
CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology


Abstract
Simple Summary
Abstract
1. Introduction
2. The DPP-4 Gene Family
DPP-4
3. DPP-4 Expression in Primary Tumors
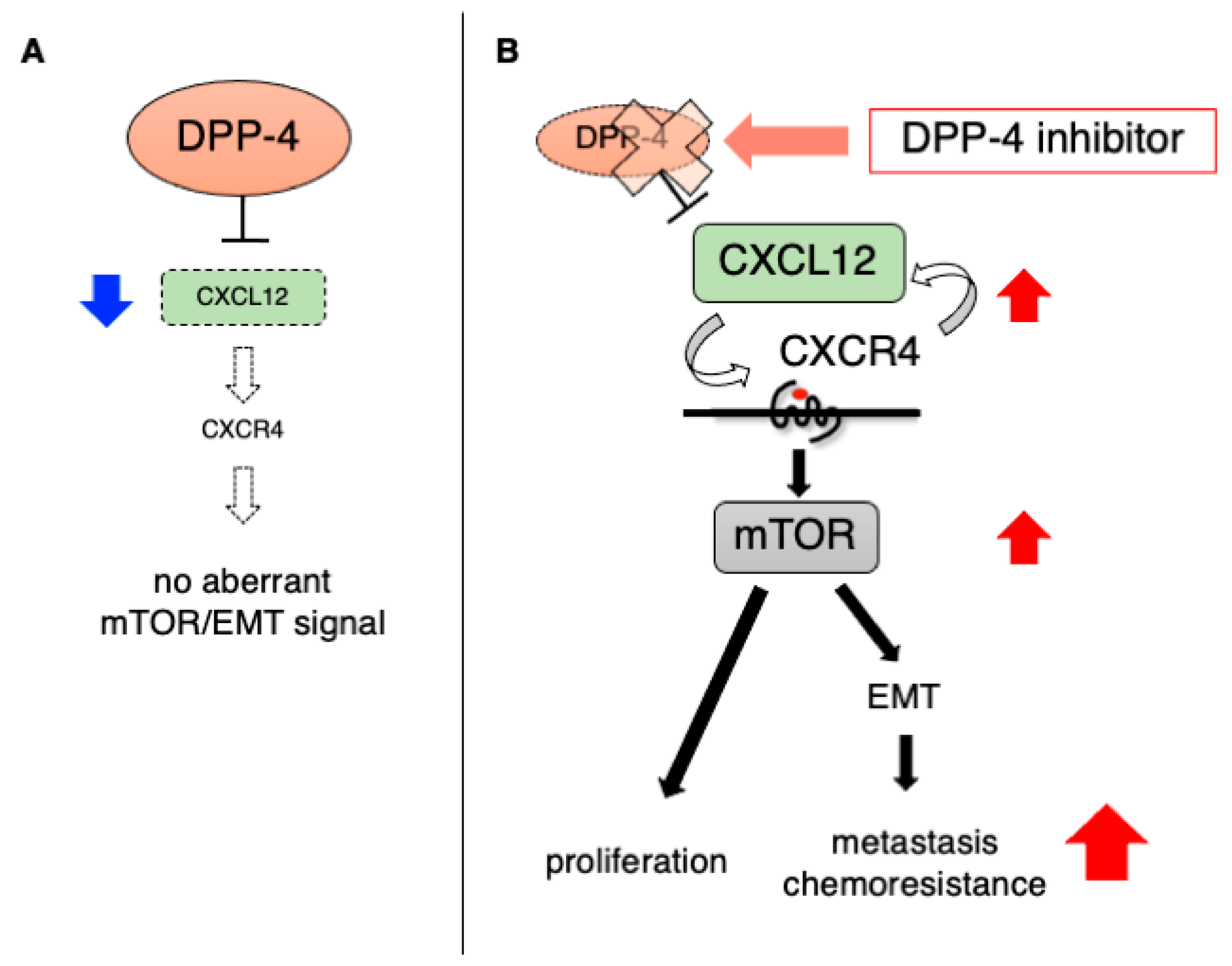
4. The Impact of DPP-4 Inhibition on Existing Tumors
5. The Influence of DPP-4 Inhibitors on Cancer: Clinical Evidence
5.1. The Impact of DPP-4 Inhibitors on Cancer Incidence in Diabetic Patients
5.2. The Influence of DPP-4 Inhibitors on Cancer Prognosis and Metastasis
6. Perspective: Co-Prescription with Metformin
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Onitilo, A.A.; Engel, J.M.; Glurich, I.; Stankowski, R.V.; Williams, G.M.; Doi, S.A. Diabetes and cancer I: Risk, survival, and implications for screening. Cancer Cause Control 2012, 23, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Barone, B.B.; Yeh, H.C.; Snyder, C.F.; Peairs, K.S.; Stein, K.B.; Derr, R.L.; Wolff, A.C.; Brancati, F.L. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. JAMA 2008, 300, 2754–2764. [Google Scholar] [CrossRef]
- Walker, J.J.; Johnson, J.A.; Wild, S.H. Diabetes treatments and cancer risk: The importance of considering aspects of drug exposure. Lancet Diabetes Endocrinol. 2013, 1, 132–139. [Google Scholar] [CrossRef]
- Sen, S.; He, Y.; Koya, D.; Kanasaki, K. Cancer biology in diabetes. J. Diabetes Investig. 2014, 5, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Janker, F.; De Meester, I.; Arni, S.; Borgeaud, N.; Yamada, Y.; Gil Bazo, I.; Weder, W.; Jungraithmayr, W. The CD26/DPP4-inhibitor vildagliptin suppresses lung cancer growth via macrophage-mediated NK cell activity. Carcinogenesis 2019, 40, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 115–134. [Google Scholar] [CrossRef]
- Bishnoi, R.; Hong, Y.R.; Shah, C.; Ali, A.; Skelton, W.P.t.; Huo, J.; Dang, N.H.; Dang, L.H. Dipeptidyl peptidase 4 inhibitors as novel agents in improving survival in diabetic patients with colorectal cancer and lung cancer: A Surveillance Epidemiology and Endpoint Research Medicare study. Cancer Med. 2019, 8, 3918–3927. [Google Scholar] [CrossRef]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4—A potential biomarker and target for cancer therapy. Pharmacol. Ther. 2019, 198, 135–159. [Google Scholar] [CrossRef]
- Davies, S.; Beckenkamp, A.; Buffon, A. CD26 a cancer stem cell marker and therapeutic target. Biomed. Pharmacother. 2015, 71, 135–138. [Google Scholar] [CrossRef]
- Varela-Calvino, R.; Rodriguez-Quiroga, M.; Dias Carvalho, P.; Martins, F.; Serra-Roma, A.; Vazquez-Iglesias, L.; Paez de la Cadena, M.; Velho, S.; Cordero, O.J. The mechanism of sitagliptin inhibition of colorectal cancer cell lines’ metastatic functionalities. IUBMB Life 2021, 73, 761–773. [Google Scholar] [CrossRef]
- Yu, D.M.; Yao, T.W.; Chowdhury, S.; Nadvi, N.A.; Osborne, B.; Church, W.B.; McCaughan, G.W.; Gorrell, M.D. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. 2010, 277, 1126–1144. [Google Scholar] [CrossRef]
- Pure, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Keane, F.M.; Gorrell, M.D. Advances in understanding the expression and function of dipeptidyl peptidase 8 and 9. Mol. Cancer Res. 2013, 11, 1487–1496. [Google Scholar] [CrossRef]
- Brunetti, M.; Holth, A.; Panagopoulos, I.; Staff, A.C.; Micci, F.; Davidson, B. Expression and clinical role of the dipeptidyl peptidases DPP8 and DPP9 in ovarian carcinoma. Virchows Arch. 2019, 474, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Taabazuing, C.Y.; Okondo, M.C.; Chui, A.J.; Rao, S.D.; Brown, F.C.; Reed, C.; Peguero, E.; de Stanchina, E.; Kentsis, A.; et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 2018, 24, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Schnapp, G.; Hoevels, Y.; Bakker, R.A.; Schreiner, P.; Klein, T.; Nar, H. A Single Second Shell Amino Acid Determines Affinity and Kinetics of Linagliptin Binding to Type 4 Dipeptidyl Peptidase and Fibroblast Activation Protein. ChemMedChem 2021, 16, 630–639. [Google Scholar] [CrossRef]
- Chien, C.H.; Huang, L.H.; Chou, C.Y.; Chen, Y.S.; Han, Y.S.; Chang, G.G.; Liang, P.H.; Chen, X. One site mutation disrupts dimer formation in human DPP-IV proteins. J. Biol. Chem. 2004, 279, 52338–52345. [Google Scholar] [CrossRef] [PubMed]
- Rohrborn, D.; Eckel, J.; Sell, H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett. 2014, 588, 3870–3877. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Kameoka, J.; Tanaka, T.; Nojima, Y.; Schlossman, S.F.; Morimoto, C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 1993, 261, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M.; Durinx, C.; Scharpe, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef]
