
The Intricate Interplay between Cell Cycle Regulators and Autophagy in Cancer




Abstract
Simple Summary
Abstract
1. Introduction
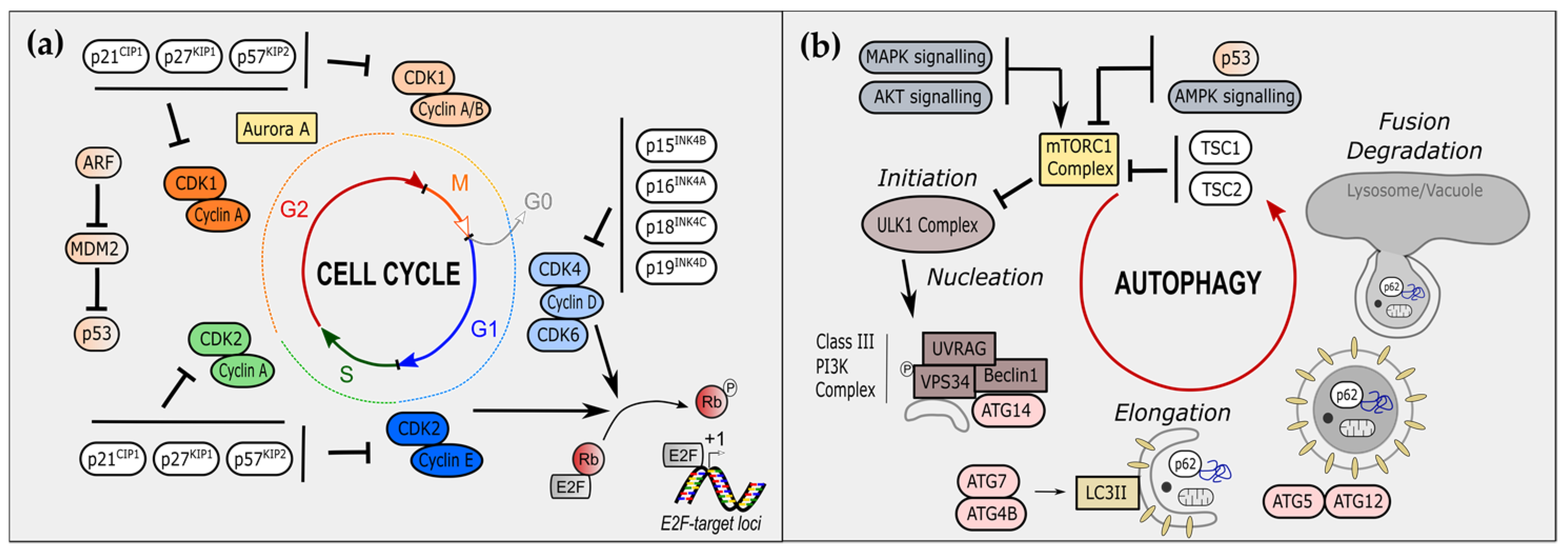
2. Cell-Cycle Regulators Modulate Autophagy
2.1. Cyclin-Dependent Kinase Inhibitors and Autophagy
2.1.1. The CIP/KIP Family and Autophagy
2.1.2. The INK Family and Autophagy
2.2. Cyclin-Dependent Kinases and Autophagy
2.2.1. S/G2/M Phases CDKs in the Control of Autophagy
2.2.2. G1 Phase CDKs in the Control of Autophagy
2.2.3. Non-Canonical CDKs and Autophagy
2.2.4. CDKs and Mitophagy
2.3. E2F Transcription Factors and Autophagy
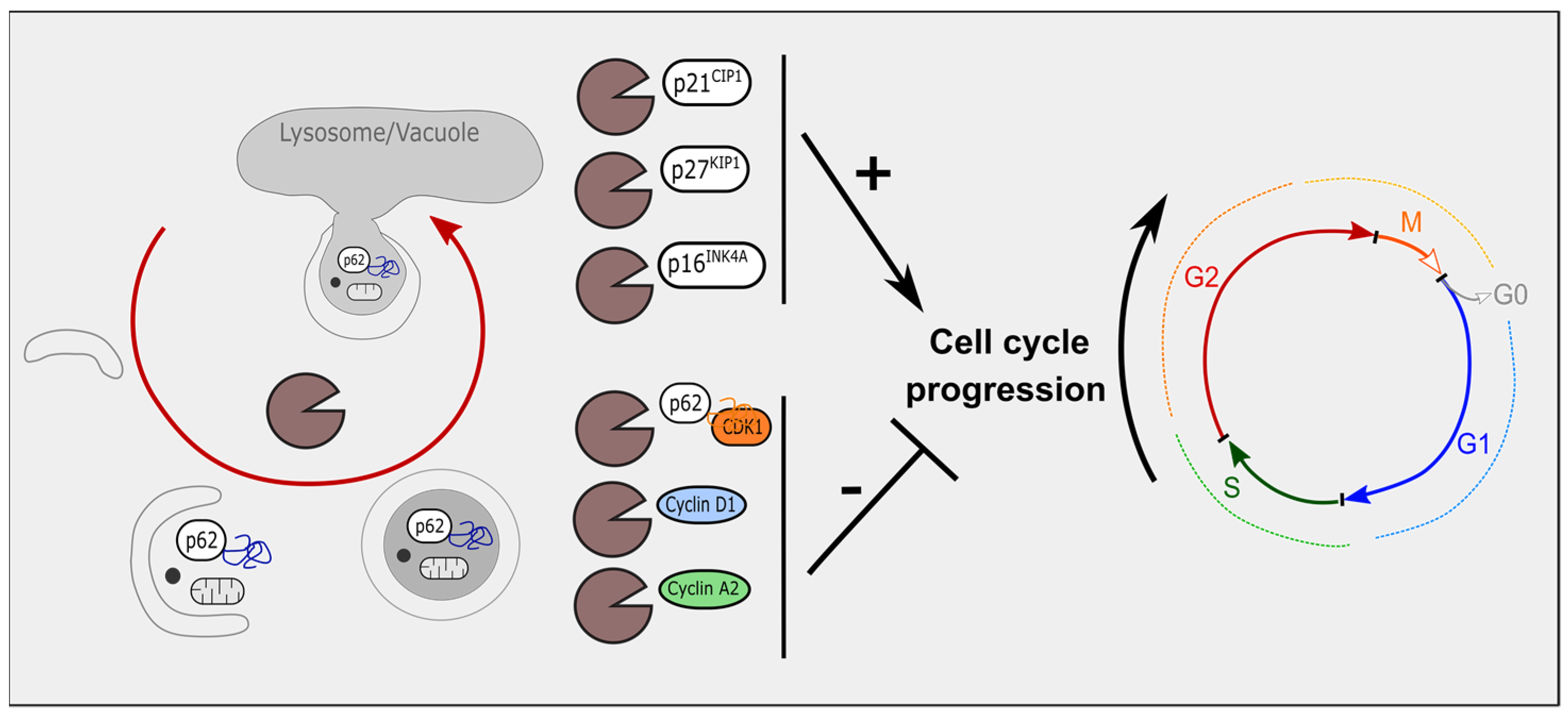
3. Autophagy Modulates Cell-Cycle Regulators
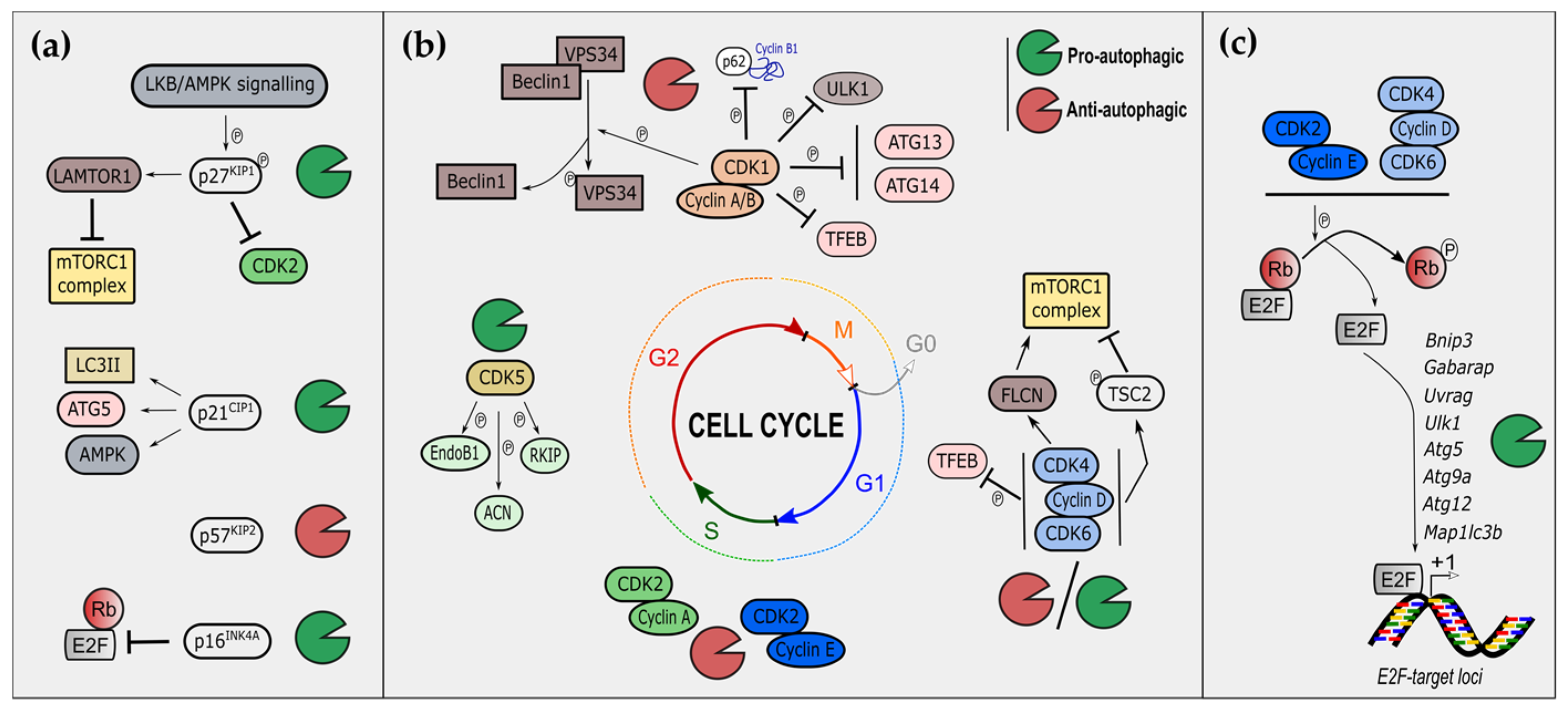
3.1. Autophagy and Cyclin-Dependent Kinase Inhibitors
3.2. Autophagy, Cyclin-Dependent Kinases and Cyclins
3.3. Mitophagy and Cell-Cycle Regulators
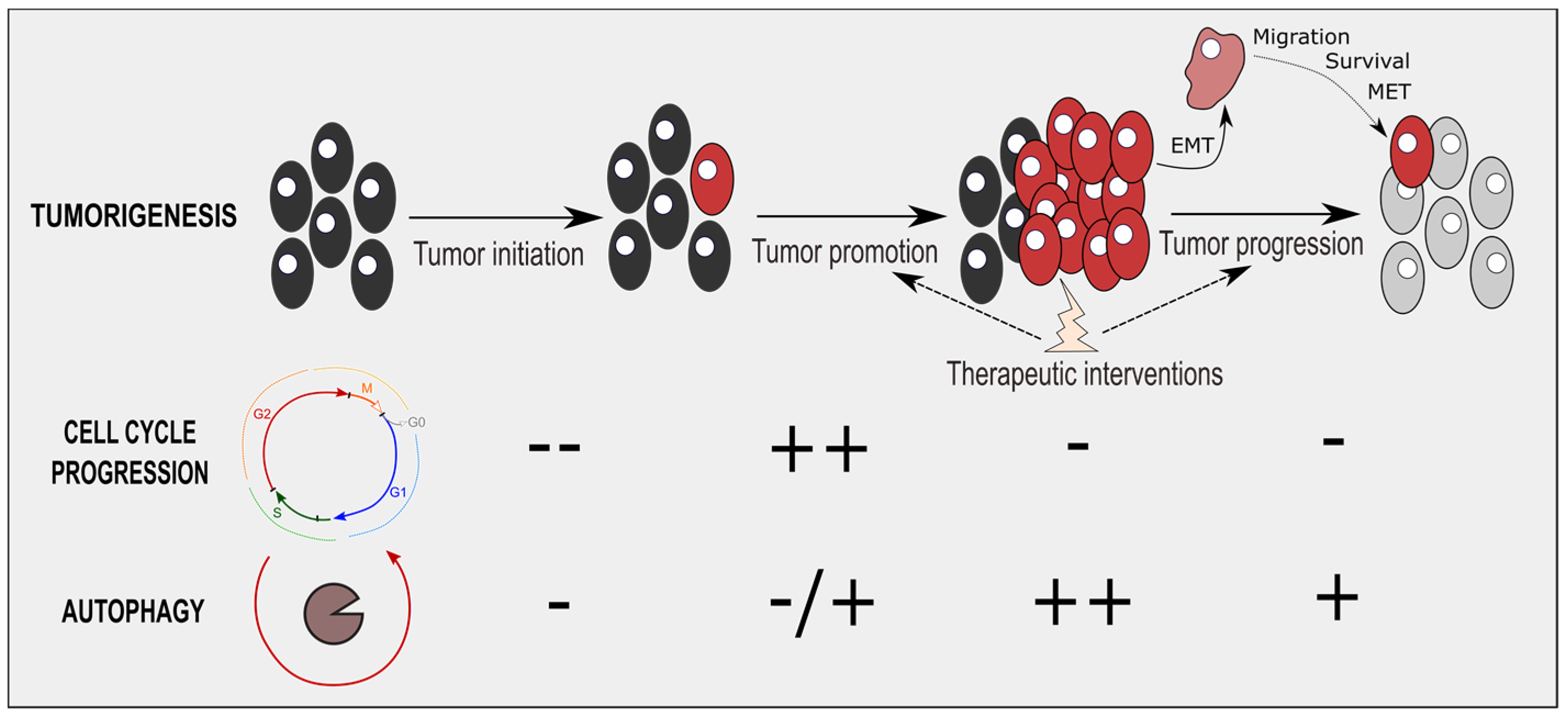
4. Therapeutic Interventions Combining Cell-Cycle and Autophagy Modulators
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACN-ACIN1 | Apoptotic chromatin condensation inducer 1 |
| AML | acute myeloid leukemia |
| AMPK | 5′-AMP-activated protein kinase |
| ATG | Autophagy Related |
| BAG3 | BCL2 associated athanogene 3 |
| BECN1 | Beclin 1 |
| BNIP3 | BCL2 adenovirus E1B 19-kDa protein-interacting protein 3 |
| CDK | Cyclin-dependent kinase |
| CEBPα | CCAAT enhancer binding brotein alpha, |
| CKI | Cyclin-dependent kinase inhibitor |
| CQ | Chloroquine |
| DIRAS3 | Distinct subgroup of the ras family member 3 |
| DNMT1 | DNA methyltransferase 1 |
| DRAM | DNA damage regulated autophagy modulator 1 |
| EGF | Epidermal growth factor |
| EMT | Epithelial–mesenchymal transition |
| EndoB1 | Endophilin B1 |
| FLCN | Folliculin |
| GABARAP | GABA type A receptor-associated protein |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GBM | Glioblastoma multiforme |
| HCQ | Hydroxychloroquine |
| HSP | Heat shock protein |
| IL | Interleukin |
| LAMTOR | Late endosomal/lysosomal adaptor |
| LKB1 | Liver kinase B1 |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Mouse double minute 2 |
| MET | Mesenchymal-epithelial transition |
| MMP | Matrix metallopeptidase |
| mTOR | Mechanistic target of rapamycin kinase |
| PDAC | pancreatic ductal adenocarcinoma |
| PDH | Pyruvate dehydrogenase |
| PDK | Pyruvate dehydrogenase kinase |
| PIK3C3 | Phosphatidylinositol 3-kinase catalytic subunit type 3 |
| PINK1 | PTEN induced kinase 1 |
| PKM | Pyruvate kinase M1/2 |
| RKIP-PEBP1 | Phosphatidylethanolamine binding brotein 1 |
| TFEB | Transcription factor EB |
| TFE3 | Transcription factor binding To IGHM enhancer 3 |
| TGFβ | Tumor suppressor transforming growth factor-β |
| ULK | Unc-51 Like autophagy activating Kinase |
| UVRAG | UV radiation resistance associated gene |
| VEGF | Vascular endothelial growth factor |
References
