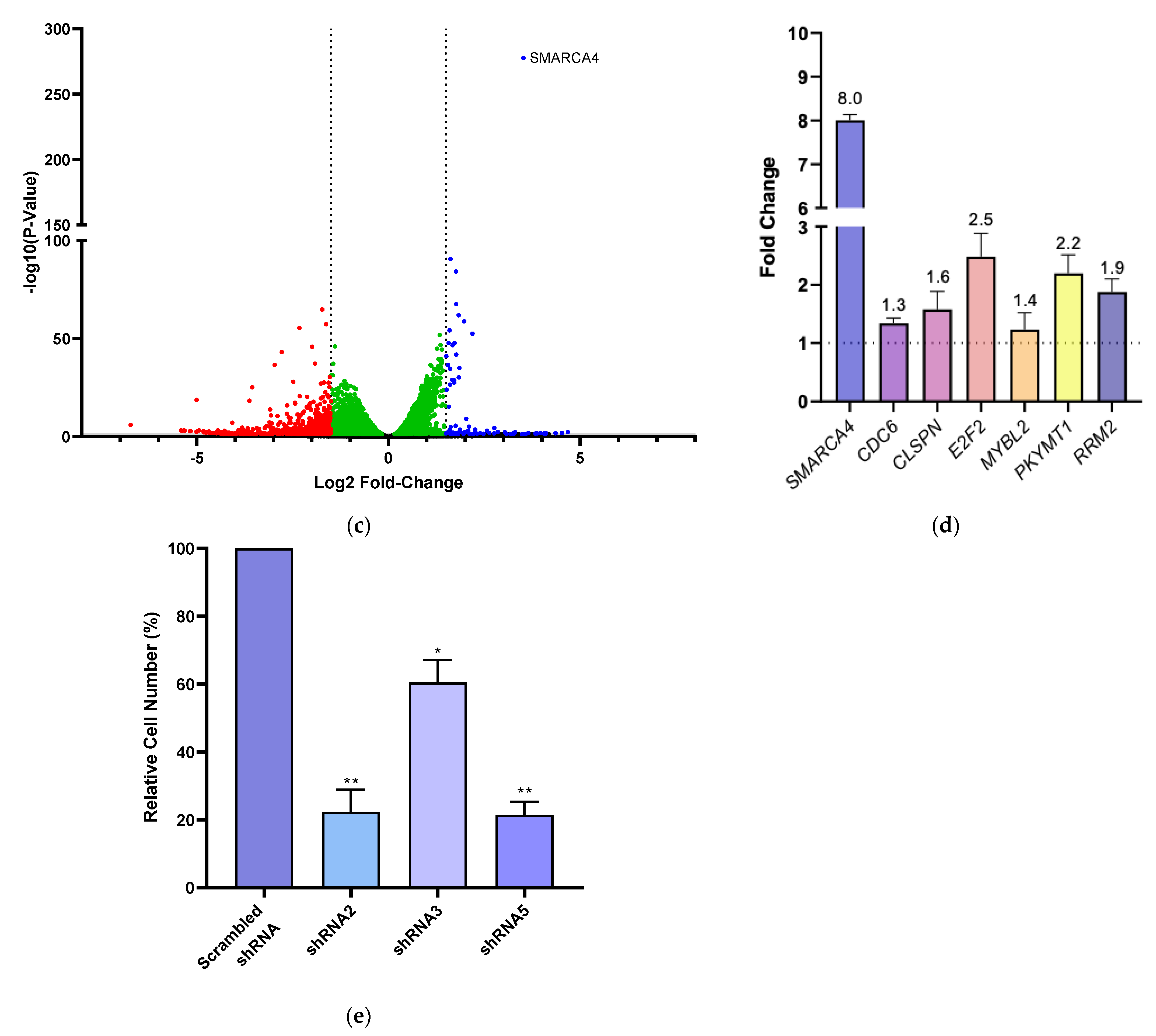
3.3. BRG1 Induces Transcription of Genes Involved in Cell Cycle Progression and Its Inhibition Results in a Loss of Cell Viability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinney, M.C.; Higgins, R.A.; Medina, E.A. Anaplastic large cell lymphoma: Twenty-five years of discovery. Arch. Pathol. Lab. Med. 2011, 135, 19–43. [Google Scholar] [CrossRef] [PubMed]
- Brugières, L.; Le Deley, M.C.; Rosolen, A.; Williams, D.; Horibe, K.; Wrobel, G.; Mann, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: Results of a randomized trial of the EICNHL Group. J. Clin. Oncol. 2009, 27, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Le Deley, M.C.; Rosolen, A.; Williams, D.M.; Horibe, K.; Wrobel, G.; Attarbaschi, A.; Zsiros, J.; Uyttebroeck, A.; Marky, I.M.; Lamant, L.; et al. Vinblastine in children and adolescents with high-risk anaplastic large-cell lymphoma: Results of the randomized ALCL99-vinblastine trial. J. Clin. Oncol. 2010, 28, 3987–3993. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Bischof, D.; Pulford, K.; Mason, D.Y.; Morris, S.W. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol. Cell Biol. 1997, 17, 2312–2325. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Turner, S.D.; Merz, H.; Yeung, D.; Alexander, D.R. CD2 promoter regulated nucleophosmin-anaplastic lymphoma kinase in transgenic mice causes B lymphoid malignancy. Anticancer Res. 2006, 26, 3275–3279. [Google Scholar]
- Turner, S.D.; Tooze, R.; Maclennan, K.; Alexander, D.R. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene 2003, 22, 7750–7761. [Google Scholar] [CrossRef]
- Giuriato, S.; Foisseau, M.; Dejean, E.; Felsher, D.W.; Al Saati, T.; Demur, C.; Ragab, A.; Kruczynski, A.; Schiff, C.; Delsol, G.; et al. Conditional TPM3-ALK and NPM-ALK transgenic mice develop reversible ALK-positive early B-cell lymphoma/leukemia. Blood 2010, 115, 4061–4070. [Google Scholar] [CrossRef] [PubMed]
- Jäger, R.; Hahne, J.; Jacob, A.; Egert, A.; Schenkel, J.; Wernert, N.; Schorle, H.; Wellmann, A. Mice transgenic for NPM-ALK develop non-Hodgkin lymphomas. Anticancer Res. 2005, 25, 3191–3196. [Google Scholar] [PubMed]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Simmons, W.J.; Dhall, G.; Howes, J.; Piva, R.; et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Crockett, D.K.; Lin, Z.; Elenitoba-Johnson, K.S.; Lim, M.S. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene 2004, 23, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, T.I.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.; Brugieres, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef]