- Kahne, T.; Lendeckel, U.; Wrenger, S.; Neubert, K.; Ansorge, S.; Reinhold, D. Dipeptidyl peptidase IV: A cell surface peptidase involved in regulating T cell growth (review). Int. J. Mol. Med. 1999, 4, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yamochi, T.; Yamochi, T.; Aytac, U.; Ohnuma, K.; McKee, K.S.; Morimoto, C.; Dang, N.H. CD26 regulates p38 mitogen-activated protein kinase-dependent phosphorylation of integrin beta1, adhesion to extracellular matrix, and tumorigenicity of T-anaplastic large cell lymphoma Karpas 299. Cancer Res. 2005, 65, 6950–6956. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 2007, 19, 495–507. [Google Scholar] [CrossRef]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef]
- Durinx, C.; Lambeir, A.M.; Bosmans, E.; Falmagne, J.B.; Berghmans, R.; Haemers, A.; Scharpe, S.; De Meester, I. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 2000, 267, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.J.; Salgado, F.J.; Nogueira, M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol. Immunother. 2009, 58, 1723–1747. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.M.; Slaitini, L.; Gysbers, V.; Riekhoff, A.G.; Kahne, T.; Knott, H.M.; De Meester, I.; Abbott, C.A.; McCaughan, G.W.; Gorrell, M.D. Soluble CD26/dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand. J. Immunol. 2011, 73, 102–111. [Google Scholar] [CrossRef]
- Lamers, D.; Famulla, S.; Wronkowitz, N.; Hartwig, S.; Lehr, S.; Ouwens, D.M.; Eckardt, K.; Kaufman, J.M.; Ryden, M.; Muller, S.; et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011, 60, 1917–1925. [Google Scholar] [CrossRef]
- Varin, E.M.; Mulvihill, E.E.; Beaudry, J.L.; Pujadas, G.; Fuchs, S.; Tanti, J.F.; Fazio, S.; Kaur, K.; Cao, X.; Baggio, L.L.; et al. Circulating Levels of Soluble Dipeptidyl Peptidase-4 Are Dissociated from Inflammation and Induced by Enzymatic DPP4 Inhibition. Cell Metab. 2019, 29, 320–334.e325. [Google Scholar] [CrossRef]
- Wesley, U.V.; McGroarty, M.; Homoyouni, A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 2005, 65, 1325–1334. [Google Scholar] [CrossRef]
- Arscott, W.T.; LaBauve, A.E.; May, V.; Wesley, U.V. Suppression of neuroblastoma growth by dipeptidyl peptidase IV: Relevance of chemokine regulation and caspase activation. Oncogene 2009, 28, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Mezawa, Y.; Daigo, Y.; Takano, A.; Miyagi, Y.; Yokose, T.; Yamashita, T.; Morimoto, C.; Hino, O.; Orimo, A. CD26 expression is attenuated by TGF-beta and SDF-1 autocrine signaling on stromal myofibroblasts in human breast cancers. Cancer Med. 2019, 8, 3936–3948. [Google Scholar] [CrossRef]
- Kawakita, E.; Yang, F.; Kumagai, A.; Takagaki, Y.; Kitada, M.; Yoshitomi, Y.; Ikeda, T.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; et al. Metformin Mitigates DPP-4 Inhibitor-Induced Breast Cancer Metastasis via Suppression of mTOR Signaling. Mol. Cancer Res. 2021, 19, 61–73. [Google Scholar] [CrossRef]
- Wesley, U.V.; Tiwari, S.; Houghton, A.N. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int. J. Cancer 2004, 109, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Cheung, A.H.; Wong, S.K.; Wan, T.M.; Ng, L.; Chow, A.K.; Cheng, N.S.; Pak, R.C.; Li, H.S.; Man, J.H.; et al. Prognostic significance of CD26 in patients with colorectal cancer. PLoS ONE 2014, 9, e98582. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Inoue, Y.; Mori, K.; Ide, S.; Imaoka, H.; Kawamura, M.; Mohri, Y.; Kusunoki, M. Prognostic relevance of stromal CD26 expression in rectal cancer after chemoradiotherapy. Int. J. Clin. Oncol. 2016, 21, 350–358. [Google Scholar] [CrossRef]
- Yu, H.; Mei, X.P.; Su, P.F.; Jin, G.Z.; Zhou, H.K. A poor prognosis in human hepatocellular carcinoma is associated with low expression of DPP4. Braz. J. Med. Biol. Res. 2020, 53, e9114. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Ino, K.; Mizutani, S.; Nawa, A.; Kikkawa, F. The expression of dipeptidyl peptidase IV (DPPIV/CD26) is associated with enhanced chemosensitivity to paclitaxel in epithelial ovarian carcinoma cells. Cancer Sci. 2010, 101, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Kikkawa, F.; Suzuki, T.; Shibata, K.; Ino, K.; Mizutani, S. Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res. 2002, 62, 2753–2757. [Google Scholar] [PubMed]
- Zhang, M.; Xu, L.; Wang, X.; Sun, B.; Ding, J. Expression levels of seprase/FAPalpha and DPPIV/CD26 in epithelial ovarian carcinoma. Oncol. Lett. 2015, 10, 34–42. [Google Scholar] [CrossRef]