- Vermeulen, K.; Bockstaele, D.R.V.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, V.; Fajas, L. Cycling through metabolism. EMBO Mol. Med. 2010, 2, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165715. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Carujo, S.; Estanyol, J.M.; Ejarque, A.; Agell, N.; Bachs, O.; Pujol, M.J. Glyceraldehyde 3-phosphate dehydrogenase is a SET-binding protein and regulates cyclin B-cdk1 activity. Oncogene 2006, 25, 4033–4042. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3343–3347. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.-D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336-349.e6. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 13, 2163–2170. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Eskelinen, E.-L.; Prescott, A.R.; Cooper, J.; Brachmann, S.M.; Wang, L.; Tang, X.; Backer, J.M.; Lucocq, J.M. Inhibition of autophagy in mitotic animal cells. Traffic 2002, 3, 878–893. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Abdi, A.; Zhivotovsky, B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy 2011, 7, 83–90. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Tajeddine, N.; Vitale, I.; Criollo, A.; Vicencio, J.M.; Hickman, J.A.; Geneste, O.; Kroemer, G. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle Georget. Tex 2007, 6, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef]
- Li, Z.; Tian, X.; Ji, X.; Wang, J.; Chen, H.; Wang, D.; Zhang, X. ULK1-ATG13 and their mitotic phospho-regulation by CDK1 connect autophagy to cell cycle. PLoS Biol. 2020, 18, e3000288. [Google Scholar] [CrossRef] [PubMed]
- Nowosad, A.; Jeannot, P.; Callot, C.; Creff, J.; Perchey, R.T.; Joffre, C.; Codogno, P.; Manenti, S.; Besson, A. p27 controls Ragulator and mTOR activity in amino acid-deprived cells to regulate the autophagy–lysosomal pathway and coordinate cell cycle and cell growth. Nat. Cell Biol. 2020, 22, 1076–1090. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Komata, T.; Kanzawa, T.; Takeuchi, H.; Germano, I.M.; Schreiber, M.; Kondo, Y.; Kondo, S. Antitumour effect of cyclin-dependent kinase inhibitors (p16INK4A, p18INK4C, p19INK4D, p21WAF1/CIP1 and p27KIP1) on malignant glioma cells. Br. J. Cancer 2003, 88, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.K.; White, J.P. The AMPK/p27Kip1 Pathway as a Novel Target to Promote Autophagy and Resilience in Aged Cells. Cells 2021, 10, 1430. [Google Scholar] [CrossRef]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the p27 E3 ligase SCFSkp2 results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Billin, A.N.; Campbell, M.E.; Russell, A.J.; Huffman, K.M.; Kraus, W.E. The AMPK/p27Kip1 Axis Regulates Autophagy/Apoptosis Decisions in Aged Skeletal Muscle Stem Cells. Stem Cell Rep. 2018, 11, 425–439. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, P.; Preet, R.; Das, D.; Satapathy, S.R.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine-mediated autophagy and apoptosis in colon cancer cells is through a p53- and p21-dependent mechanism. Oncol. Res. 2012, 20, 81–91. [Google Scholar] [CrossRef]