- Prokoph, N.; Probst, N.A.; Lee, L.C.; Monahan, J.M.; Matthews, J.D.; Liang, H.C.; Bahnsen, K.; Montes-Mojarro, I.A.; Karaca-Atabay, E.; Sharma, G.G.; et al. IL10RA Modulates Crizotinib Sensitivity in NPM1-ALK-positive Anaplastic Large Cell Lymphoma. Blood 2020, 136, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Pellegrino, E.; Mattioli, M.; Agnelli, L.; Lombardi, L.; Boccalatte, F.; Costa, G.; Ruggeri, B.A.; Cheng, M.; Chiarle, R.; et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Investig. 2006, 116, 3171–3182. [Google Scholar] [CrossRef]
- Piva, R.; Chiarle, R.; Manazza, A.D.; Taulli, R.; Simmons, W.; Ambrogio, C.; D’Escamard, V.; Pellegrino, E.; Ponzetto, C.; Palestro, G.; et al. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood 2006, 107, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, M.F.D.; Betz, B.L.; Rorie, C.J.; Reisman, D.N.; Wang, W.; Weissman, B.E. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol. 2001, 186, 136–145. [Google Scholar] [CrossRef]
- Reisman, D.N.; Sciarrotta, J.; Wang, W.; Funkhouser, W.K.; Weissman, B.E. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: Correlation with poor prognosis. Cancer Res. 2003, 63, 560–566. [Google Scholar] [PubMed]
- Laimer, D.; Dolznig, H.; Kollmann, K.; Vesely, P.W.; Schlederer, M.; Merkel, O.; Schiefer, A.I.; Hassler, M.R.; Heider, S.; Amenitsch, L.; et al. IPDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat. Med. 2012, 18, 1699–1704. [Google Scholar] [CrossRef]
- Fukuoka, J.; Fujii, T.; Shih, J.H.; Dracheva, T.; Meerzaman, D.; Player, A.; Hong, K.; Settnek, S.; Gupta, A.; Buetow, K.; et al. Chromatin remodeling factors and BRM/BRG1 expression as prognostic indicators in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 4314–4324. [Google Scholar] [CrossRef]
- Reisman, D.N.; Strobeck, M.W.; Betz, B.L.; Sciariotta, J.; Funkhouser, W., Jr.; Murchardt, C.; Yaniv, M.; Sherman, L.S.; Knudsen, E.S.; Weissman, B.E. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: Differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene 2002, 21, 1196–1207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bultman, S.J.; Herschkowitz, J.I.; Godfrey, V.; Gebuhr, T.C.; Yaniv, M.; Perou, C.M.; Magnuson, T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 2008, 27, 460–468. [Google Scholar] [CrossRef]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Glaros, S.; Cirrincione, G.M.; Palanca, A.; Metzger, D.; Reisman, D. Targeted knockout of BRG1 potentiates lung cancer development. Cancer Res. 2008, 68, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Reisman, D.N.; Sciarrotta, J.; Bouldin, T.W.; Weissman, B.E.; Funkhouser, W.K. The expression of the SWI/SNF ATPase subunits BRG1 and BRM in normal human tissues. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.R.; Love, I.M.; Imbalzano, A.N.; Grossman, S.R.; Androphy, E.J. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene 2009, 28, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Tando, T.; Ishizaka, A.; Watanabe, H.; Ito, T.; Iida, S.; Haraguchi, T.; Mizutani, T.; Izumi, T.; Isobe, T.; Akiyama, T.; et al. Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-kappaB pathway. J. Biol. Chem. 2010, 285, 21951–21960. [Google Scholar] [CrossRef] [PubMed]
- Motegi, A.; Fujimoto, J.; Kotani, M.; Sakuraba, H.; Yamamoto, T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J. Cell Sci. 2004, 117, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Mazot, P.; Cazes, A.; Dingli, F.; Degoutin, J.; Irinopoulou, T.; Boutterin, M.C.; Lombard, B.; Loew, D.; Hallberg, B.; Palmer, R.H.; et al. Internalization and down-regulation of the ALK receptor in neuroblastoma cell lines upon monoclonal antibodies treatment. PLoS ONE 2012, 7, e33581. [Google Scholar] [CrossRef]
- Wu, C.; Molavi, O.; Zhang, H.; Gupta, N.; Alshareef, A.; Bone, K.M.; Gopal, K.; Wu, F.; Lewis, J.T.; Douglas, D.N.; et al. STAT1 is phosphorylated and downregulated by the oncogenic tyrosine kinase NPM-ALK in ALK-positive anaplastic large-cell lymphoma. Blood 2015, 126, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Dalla Rosa, H.; Vignes, N.; Rosolen, A. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: Role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 2004, 64, 3256–3264. [Google Scholar] [CrossRef][Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′ to 3′) |
|---|---|
| BRG1 Forward | TGCTGCGGCCCTTCTTGCTC |
| BRG1 Reverse | GGTGCCGCCTTTGCCCTTCT |
| CDC6 Forward | ACCTATGCAACACTCCCCATT |
| CDC6 Reverse | TGGCTAGTTCTCTTTTGCTAGGA |
| CLSPN Forward | AAGACAGTGATTCCGAAACAGAG |
| CLSPN Reverse | TGCGCTTCAAGATTTTCCTGA |
| E2F2 Forward | CGTCCCTGAGTTCCCAACC |
| E2F2 Reverse | GCGAAGTGTCATACCGAGTCTT |
| MYBL2 Forward | CCGGAGCAGAGGGATAGCA |
| MYBL2 Reverse | CAGTGCGGTTAGGGAAGTGG |
| PKYMT2 Forward | GCCTGCCAACATCTTCCTG |
| PKYMT2 Reverse | CCCAGACTGAACACATCCGC |
| RRM2 Forward | CACGGAGCCGAAAACTAAAGC |
| RRM2 Reverse | TCTGCCTTCTTATACATCTGCCA |
| NPM-ALK Forward | CTGTACAGCCAACGGTTTCCC |
| NPM-ALK Reverse | GGCCCAGACCCGAATGAGG |
| GAPDH Forward | CCACTCCTCCACCTTTGAC |
| GAPDH Reverse | ACCCTGTTGCTGTAGCCA |
| Mouse GAPDH Forward | CATCACTGCCACCCAGAAGACTG |
| Mouse GAPDH Reverse | ATGCCAGTGAGCTTCCCGTTCAG |
| Mouse BRG1 Forward | GAAAGTGGCTCTGAAGAGGAGG |
| Mouse BRG1 Reverse | TCCACCTCAGAGACATCATCGC |
| Mouse HPRT Forward | CTGGTGAAAAGGACCTCTCGAAG |
| Mouse HPRT Reverse | CCAGTTTCACTAATGACACAAACG |
| Brg1 shRNA | TRC Construct | Sequence |
|---|---|---|