- Yan, L.; Tian, X.; Ye, C.; Guan, X.; Dong, B.; Zhao, M.; Wu, J.; Hao, C. CD26 as a Promising Biomarker for Predicting Prognosis in Patients with Pancreatic Tumors. Onco Targets Ther. 2020, 13, 12615–12623. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Qi, L.; Bo, X.J.; Liu, G.D.; Wang, J.M.; Li, G. Expression of CD26 and CXCR4 in prostate carcinoma and its relationship with clinical parameters. J. Res. Med. Sci. 2013, 18, 647–652. [Google Scholar]
- Song, Y.; Zhou, M.; Cao, Y.; Qi, J.; Geng, J.; Liu, X. Expression of GLP-1 receptor and CD26 in human thyroid C-cells: The association of thyroid C-cell tumorigenesis with incretin-based medicine. Oncol. Lett. 2017, 13, 2684–2690. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.I.; Yeh, B.W.; Li, W.M.; Chan, T.C.; Chang, I.W.; Huang, C.N.; Li, C.C.; Ke, H.L.; Yeh, H.C.; Wu, W.J.; et al. DPP4/CD26 overexpression in urothelial carcinoma confers an independent prognostic impact and correlates with intrinsic biological aggressiveness. Oncotarget 2017, 8, 2995–3008. [Google Scholar] [CrossRef]
- Aldinucci, D.; Poletto, D.; Lorenzon, D.; Nanni, P.; Degan, M.; Olivo, K.; Rapana, B.; Pinto, A.; Gattei, V. CD26 expression correlates with a reduced sensitivity to 2’-deoxycoformycin-induced growth inhibition and apoptosis in T-cell leukemia/lymphomas. Clin. Cancer Res. 2004, 10, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Cro, L.; Morabito, F.; Zucal, N.; Fabris, S.; Lionetti, M.; Cutrona, G.; Rossi, F.; Gentile, M.; Ferrario, A.; Ferrarini, M.; et al. CD26 expression in mature B-cell neoplasia: Its possible role as a new prognostic marker in B-CLL. Hematol. Oncol. 2009, 27, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, M.; Lewandowski, K.; Czyz, A.; Kiernicka-Parulska, J.; Przybylowicz-Chalecka, A.; Jarmuz-Szymczak, M.; Lewandowska, M.; Komarnicki, M. The prognostic significance of surface dipeptidylpeptidase IV (CD26) expression in B-cell chronic lymphocytic leukemia. Leuk. Res. 2016, 47, 166–171. [Google Scholar] [CrossRef]
- Moffitt, L.R.; Bilandzic, M.; Wilson, A.L.; Chen, Y.; Gorrell, M.D.; Oehler, M.K.; Plebanski, M.; Stephens, A.N. Hypoxia Regulates DPP4 Expression, Proteolytic Inactivation, and Shedding from Ovarian Cancer Cells. Int J. Mol. Sci 2020, 21, 8110. [Google Scholar] [CrossRef]
- Beckenkamp, A.; Willig, J.B.; Santana, D.B.; Nascimento, J.; Paccez, J.D.; Zerbini, L.F.; Bruno, A.N.; Pilger, D.A.; Wink, M.R.; Buffon, A. Differential Expression and Enzymatic Activity of DPPIV/CD26 Affects Migration Ability of Cervical Carcinoma Cells. PLoS ONE 2015, 10, e0134305. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra351. [Google Scholar] [CrossRef]
- He, L.; Zhang, T.; Sun, W.; Qin, Y.; Wang, Z.; Dong, W.; Zhang, H. The DPP-IV inhibitor saxagliptin promotes the migration and invasion of papillary thyroid carcinoma cells via the NRF2/HO1 pathway. Med. Oncol. 2020, 37, 97. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar] [CrossRef]
- Sun, Y.X.; Pedersen, E.A.; Shiozawa, Y.; Havens, A.M.; Jung, Y.; Wang, J.; Pienta, K.J.; Taichman, R.S. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL12. Clin. Exp. Metastasis 2008, 25, 765–776. [Google Scholar] [CrossRef]
- Yang, F.; Takagaki, Y.; Yoshitomi, Y.; Ikeda, T.; Li, J.; Kitada, M.; Kumagai, A.; Kawakita, E.; Shi, S.; Kanasaki, K.; et al. Inhibition of Dipeptidyl Peptidase-4 Accelerates Epithelial-Mesenchymal Transition and Breast Cancer Metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019, 79, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus inhibits breast cancer cell growth through PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 7163–7169. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kang, S.E.; Ahn, K.S.; Um, J.Y.; Yang, W.M.; Yun, M.; Lee, S.G. Inhibition of the PI3K-AKT-mTOR pathway suppresses the adipocyte-mediated proliferation and migration of breast cancer cells. J. Cancer 2020, 11, 2552–2559. [Google Scholar] [CrossRef] [PubMed]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J.; et al. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef]
- Zhang, G.; Kim, S.; Gu, X.; Yu, S.P.; Wei, L. DPP-4 Inhibitor Linagliptin is Neuroprotective in Hyperglycemic Mice with Stroke via the AKT/mTOR Pathway and Anti-apoptotic Effects. Neurosci. Bull. 2020, 36, 407–418. [Google Scholar] [CrossRef]
- El-Marasy, S.A.; Abdel-Rahman, R.F.; Abd-Elsalam, R.M. Neuroprotective effect of vildagliptin against cerebral ischemia in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 1133–1145. [Google Scholar] [CrossRef]
- Li, X.; Ma, G.; Ma, Q.; Li, W.; Liu, J.; Han, L.; Duan, W.; Xu, Q.; Liu, H.; Wang, Z.; et al. Neurotransmitter substance P mediates pancreatic cancer perineural invasion via NK-1R in cancer cells. Mol. Cancer Res. 2013, 11, 294–302. [Google Scholar] [CrossRef]