- Manu, K.A.; Cao, P.H.A.; Chai, T.F.; Casey, P.J.; Wang, M. p21cip1/waf1 Coordinates Autophagy, Proliferation and Apoptosis in Response to Metabolic Stress. Cancers 2019, 11, 1112. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wan, C.-X.; Huang, S.-H.; Wang, H.-B.; Fan, D.; Wu, H.-M.; Wu, Q.-Q.; Ma, Z.-G.; Deng, W.; Tang, Q.-Z. Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy. Cell Death Dis. 2019, 10, 403. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, M.; Liu, L.; Yang, J.; Wang, H.; Wan, C.; Deng, W.; Tang, Q. Autophagy is involved in the protective effect of p21 on LPS-induced cardiac dysfunction. Cell Death Dis. 2020, 11, 554. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Y.; Li, Q.; Jing, L.; Wu, T.; Han, L.-L.; Wang, Y.; Yu, S.-Z.; Nan, K.-J.; Guo, H. P57-mediated autophagy promotes the efficacy of EGFR inhibitors in hepatocellular carcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 147–157. [Google Scholar] [CrossRef]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Martin, V.; Gomez-Manzano, C.; Johnson, D.G.; Alonso, M.; White, E.; Xu, J.; McDonnell, T.J.; Shinojima, N.; Fueyo, J. The RB-E2F1 Pathway Regulates Autophagy. Cancer Res. 2010, 70, 7882–7893. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Cassidy, L.D.; Young, A.R.J.; Young, C.N.J.; Soilleux, E.J.; Fielder, E.; Weigand, B.M.; Lagnado, A.; Brais, R.; Ktistakis, N.T.; Wiggins, K.A.; et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat. Commun. 2020, 11, 307. [Google Scholar] [CrossRef]
- Coryell, P.R.; Goraya, S.K.; Griffin, K.A.; Redick, M.A.; Sisk, S.R.; Purvis, J.E. Autophagy regulates the localization and degradation of p16INK4a. Aging Cell 2020, 19, e13171. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell. Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef]
- Furuya, T.; Kim, M.; Lipinski, M.; Li, J.; Kim, D.; Lu, T.; Shen, Y.; Rameh, L.; Yankner, B.; Tsai, L.-H.; et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol. Cell 2010, 38, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Amanchy, R.; Diaz-Meco, M.T.; Moscat, J. Phosphorylation of p62 by cdk1 Controls the Timely Transit of Cells through Mitosis and Tumor Cell Proliferation. Mol. Cell. Biol. 2011, 31, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Odle, R.I.; Walker, S.A.; Oxley, D.; Kidger, A.M.; Balmanno, K.; Gilley, R.; Okkenhaug, H.; Florey, O.; Ktistakis, N.T.; Cook, S.J. An mTORC1-to-CDK1 Switch Maintains Autophagy Suppression during Mitosis. Mol. Cell 2020, 77, 228-240.e7. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, M.; Vernier, M.; Mignacca, L.; Lessard, F.; Huot, G.; Moiseeva, O.; Bourdeau, V.; Ferbeyre, G. A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer Res. 2016, 76, 3252–3264. [Google Scholar] [CrossRef]
- Iriyama, N.; Hino, H.; Moriya, S.; Hiramoto, M.; Hatta, Y.; Takei, M.; Miyazawa, K. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk. Lymphoma 2018, 59, 1439–1450. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Martínez-Carreres, L.; Puyal, J.; Leal-Esteban, L.C.; Orpinell, M.; Castillo-Armengol, J.; Giralt, A.; Dergai, O.; Moret, C.; Barquissau, V.; Nasrallah, A.; et al. CDK4 Regulates Lysosomal Function and mTORC1 Activation to Promote Cancer Cell Survival. Cancer Res. 2019, 79, 5245–5259. [Google Scholar] [CrossRef]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar] [CrossRef]