| 1 | TRCN0000015548 | CCATATTTATACAGCAGAGAACTCGAGTTCTCTGCTGTATAAATATGG |
| 2 | TRCN0000015549 | CCGGCCCGTGGACTTCAAGAAGATACTCGAGTATCTTCTTGAAGTCCACGGG |
| 3 | TRCN0000015550 | CCGGGCCAAGCAAGATGTCGATGATCTCGAGATCATCGACATCTTGCTTGGCTTTTT |
| 5 | TRCN0000015552 | CCGGCGGCAGACACTGTGATCATTTCTCGAGAAATGATCACAGTGTCTGCCGTTTTT |
| Pathway | Gene Count | Entities Total |
|---|---|---|
| Cell cycle | 12 | 734 |
| Cell cycle, mitotic | 10 | 596 |
| Cell cycle checkpoints | 6 | 280 |
| Mitotic G1 phase and G1/S transition | 8 | 173 |
| G2/M checkpoints | 5 | 154 |
| G1/S checkpoints | 5 | 150 |
| DNA replication | 3 | 142 |
| DNA replication pre-initiation | 3 | 88 |
| Apoptotic execution phase | 3 | 54 |
| Transcriptional regulation by E2F6 | 2 | 46 |
| Pathway | Gene Count | Entities Total |
|---|---|---|
| Reproduction | 8 | 123 |
| HDAC deacetylase histones | 7 | 67 |
| Amyloid fibre formation | 7 | 88 |
| Meiosis | 7 | 92 |
| RNA polymerase I promoter opening | 6 | 34 |
| DNA methylation | 6 | 36 |
| PRC2 methylates histones and DNA | 6 | 44 |
| SIRT1 negatively regulated rRNA expression | 6 | 45 |
| ERCC6 (CSB) and EHMT2 (G9a) positively regulate rRNA expression | 6 | 48 |
| Activated PKN1 stimulates transcription of Androgen Receptor (AR) regulated genes KLK2 and KLK3 | 6 | 49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garland, G.D.; Ducray, S.P.; Jahangiri, L.; Pucci, P.; Amos Burke, G.A.; Monahan, J.; Lai, R.; Merkel, O.; Schiefer, A.-I.; Kenner, L.; et al. BRG1 and NPM-ALK Are Co-Regulated in Anaplastic Large-Cell Lymphoma; BRG1 Is a Potential Therapeutic Target in ALCL. Cancers 2022, 14, 151. https://doi.org/10.3390/cancers14010151
Garland GD, Ducray SP, Jahangiri L, Pucci P, Amos Burke GA, Monahan J, Lai R, Merkel O, Schiefer A-I, Kenner L, et al. BRG1 and NPM-ALK Are Co-Regulated in Anaplastic Large-Cell Lymphoma; BRG1 Is a Potential Therapeutic Target in ALCL. Cancers. 2022; 14(1):151. https://doi.org/10.3390/cancers14010151
Chicago/Turabian StyleGarland, Gavin D., Stephen P. Ducray, Leila Jahangiri, Perla Pucci, G. A. Amos Burke, Jack Monahan, Raymond Lai, Olaf Merkel, Ana-Iris Schiefer, Lukas Kenner, and et al. 2022. "BRG1 and NPM-ALK Are Co-Regulated in Anaplastic Large-Cell Lymphoma; BRG1 Is a Potential Therapeutic Target in ALCL" Cancers 14, no. 1: 151. https://doi.org/10.3390/cancers14010151
APA StyleGarland, G. D., Ducray, S. P., Jahangiri, L., Pucci, P., Amos Burke, G. A., Monahan, J., Lai, R., Merkel, O., Schiefer, A.-I., Kenner, L., Bannister, A. J., & Turner, S. D. (2022). BRG1 and NPM-ALK Are Co-Regulated in Anaplastic Large-Cell Lymphoma; BRG1 Is a Potential Therapeutic Target in ALCL. Cancers, 14(1), 151. https://doi.org/10.3390/cancers14010151