- Cheng, Y.; Tang, X.Y.; Li, Y.X.; Zhao, D.D.; Cao, Q.H.; Wu, H.X.; Yang, H.B.; Hao, K.; Yang, Y. Depression-Induced Neuropeptide Y Secretion Promotes Prostate Cancer Growth by Recruiting Myeloid Cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Bronger, H.; Karge, A.; Dreyer, T.; Zech, D.; Kraeft, S.; Avril, S.; Kiechle, M.; Schmitt, M. Induction of cathepsin B by the CXCR3 chemokines CXCL9 and CXCL10 in human breast cancer cells. Oncol. Lett. 2017, 13, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Nam, J.S.; Lim, Y.; Lee, Y.H. TNFalpha-exposed bone marrow-derived mesenchymal stem cells promote locomotion of MDA-MB-231 breast cancer cells through transcriptional activation of CXCR3 ligand chemokines. J. Biol. Chem. 2010, 285, 30731–30740. [Google Scholar] [CrossRef]
- Zipin-Roitman, A.; Meshel, T.; Sagi-Assif, O.; Shalmon, B.; Avivi, C.; Pfeffer, R.M.; Witz, I.P.; Ben-Baruch, A. CXCL10 promotes invasion-related properties in human colorectal carcinoma cells. Cancer Res. 2007, 67, 3396–3405. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, J.; Wang, T.; Zhang, X.; Liu, D. CXCL10/CXCR3 axis promotes the invasion of gastric cancer via PI3K/AKT pathway-dependent MMPs production. Biomed. Pharmacother. 2016, 82, 479–488. [Google Scholar] [CrossRef]
- Li, S.; Fan, Y.; Kumagai, A.; Kawakita, E.; Kitada, M.; Kanasaki, K.; Koya, D. Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance through the CXCL12/CXCR4/mTOR/TGFbeta Signaling Pathway in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 805. [Google Scholar] [CrossRef]
- Mizokami, Y.; Kajiyama, H.; Shibata, K.; Ino, K.; Kikkawa, F.; Mizutani, S. Stromal cell-derived factor-1alpha-induced cell proliferation and its possible regulation by CD26/dipeptidyl peptidase IV in endometrial adenocarcinoma. Int. J. Cancer 2004, 110, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Koma, Y.I.; Arai, N.; Kido, A.; Urakawa, N.; Nishio, M.; Shigeoka, M.; Yokozaki, H. CCL3-CCR5 axis contributes to progression of esophageal squamous cell carcinoma by promoting cell migration and invasion via Akt and ERK pathways. Lab. Investig. 2020, 100, 1140–1157. [Google Scholar] [CrossRef]
- da Silva, J.M.; Moreira Dos Santos, T.P.; Sobral, L.M.; Queiroz-Junior, C.M.; Rachid, M.A.; Proudfoot, A.E.I.; Garlet, G.P.; Batista, A.C.; Teixeira, M.M.; Leopoldino, A.M.; et al. Relevance of CCL3/CCR5 axis in oral carcinogenesis. Oncotarget 2017, 8, 51024–51036. [Google Scholar] [CrossRef] [PubMed]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife 2019, 8. [Google Scholar] [CrossRef]
- An, G.; Wu, F.; Huang, S.; Feng, L.; Bai, J.; Gu, S.; Zhao, X. Effects of CCL5 on the biological behavior of breast cancer and the mechanisms of its interaction with tumorassociated macrophages. Oncol. Rep. 2019, 42, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Chen, L.; Ma, L.; Wang, D.; Shao, B.; Wu, J.; Wu, H.; Jin, Y. Expression and prognostic significance of CCL11/CCR3 in glioblastoma. Oncotarget 2016, 7, 32617–32627. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Li, X.; Bi, Y.; Zheng, Y.; Wang, J.; Li, X.; Huang, Z.; Chen, L.; Huang, Y.; Huang, Y. CCL14 is a prognostic biomarker and correlates with immune infiltrates in hepatocellular carcinoma. Aging (Albany NY) 2020, 12, 784–807. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Ling, Y.; Huang, L.; Huang, H.; Chen, X.; Xiao, Y.; Zhu, Z.; Chen, J. C-C motif chemokine 14 as a novel potential biomarker for predicting the prognosis of epithelial ovarian cancer. Oncol. Lett. 2020, 19, 2875–2883. [Google Scholar] [CrossRef]
- Li, Q.; Shi, L.; Gui, B.; Yu, W.; Wang, J.; Zhang, D.; Han, X.; Yao, Z.; Shang, Y. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011, 71, 6899–6908. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Li, S.; Yue, M.; Li, C.; Kang, Z. Lysine demethylase 5B suppresses CC chemokine ligand 14 to promote progression of colorectal cancer through the Wnt/beta-catenin pathway. Life Sci. 2021, 264, 118726. [Google Scholar] [CrossRef]
- Zhu, M.; Xu, W.; Wei, C.; Huang, J.; Xu, J.; Zhang, Y.; Zhao, Y.; Chen, J.; Dong, S.; Liu, B.; et al. CCL14 serves as a novel prognostic factor and tumor suppressor of HCC by modulating cell cycle and promoting apoptosis. Cell Death Dis. 2019, 10, 796. [Google Scholar] [CrossRef]
- Huang, Y.H.; Chang, C.Y.; Kuo, Y.Z.; Fang, W.Y.; Kao, H.Y.; Tsai, S.T.; Wu, L.W. Cancer-associated fibroblast-derived interleukin-1beta activates protumor C-C motif chemokine ligand 22 signaling in head and neck cancer. Cancer Sci. 2019, 110, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Yaguchi, T.; Ohmura, G.; Ohta, S.; Kobayashi, A.; Kawamura, N.; Fujita, T.; Nakano, H.; Shimada, T.; Takahashi, T.; et al. Autocrine and paracrine loops between cancer cells and macrophages promote lymph node metastasis via CCR4/CCL22 in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, 2755–2766. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults with Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef]