- Yin, Q.; Jian, Y.; Xu, M.; Huang, X.; Wang, N.; Liu, Z.; Li, Q.; Li, J.; Zhou, H.; Xu, L.; et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J. Cell Biol. 2020, 219, e201911036. [Google Scholar] [CrossRef] [PubMed]
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef]
- Wong, A.S.L.; Lee, R.H.K.; Cheung, A.Y.; Yeung, P.K.; Chung, S.K.; Cheung, Z.H.; Ip, N.Y. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 2011, 13, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Shu, Y.; Gao, C.; Wang, X.; Qi, G.; Zhang, P.; Li, M.; Shi, J.; Tian, B. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2870–2880. [Google Scholar] [CrossRef]
- Su, L.-Y.; Li, H.; Lv, L.; Feng, Y.-M.; Li, G.-D.; Luo, R.; Zhou, H.-J.; Lei, X.-G.; Ma, L.; Li, J.-L.; et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy 2015, 11, 1745–1759. [Google Scholar] [CrossRef]
- Nandi, N.; Tyra, L.K.; Stenesen, D.; Krämer, H. Stress-induced Cdk5 activity enhances cytoprotective basal autophagy in Drosophila melanogaster by phosphorylating acinus at serine437. eLife 2017, 6, e30760. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chow, H.-M.; Liu, Y.; Wu, D.; Shi, M.; Li, J.; Wen, L.; Gao, Y.; Chen, G.; Zhuang, K.; et al. Cyclin-Dependent Kinase 5–Dependent BAG3 Degradation Modulates Synaptic Protein Turnover. Biol. Psychiatry 2020, 87, 756–769. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Paparidis, N.F.; Canduri, F. The Emerging Picture of CDK11: Genetic, Functional and Medicinal Aspects. Curr. Med. Chem. 2018, 25, 880–888. [Google Scholar] [CrossRef]
- Wilkinson, S.; Croft, D.R.; O’Prey, J.; Meedendorp, A.; O’Prey, M.; Dufès, C.; Ryan, K.M. The cyclin-dependent kinase PITSLRE/CDK11 is required for successful autophagy. Autophagy 2011, 7, 1295–1301. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Wang, C.; Li, Z.; Lu, Y.; Du, R.; Katiyar, S.; Yang, J.; Fu, M.; Leader, J.E.; Quong, A.; Novikoff, P.M.; et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc. Natl. Acad. Sci. USA 2006, 103, 11567–11572. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Ciro, M.; Cocito, A.; Helin, K. E2F target genes: Unraveling the biology. Trends Biochem. Sci. 2004, 29, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Balciunaite, E.; Blais, A.; Spektor, A.; Scarpulla, R.C.; Young, R.; Kluger, Y.; Dynlacht, B.D. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 2004, 16, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Mestres-Arenas, A.; Fajas, L.; Leal-Esteban, L.C. The multifaceted role of cell cycle regulators in the coordination of growth and metabolism. FEBS J. 2021, 288, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar] [CrossRef]
- Weinmann, A.S.; Bartley, S.M.; Zhang, T.; Zhang, M.Q.; Farnham, P.J. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell. Biol. 2001, 21, 6820–6832. [Google Scholar] [CrossRef]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef]
- Ruan, C.; Wang, C.; Gong, X.; Zhang, Y.; Deng, W.; Zhou, J.; Huang, D.; Wang, Z.; Zhang, Q.; Guo, A.; et al. An integrative multi-omics approach uncovers the regulatory role of CDK7 and CDK4 in autophagy activation induced by silica nanoparticles. Autophagy 2021, 17, 1426–1447. [Google Scholar] [CrossRef]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef]