- Leiter, L.A.; Teoh, H.; Mosenzon, O.; Cahn, A.; Hirshberg, B.; Stahre, C.A.; Hoekstra, J.B.; Alvarsson, M.; Im, K.; Scirica, B.M.; et al. Frequency of cancer events with saxagliptin in the SAVOR-TIMI 53 trial. Diabetes Obes. Metab. 2016, 18, 186–190. [Google Scholar] [CrossRef]
- Matthews, D.R.; Paldanius, P.M.; Proot, P.; Chiang, Y.; Stumvoll, M.; Del Prato, S.; VERIFY Study Group. Glycaemic durability of an early combination therapy with vildagliptin and metformin versus sequential metformin monotherapy in newly diagnosed type 2 diabetes (VERIFY): A 5-year, multicentre, randomised, double-blind trial. Lancet 2019, 394, 1519–1529. [Google Scholar] [CrossRef]
- Elashoff, M.; Matveyenko, A.V.; Gier, B.; Elashoff, R.; Butler, P.C. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011, 141, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.Y.; Shih, S.R.; Tseng, C.H. A review on the association between glucagon-like peptide-1 receptor agonists and thyroid cancer. Exp. Diabetes Res. 2012, 2012, 924168. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Lee, K.Y.; Tseng, F.H. An updated review on cancer risk associated with incretin mimetics and enhancers. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2015, 33, 67–124. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Sitagliptin use and thyroid cancer risk in patients with type 2 diabetes. Oncotarget 2016, 7, 24871–24879. [Google Scholar] [CrossRef] [PubMed]
- Dore, D.D.; Seeger, J.D.; Chan, K.A. Incidence of health insurance claims for thyroid neoplasm and pancreatic malignancy in association with exenatide: Signal refinement using active safety surveillance. Ther. Adv. Drug Saf. 2012, 3, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Romley, J.A.; Goldman, D.P.; Solomon, M.; McFadden, D.; Peters, A.L. Exenatide therapy and the risk of pancreatitis and pancreatic cancer in a privately insured population. Diabetes Technol. Ther. 2012, 14, 904–911. [Google Scholar] [CrossRef]
- Funch, D.; Gydesen, H.; Tornoe, K.; Major-Pedersen, A.; Chan, K.A. A prospective, claims-based assessment of the risk of pancreatitis and pancreatic cancer with liraglutide compared to other antidiabetic drugs. Diabetes Obes. Metab. 2014, 16, 273–275. [Google Scholar] [CrossRef]
- Gokhale, M.; Buse, J.B.; Gray, C.L.; Pate, V.; Marquis, M.A.; Sturmer, T. Dipeptidyl-peptidase-4 inhibitors and pancreatic cancer: A cohort study. Diabetes Obes. Metab. 2014, 16, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Sitagliptin and pancreatic cancer risk in patients with type 2 diabetes. Eur. J. Clin. Investig. 2016, 46, 70–79. [Google Scholar] [CrossRef]
- Knapen, L.M.; van Dalem, J.; Keulemans, Y.C.; van Erp, N.P.; Bazelier, M.T.; De Bruin, M.L.; Leufkens, H.G.; Croes, S.; Neef, C.; de Vries, F.; et al. Use of incretin agents and risk of pancreatic cancer: A population-based cohort study. Diabetes Obes. Metab. 2016, 18, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, L. Incretin-based drugs and adverse pancreatic events: Almost a decade later and uncertainty remains. Diabetes Care 2015, 38, 951–953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Egan, A.G.; Blind, E.; Dunder, K.; de Graeff, P.A.; Hummer, B.T.; Bourcier, T.; Rosebraugh, C. Pancreatic safety of incretin-based drugs—FDA and EMA assessment. N. Engl. J. Med. 2014, 370, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Abd El Aziz, M.; Cahyadi, O.; Meier, J.J.; Schmidt, W.E.; Nauck, M.A. Incretin-based glucose-lowering medications and the risk of acute pancreatitis and malignancies: A meta-analysis based on cardiovascular outcomes trials. Diabetes Obes. Metab. 2020, 22, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Boniol, M.; Franchi, M.; Bota, M.; Leclercq, A.; Guillaume, J.; van Damme, N.; Corrao, G.; Autier, P.; Boyle, P. Incretin-Based Therapies and the Short-term Risk of Pancreatic Cancer: Results From Two Retrospective Cohort Studies. Diabetes Care 2018, 41, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sun, J.; Han, M.; Cho, Y.; Lee, J.Y.; Nam, C.M.; Kang, E.S. Nationwide Trends in Pancreatitis and Pancreatic Cancer Risk Among Patients with Newly Diagnosed Type 2 Diabetes Receiving Dipeptidyl Peptidase 4 Inhibitors. Diabetes Care 2019, 42, 2057–2064. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, D.J.; Shin, S. Incident cancer risk in dipeptidyl peptidase-4 inhibitor-treated patients with type 2 diabetes mellitus. Cancer Manag. Res. 2019, 11, 7427–7438. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; Douros, A.; Yin, H.; Yu, O.H.; Faillie, J.L.; Montastruc, F.; Platt, R.W.; Bouganim, N.; Azoulay, L. Incretin based drugs and risk of cholangiocarcinoma among patients with type 2 diabetes: Population based cohort study. BMJ 2018, 363, k4880. [Google Scholar] [CrossRef]
- Marzioni, M.; Alpini, G.; Saccomanno, S.; Candelaresi, C.; Venter, J.; Rychlicki, C.; Fava, G.; Francis, H.; Trozzi, L.; Glaser, S.; et al. Glucagon-like peptide-1 and its receptor agonist exendin-4 modulate cholangiocyte adaptive response to cholestasis. Gastroenterology 2007, 133, 244–255. [Google Scholar] [CrossRef]
- Chen, B.D.; Zhao, W.C.; Dong, J.D.; Sima, H. Expression of GLP-1R protein and its clinical role in intrahepatic cholangiocarcinoma tissues. Mol. Biol. Rep. 2014, 41, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Alpini, G.; Saccomanno, S.; Candelaresi, C.; Venter, J.; Rychlicki, C.; Fava, G.; Francis, H.; Trozzi, L.; Benedetti, A. Exendin-4, a glucagon-like peptide 1 receptor agonist, protects cholangiocytes from apoptosis. Gut 2009, 58, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; Yin, H.; Yu, O.H.Y.; Pollak, M.N.; Azoulay, L. Incretin-based Drugs and the Incidence of Colorectal Cancer in Patients with Type 2 Diabetes. Epidemiology 2018, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Dicembrini, I.; Nreu, B.; Montereggi, C.; Mannucci, E.; Monami, M. Risk of cancer in patients treated with dipeptidyl peptidase-4 inhibitors: An extensive meta-analysis of randomized controlled trials. Acta Diabetol. 2020, 57, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Ueki, K.; Tanizawa, Y.; Nakamura, J.; Yamada, Y.; Inagaki, N.; Watada, H.; Shimomura, I.; Nishimura, R.; Miyoshi, H.; Abiko, A.; et al. Long-term safety and efficacy of alogliptin, a DPP-4 inhibitor, in patients with type 2 diabetes: A 3-year prospective, controlled, observational study (J-BRAND Registry). BMJ Open Diabetes Res. Care 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, J.; Yuan, Y.; Zou, Z.; Lai, X.; Rahmani, D.M.; Wang, F.; Xi, Y.; Huang, Q.; Bu, S. Dipeptidyl peptidase-4 inhibitors and cancer risk in patients with type 2 diabetes: A meta-analysis of randomized clinical trials. Sci. Rep. 2017, 7, 8273. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Yu, S.; Yang, Z.; Wu, S.; Gao, L.; Wang, H.; Zhang, Y.; Zhan, S.; Ji, L.; Sun, F. Effect of incretin-based therapies on cancers of digestive system among 101 595 patients with type 2 diabetes mellitus: A systematic review and network meta-analysis combining 84 trials with a median duration of 30 weeks. BMJ Open Diabetes Res. Care 2019, 7, e000728. [Google Scholar] [CrossRef] [PubMed]
- Stattin, P.; Bjor, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef]
- Shah, C.; Hong, Y.R.; Bishnoi, R.; Ali, A.; Skelton, W.P.t.; Dang, L.H.; Huo, J.; Dang, N.H. Impact of DPP4 Inhibitors in Survival of Patients With Prostate, Pancreas, and Breast Cancer. Front. Oncol. 2020, 10, 405. [Google Scholar] [CrossRef]
- Ali, A.; Fuentes, A.; Skelton, W.I.; Wang, Y.; McGorray, S.; Shah, C.; Bishnoi, R.; Dang, L.H.; Dang, N.H. A multi-center retrospective analysis of the effect of DPP4 inhibitors on progression-free survival in advanced airway and colorectal cancers. Mol. Clin. Oncol. 2019, 10, 118–124. [Google Scholar] [CrossRef]
- Noh, Y.; Jeon, S.M.; Shin, S. Association between glucose-lowering treatment and cancer metastasis among patients with preexisting type 2 diabetes and incident malignancy. Int. J. Cancer 2019, 144, 1530–1539. [Google Scholar] [CrossRef]
- Rathmann, W.; Kostev, K. Association of dipeptidyl peptidase 4 inhibitors with risk of metastases in patients with type 2 diabetes and breast, prostate or digestive system cancer. J. Diabetes Complicat. 2017, 31, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Vihervuori, V.J.; Talala, K.; Taari, K.; Lahtela, J.; Tammela, T.; Auvinen, A.; Raittinen, P.V.H.; Murtola, T.J. Antidiabetic drugs and prostate cancer prognosis in a Finnish population-based cohort. Cancer Epidemiol. Biomark. Prev. 2021. [Google Scholar] [CrossRef]
- Villarreal-Garza, C.; Shaw-Dulin, R.; Lara-Medina, F.; Bacon, L.; Rivera, D.; Urzua, L.; Aguila, C.; Ramirez-Morales, R.; Santamaria, J.; Bargallo, E.; et al. Impact of diabetes and hyperglycemia on survival in advanced breast cancer patients. Exp. Diabetes Res. 2012, 2012, 732027. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ning, N.Y.; Rao, Q.X.; Chen, R.; Wang, L.J.; Lin, Z.Q. Pretreatment glycemic control status is an independent prognostic factor for cervical cancer patients receiving neoadjuvant chemotherapy for locally advanced disease. BMC Cancer 2017, 17, 517. [Google Scholar] [CrossRef] [PubMed]