- Korah, J.; Canaff, L.; Lebrun, J.-J. The Retinoblastoma Tumor Suppressor Protein (pRb)/E2 Promoter Binding Factor 1 (E2F1) Pathway as a Novel Mediator of TGFβ-induced Autophagy. J. Biol. Chem. 2016, 291, 2043–2054. [Google Scholar] [CrossRef]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Jurado, E.G.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Manzano, C.; Mitlianga, P.; Fueyo, J.; Lee, H.Y.; Hu, M.; Spurgers, K.B.; Glass, T.L.; Koul, D.; Liu, T.J.; McDonnell, T.J.; et al. Transfer of E2F-1 to human glioma cells results in transcriptional up-regulation of Bcl-2. Cancer Res. 2001, 61, 6693–6697. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.-L.; Bast, R.C. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092. [Google Scholar] [CrossRef]
- Sutton, M.N.; Huang, G.Y.; Zhou, J.; Mao, W.; Langley, R.; Lu, Z.; Bast, R.C. Amino Acid Deprivation-Induced Autophagy Requires Upregulation of DIRAS3 through Reduction of E2F1 and E2F4 Transcriptional Repression. Cancers 2019, 11, 603. [Google Scholar] [CrossRef]
- Johnson, D.G.; Degregori, J. Putting the Oncogenic and Tumor Suppressive Activities of E2F into Context. Curr. Mol. Med. 2006, 6, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Hashemolhosseini, S.; Nagamine, Y.; Morley, S.J.; Desrivières, S.; Mercep, L.; Ferrari, S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J. Biol. Chem. 1998, 273, 14424–14429. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, S.; Sakaida, H.; Hori, T.; Maeda, M.; Uchiyama, T. The Upregulation of p27Kip1 by Rapamycin Results in G1 Arrest in Exponentially Growing T-Cell Lines. Blood 1998, 91, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Valle, F.; Badura, M.L.; Braunstein, S.; Narasimhan, M.; Schneider, R.J. Mitotic Raptor Promotes mTORC1 Activity, G2/M Cell Cycle Progression, and Internal Ribosome Entry Site-Mediated mRNA Translation. Mol. Cell. Biol. 2010, 30, 3151–3164. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chen, Y.; Yan, C.; Guo, X.; Jiang, T.; Guli, A.; Song, X.; Wan, Q.; Shu, Q.; Ding, S. Phosphoproteome Profiling Revealed the Importance of mTOR Inhibition on CDK1 Activation to Further Regulate Cell Cycle Progression. J. Proteome Res. 2021, 20, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef]
- Wan, Q.; Chen, H.; Li, X.; Yan, L.; Sun, Y.; Wang, J. Artesunate inhibits fibroblasts proliferation and reduces surgery-induced epidural fibrosis via the autophagy-mediated p53/p21waf1/cip1 pathway. Eur. J. Pharmacol. 2019, 842, 197–207. [Google Scholar] [CrossRef]
- Hwang, J.R.; Kim, W.Y.; Cho, Y.-J.; Ryu, J.-Y.; Choi, J.-J.; Jeong, S.Y.; Kim, M.-S.; Kim, J.H.; Paik, E.S.; Lee, Y.-Y.; et al. Chloroquine reverses chemoresistance via upregulation of p21WAF1/CIP1 and autophagy inhibition in ovarian cancer. Cell Death Dis. 2020, 11, 1034. [Google Scholar] [CrossRef]
- Pisonero-Vaquero, S.; Soldati, C.; Cesana, M.; Ballabio, A.; Medina, D.L. TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response. Cells 2020, 9, 1186. [Google Scholar] [CrossRef]
- Jia, W.; He, M.-X.; McLeod, I.X.; Guo, J.; Ji, D.; He, Y.-W. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 2015, 11, 2335–2345. [Google Scholar] [CrossRef]
- Herrero-Ruiz, J.; Mora-Santos, M.; Giráldez, S.; Sáez, C.; Japón, M.Á.; Tortolero, M.; Romero, F. βTrCP controls the lysosome-mediated degradation of CDK1, whose accumulation correlates with tumor malignancy. Oncotarget 2014, 5, 7563–7574. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Moreno, M.; Giráldez, S.; Sáez, C.; Japón, M.Á.; Tortolero, M.; Romero, F. Both p62/SQSTM1-HDAC6-dependent autophagy and the aggresome pathway mediate CDK1 degradation in human breast cancer. Sci. Rep. 2017, 7, 10078. [Google Scholar] [CrossRef] [PubMed]
- Pirtoli, L.; Belmonte, G.; Toscano, M.; Tini, P.; Miracco, C. Cyclin D1 Co-localizes with Beclin-1 in Glioblastoma Recurrences: A Clue to a Therapy-induced, Autophagy-mediated Degradative Mechanism? Anticancer Res. 2016, 36, 4057–4062. [Google Scholar] [PubMed]
- Wu, S.-Y.; Lan, S.-H.; Wu, S.-R.; Chiu, Y.-C.; Lin, X.-Z.; Su, I.-J.; Tsai, T.-F.; Yen, C.-J.; Lu, T.-H.; Liang, F.-W.; et al. Hepatocellular carcinoma–related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology 2018, 68, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ding, X.-F.; Bouamar, H.; Pressley, K.; Sun, L.-Z. Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol. Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef]
- Loukil, A.; Zonca, M.; Rebouissou, C.; Baldin, V.; Coux, O.; Biard-Piechaczyk, M.; Blanchard, J.-M.; Peter, M. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J. Cell Sci. 2014, 127, 2145–2150. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef]
- Lin, H.; Xiong, H.; Su, Z.; Pang, J.; Lai, L.; Zhang, H.; Jian, B.; Zhang, W.; Zheng, Y. Inhibition of DRP-1-Dependent Mitophagy Promotes Cochlea Hair Cell Senescence and Exacerbates Age-Related Hearing Loss. Front. Cell. Neurosci. 2019, 13, 550. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Y.; Bai, X.; Yin, Y.; Dai, L.; Zhou, H.; Wu, Q.; Wang, Y. Deubiquitinating enzyme USP30 negatively regulates mitophagy and accelerates myocardial cell senescence through antagonism of Parkin. Cell Death Discov. 2021, 7, 187. [Google Scholar] [CrossRef]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Domenico, J.; Gelfand, E.W. Cyclin-Dependent Kinase 6 Inhibits Proliferation of Human Mammary Epithelial Cells1 1 Supported in part by grants from the Denver Metropolitan Chapter of the Susan G. Komen Breast Cancer Foundation and the Cancer League of Colorado, American Cancer Society (grant IM-746), NIH (grants HL-36577 and AI-42246), and University of Colorado Cancer Center (grant CA46934). Mol. Cancer Res. 2004, 2, 105–114. [Google Scholar] [PubMed]
- Tay, S.-P.; Yeo, C.W.S.; Chai, C.; Chua, P.-J.; Tan, H.-M.; Ang, A.X.Y.; Yip, D.L.H.; Sung, J.-X.; Tan, P.H.; Bay, B.-H.; et al. Parkin Enhances the Expression of Cyclin-dependent Kinase 6 and Negatively Regulates the Proliferation of Breast Cancer Cells. J. Biol. Chem. 2010, 285, 29231–29238. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.-H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225-235.e5. [Google Scholar] [CrossRef]