- Alpertunga, I.; Sadiq, R.; Pandya, D.; Lo, T.; Dulgher, M.; Evans, S.; Bennett, B.; Rennert, N.; Frank, R.C. Glycemic Control as an Early Prognostic Marker in Advanced Pancreatic Cancer. Front. Oncol. 2021, 11, 571855. [Google Scholar] [CrossRef]
- Pillozzi, S.; Bernini, A.; Spiga, O.; Lelli, B.; Petroni, G.; Bracci, L.; Niccolai, N.; Arcangeli, A. Peptides and small molecules blocking the CXCR4/CXCL12 axis overcome bone marrowinduced chemoresistance in acute leukemias. Oncol. Rep. 2019, 41, 312–324. [Google Scholar] [CrossRef]
- Zhang, F.; Cui, J.Y.; Gao, H.F.; Yu, H.; Gao, F.F.; Chen, J.L.; Chen, L. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 2020, 16, 2619–2633. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Col, N.F.; Ochs, L.; Springmann, V.; Aragaki, A.K.; Chlebowski, R.T. Metformin and breast cancer risk: A meta-analysis and critical literature review. Breast Cancer Res. Treat. 2012, 135, 639–646. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Qu, B.; Huang, X.; Song, Y.; Gao, P.; Shi, J.; Zhou, C.; Wang, Z. The potential adjunctive benefit of adding metformin to standard treatment in inoperable cancer patients: A meta-analysis of randomized controlled trials. Ann. Transl. Med. 2020, 8, 1404. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y. Metformin attenuates cells stemness and epithelialmesenchymal transition in colorectal cancer cells by inhibiting the Wnt3a/betacatenin pathway. Mol. Med. Rep. 2019, 19, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Moon, S.J.; Baek, J.A.; Lee, E.J.; Jung, K.A.; Kim, E.K.; Kim, D.S.; Lee, J.H.; Kwok, S.K.; Min, J.K.; et al. Metformin Augments Anti-Inflammatory and Chondroprotective Properties of Mesenchymal Stem Cells in Experimental Osteoarthritis. J. Immunol. 2019, 203, 127–136. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tumor Type | Possible Association Between DPP-4/CD26 Expression in Tumors and Tumor Progression | Reference |
|---|---|---|
| Breast cancer | Decreased stromal CD26 expression in tumors is associated with poor outcomes for BC patients. | [37] |
| DPP-4 knockdown in a primary mammary tumor induces tumor growth and metastasis in vivo. | [38] | |
| Colorectal cancer | High expression levels of CD26 in tumors is associated with distant metastasis and worse overall survival in CRC patients. | [40] |
| Expression of stromal CD26 after preoperative CRT is associated with tumor recurrence and prognosis in rectal cancer patients. | [41] | |
| Hepatocellular carcinoma | Low levels of DPP-4 expression in tumors are linked to the aggressiveness of HCC and poor overall survival in HCC patients. | [42] |
| Lung cancer | Restoration of DPP-4 expression in NSCLC cells contributes to inhibiting cell progression in vitro and in vivo. | [39] |
| Ovarian cancer | DPP-4 overexpression contributes to prolonged survival, decreased invasive activity, and increased chemosensitivity in vitro and in vivo. | [43,44] |
| DPP-4 expression is associated with lymph node metastasis and a worse stage in tumor samples of ovarian cancer patients. | [45] | |
| Pancreatic tumor | CD26 expression is significantly increased in tumors and its level correlates with overall survival in PDAC patients. | [46] |
| Prostate cancer | CD26 expression in prostate cancer tissues is correlated with CXCR4, PSA level, tumor residue, cancer stage, and tumor size. | [47] |
| Thyroid cancer | CD26 expression is negatively correlated with GLP-1R expression and has no significant association with the survival of patients with medullary thyroid carcinoma. | [48] |
| Urothelial carcinoma | DPP-4 overexpression in tumors is associated with the clinical aggressiveness of UCs. | [49] |
| Hematological malignancies | CD26 expression correlates with a poor response to 2′-deoxycoformycin in T-cell leukemia/lymphomas. | [50] |
| CD26 expression is associated with an unfavorable clinical outcome in B-CLL patients. | [51] | |
| CD26 expression on B-CLL cells is associated with the tumor mass and influences time to treatment. | [52] |
| Substrates | Consequences of Processing by DPP-4 | Receptor Recognition | Possible Activity of Substrates in Tumor Progression | Reference |
|---|---|---|---|---|
| Substance P | inactivation | - | promotes pancreatic cancer cell proliferation and invasion | [70] |
| Neuropeptide Y | inactivation | - | promotes myeloid cell infiltration and increases IL-6 levels in prostate cancer cell | [71] |
| CXCL9/Mig | inactivation | CXCR3 decreased | enhances breast cancer cell invasiveness/migration and reduces immune cell infiltration | [72,73] |
| CXCL10/IP-10 | inactivation | CXCR3 decreased | enhances breast cancer cell invasiveness/migration and reduces immune cell infiltration | [72,73] |
| promotes colorectal carcinoma cell invasiveness | [74] | |||
| promotes gastric cancer cell invasion | [75] | |||
| CXCL11/I-TAC | inactivation | CXCR3 decreased | enhances breast cancer cell migration | [73] |
| CXCL12/SDF-1 | inactivation | CXCR4 decreased | induces breast/mammary cancer cell migration, metastasis and chemoresistance | [38,62,76] |
| induces prostate cancer cell invasion and metastasis | [61] | |||
| effects on endometrial adenocarcinoma cell proliferation | [77] | |||
| CCL3/LD78β | inactivation | CCR3 decreased | promotes migration and invasion of esophageal squamous cell carcinoma | [78] |
| activation | CCR1/CCR5 increased | induces proliferation and invasion of oral squamous cell carcinoma | [79] | |
| CCL5/RANTES | inactivation | CCR1/CCR3 decreased | promotes breast cancer migration, invasion, metastasis and recurrence | [80,81,82] |
| activation | CCR5 increased | promotes prostate cancer cell migration and metastasis | [83] | |
| promotes pancreatic cancer cell invasion and migration | [84] | |||
| CCL11/eotaxin | inactivation | CCR3 decreased | promotes the proliferation, migration and invasion of glioblastoma cells | [85] |
| CCL14/HCC-1 | inactivation | CCR1/CCR3/CCR5 decreased | potential prognostic biomarker of hepatocellular and ovarian cancer | [86,87] |
| promotes angiogenesis and metastasis of breast cancer | [88] | |||
| suppress the tumor progression of colorectal and hepatocellular carcinoma | [89,90] | |||
| CCL22/MDC | inactivation | CCR4 decreased | increases oral cancer cell proliferation, invasion and migration | [91] |
| stimulates the migration of Tregs and impairs antitumor immunity in ovarian cancer | [92] | |||
| promotes lymph node metastasis of CCR4+ head and neck squamous cell carcinoma | [93] |
| SAVOR-TIMI 53 [96,100] | EXAMINE [97] | TECOS [98] | CARMELINA [99] | VERIFY [101] | |
|---|---|---|---|---|---|
| DPP-4 inhibitor | Saxagliptin | Alogliptin | Sitagliptin | Linagliptin | Vildagliptin + metformin |
| Follow-up (years) | 2.1 | 1.5 | 3.0 | 2.2 | up to 5 |
| Patients | 16,492 | 5380 | 14,671 | 6979 | 2001 |
| Age (years) | 65.1 | 61.0 | 65.5 | 66.1 | 54 |
| Body-mass index ⁑—mean | 31.1 | 28.7 | 30.2 | 31.4 | 31.2 |
| Glycated hemoglobin—mean (%) | 8.0 ± 1.4 | 8.0 ± 1.1 | 7.2 ± 0.5 | 7.9 | 6.7 |
| Duration of diabetes (years) | 10.3 | 7.1 | 11.6 | 15.0 | 3.3 (months) |
| Metformin use—no. (%) | |||||
| Placebo—no. (%) | 5684 (69.2) | 1805 (67.4) | 6030 (82.2) | 1927 (55.3) | all |
| DPP-4 inhibitors—no. (%) | 5789 (69.9) | 1757 (65.0) | 5936 (81.0) | 1881 (53.8) | all |
| Inclusion criteria | Established CVD, multiple risk factors for VD | An ACS with 15 to 90 days before randomization | Established CVD | High CV and renal risk | Diagnosed for type 2 diabetes within 2 years prior to enrolment |
| Primary outcome | 3P-MACE | 3P-MACE | 4P-MACE | 3P-MACE | The time from randomization to initial treatment failure (*) |
| Incidence of cancer | ** | ||||
| Placebo—no. (%) | 362 (4.4) | 51 (1.9) | 371 (5.1) | 134 (3.8) | 54 (5.4) ※ |
| DPP-4 inhibitors—no. (%) | 327 (3.9) | 55 (2.0) | 341 (4.7) | 116 (3.3) | 62 (6.2) ※※ |
| P value | 0.15 | 0.77 | unknown | unknown | unknown |
| Pancreatic cancer | no reports | ||||
| Placebo—no. (%) | 5 | - | 10 (0.1) | 4 (0.1) | 2 (0.3) ※ |
| DPP-4 inhibitors—no. (%) | 12 | - | 9 (0.1) | 11 (0.3) | 3 (0.3) ※※ |
| Other cancers—no. (%) | unknown | unknown | unknown | Colon cancer | Prostate cancer |
| Placebo: 8 (0.2) | Met: 0 | ||||
| Linagliptin: 6 (0.2) | Vildagliptin + Met: 6 (0.6) | ||||
| Gastric cancer | Breast cancer | ||||
| Placebo: 3 (0.1) | Met: 1 (0.1) | ||||
| Linagliptin: 0 | Vildagliptin + Met: 3 (0.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawakita, E.; Koya, D.; Kanasaki, K. CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology. Cancers 2021, 13, 2191. https://doi.org/10.3390/cancers13092191
Kawakita E, Koya D, Kanasaki K. CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology. Cancers. 2021; 13(9):2191. https://doi.org/10.3390/cancers13092191
Chicago/Turabian StyleKawakita, Emi, Daisuke Koya, and Keizo Kanasaki. 2021. "CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology" Cancers 13, no. 9: 2191. https://doi.org/10.3390/cancers13092191
APA StyleKawakita, E., Koya, D., & Kanasaki, K. (2021). CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology. Cancers, 13(9), 2191. https://doi.org/10.3390/cancers13092191