- Cuyàs, E.; Corominas-Faja, B.; Joven, J.; Menendez, J.A. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Cell Cycle Control. 2014, 1170, 113–144. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Shi, T.-T.; Yu, X.-X.; Yan, L.-J.; Xiao, H.-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [CrossRef]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef]
- Small, J.; Washburn, E.; Millington, K.; Zhu, J.; Holder, S.L. The addition of abemaciclib to sunitinib induces regression of renal cell carcinoma xenograft tumors. Oncotarget 2017, 8, 95116–95134. [Google Scholar] [CrossRef]
- Okada, Y.; Kato, S.; Sakamoto, Y.; Oishi, T.; Ishioka, C. Synthetic lethal interaction of CDK inhibition and autophagy inhibition in human solid cancer cell lines. Oncol. Rep. 2017, 38, 31–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ji, Y.; Liu, X.; Li, J.; Xie, X.; Huang, M.; Jiang, J.; Liao, Y.-P.; Donahue, T.; Meng, H. Use of ratiometrically designed nanocarrier targeting CDK4/6 and autophagy pathways for effective pancreatic cancer treatment. Nat. Commun. 2020, 11, 4249. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.; Liang, M.-L.; Zheng, J.-H.; Lin, Y.-C.; Yang, Y.-C.; Vo, T.-H.; Liou, J.-P.; Yen, Y.; Chen, C.-H. Combining an Autophagy Inhibitor, MPT0L145, with Abemaciclib Is a New Therapeutic Strategy in GBM Treatment. Cancers 2021, 13, 6117. [Google Scholar] [CrossRef]
- Nakatani, K.; Matsuo, H.; Harata, Y.; Higashitani, M.; Koyama, A.; Noura, M.; Nishinaka-Arai, Y.; Kamikubo, Y.; Adachi, S. Inhibition of CDK4/6 and autophagy synergistically induces apoptosis in t(8;21) acute myeloid leukemia cells. Int. J. Hematol. 2021, 113, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Nakatani, K.; Harata, Y.; Higashitani, M.; Ito, Y.; Inagami, A.; Noura, M.; Nakahata, T.; Adachi, S. Efficacy of a combination therapy targeting CDK4/6 and autophagy in a mouse xenograft model of t(8;21) acute myeloid leukemia. Biochem. Biophys. Rep. 2021, 27, 101099. [Google Scholar] [CrossRef] [PubMed]
- Singareeka Raghavendra, A.; Kwiatkowski, D.; Damodaran, S.; Kettner, N.M.; Ramirez, D.L.; Gombos, D.S.; Hunt, K.; Shen, Y.; Keyomarsi, K.; Tripathy, D. Phase I safety and efficacy study of autophagy inhibition with hydroxychloroquine to augment the antiproliferative and biological effects of preoperative palbociclib plus letrozole for estrogen receptor-positive, HER2-negative metastatic breast cancer (MBC). J. Clin. Oncol. 2021, 39, 1067. [Google Scholar] [CrossRef]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef]
- Xu, L.-Z.; Long, Z.-J.; Peng, F.; Liu, Y.; Xu, J.; Wang, C.; Jiang, L.; Guo, T.; Kamran, M.; Li, S.-S.; et al. Aurora kinase A suppresses metabolic stress-induced autophagic cell death by activating mTOR signaling in breast cancer cells. Oncotarget 2014, 5, 7498–7511. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | Cancer Type | Refs |
|---|---|---|
| Breast cancer, solid tumors | [56] |
| Solid tumors | [120] |
| Pancreatic cancer | [121] |
| Brain cancer (glioblastoma multiforme) | [122] |
| t(8;21) Acute myeloid leukemia | [123] |
| [124] | ||
| Estrogen receptor-positive, HER2-negative metastatic breast cancer | [125] |
| Clinical Trial | Official Title | Condition | Intervention | Study Description | ClinicalTrials.gov Identifiers |
|---|---|---|---|---|---|
| PALAVY | A phase II trial of Avelumab or hydroxychloroquine with or without palbociclib to eliminate dormant breast cancer | Early-stage ER + breast cancer | HCQ, Avelumab, palbociclib | Randomized, open label phase II clinical trial that will assess the safety and early efficacy of hydroxychloroquine or Avelumab, with or without palbociclib, in early-stage ER+ breast cancer patients who are found to harbor disseminated tumor cells (DTCs) in the bone marrow after definitive surgery and standard adjuvant therapy. | NCT04841148 |
| ABBY | A phase II pilot trial of abemaciclib or abemaciclib and hydroxychloroquine to target minimal residual disease in breast cancer patients | Invasive breast cancer | abemaciclib HCQ | Randomized, open label phase II clinical trial that is testing whether the use of hydroxychloroquine and abemaciclib can reduce the number or eliminate DTCs in bone marrow. | NCT04523857 |
| Hydroxychloroquine, abemaciclib, and endocrine therapy in hormone receptor positive (HR+)/Her 2 negative breast cancer |
HR+/Her 2- advanced breast cancer, advanced solid tumors | Abemaciclib HCQ | Non-randomized, open label phase I clinical trial that assessed safety, tolerability, and efficacy of abemaciclib combined with the autophagy inhibitor hydroxychloroquine in advanced solid tumors and HR+/Her 2-advanced breast cancer | NCT04316169 | |
| Phase I/II safety and efficacy study of autophagy inhibition with hydroxychloroquine to augment the antiproliferative and biological effects of pre-operative palbociclib plus letrozole for estrogen receptor-positive and HER2-negative breast cancer | HR+/HER2-bbreast cancer | HCQ, letrozole, palbociclib | Open label phase I/II clinical trial investigating the side effects and best dose of hydroxychloroquine when given together with palbociclib and letrozole before surgery in treating patients with estrogen receptor positive, HER2-negative breast cancer. | NCT03774472 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziegler, D.V.; Huber, K.; Fajas, L. The Intricate Interplay between Cell Cycle Regulators and Autophagy in Cancer. Cancers 2022, 14, 153. https://doi.org/10.3390/cancers14010153
Ziegler DV, Huber K, Fajas L. The Intricate Interplay between Cell Cycle Regulators and Autophagy in Cancer. Cancers. 2022; 14(1):153. https://doi.org/10.3390/cancers14010153
Chicago/Turabian StyleZiegler, Dorian V., Katharina Huber, and Lluis Fajas. 2022. "The Intricate Interplay between Cell Cycle Regulators and Autophagy in Cancer" Cancers 14, no. 1: 153. https://doi.org/10.3390/cancers14010153
APA StyleZiegler, D. V., Huber, K., & Fajas, L. (2022). The Intricate Interplay between Cell Cycle Regulators and Autophagy in Cancer. Cancers, 14(1), 153. https://doi.org/10.3390/cancers14010153