
Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC)



Abstract
Simple Summary
Abstract
1. Non-Medullary Thyroid Carcinoma (NMTC)
2. Familial Non-Medullary Thyroid Carcinoma (FNMTC)
3. Genetics of Familial NMTC (FNMTC)
3.1. Predisposing Loci Identified with Linkage Analysis and Candidate Gene Screening or Whole Exome Sequencing
3.1.1. fPTC/PRN1 (1q21)
3.1.2. NMTC1 (2q21)
3.1.3. SRGAP1 (12p14)
3.1.4. MNG1 (14q32)
3.1.5. miR-886-3p and miR-20a
3.1.6. SRRM2
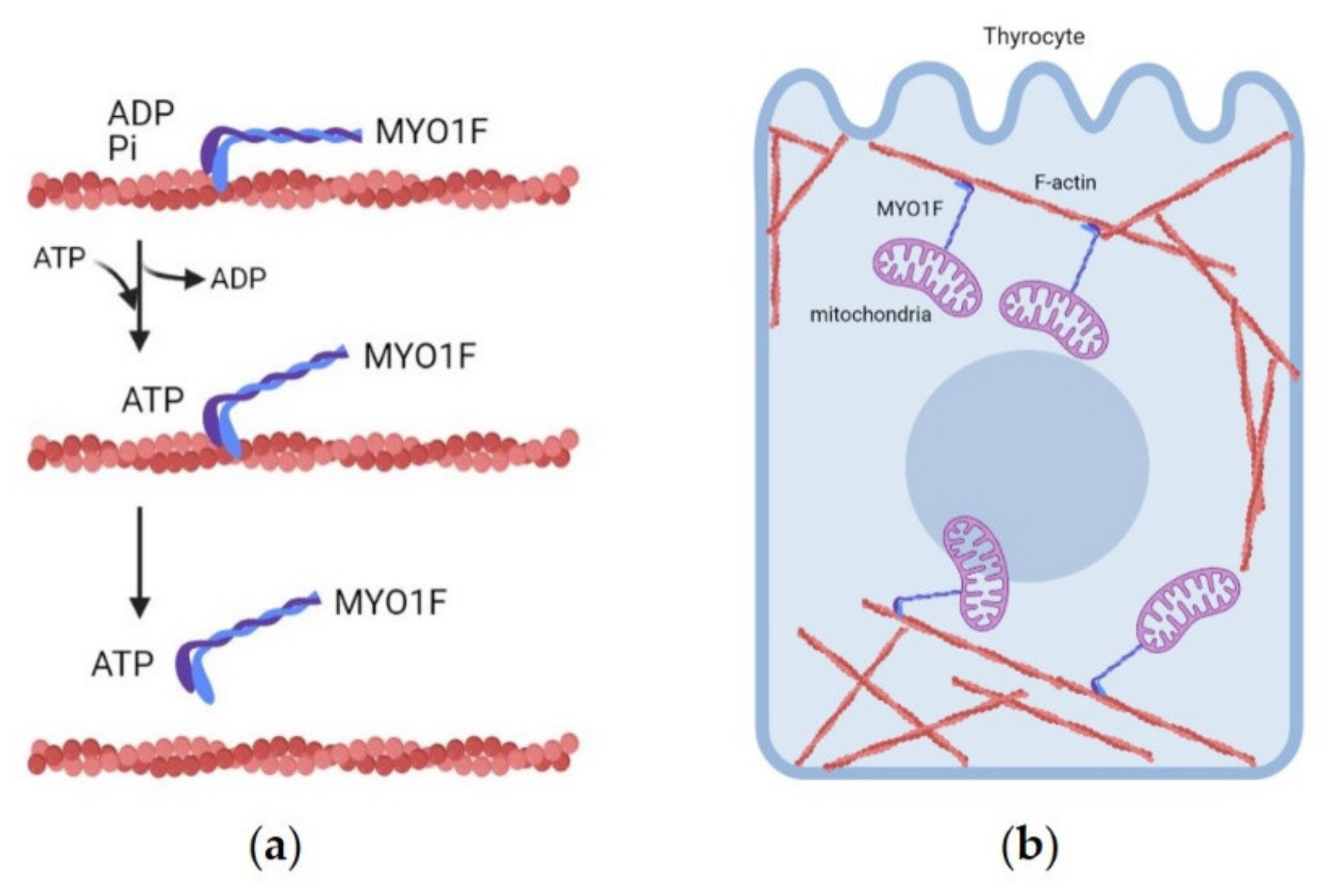
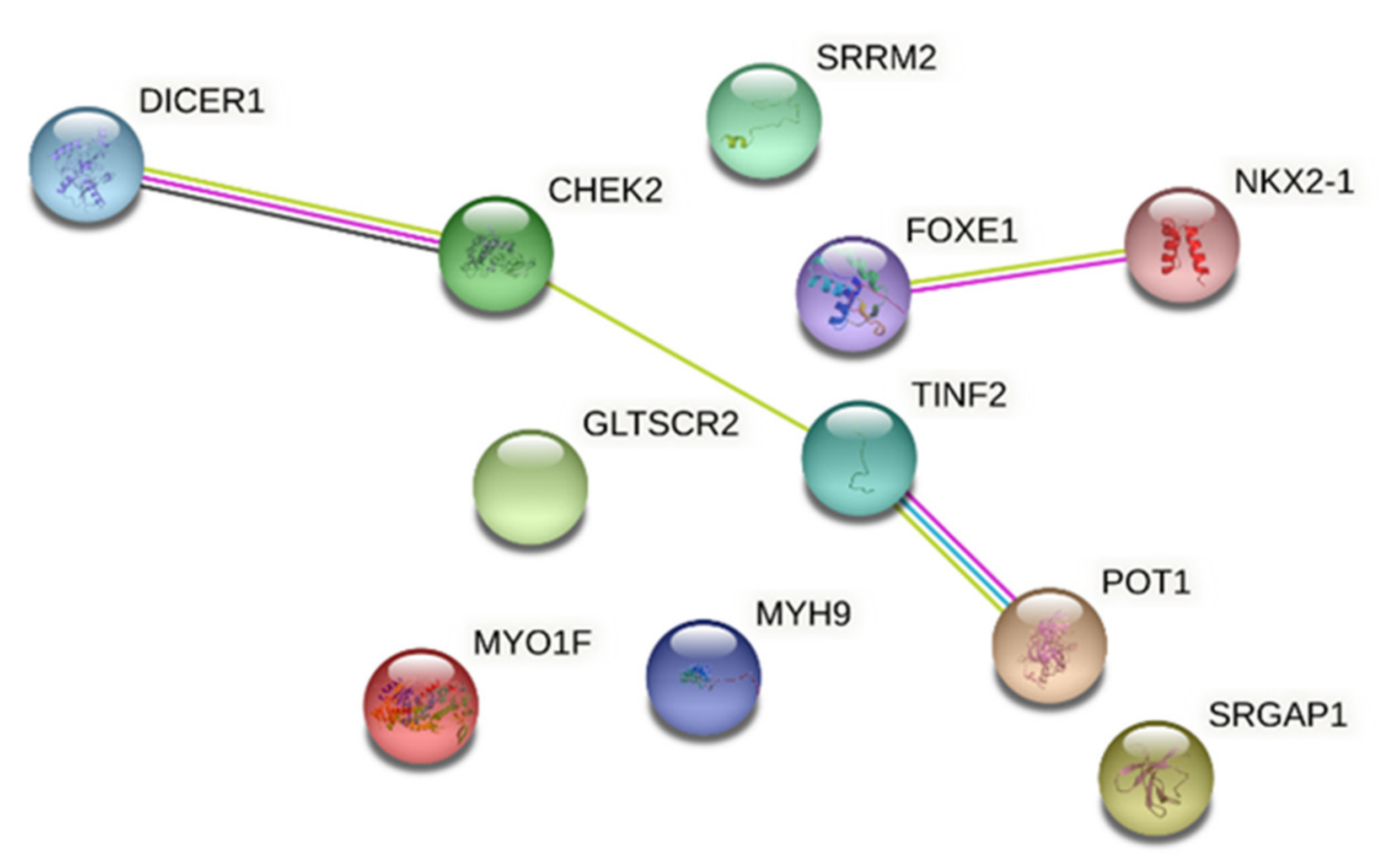
3.1.7. MYO1F (19p13.2 Locus)
3.1.8. FTEN (8p23.1-p22) and 8q24
3.1.9. MAP2K5
3.1.10. NOP53
3.2. Predisposing Loci Identified via Genome-Wide Association Studies (GWAS)
3.2.1. q22.33 Locus
FOXE1
PTCS2-MYH9
3.2.2. NKX2-1 (14q13.3 Locus)
3.2.3. DIRC3 (2q35 Locus)
3.3. Whole Genome Sequencing (WGS) Analysis
3.3.1. CHEK2
3.3.2. Telomere Abnormalities
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schlumberger, M.J. Papillary and Follicular Thyroid Carcinoma. N. Engl. J. Med. 1998, 338, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Kim, J.; Gosnell, J.E.; Roman, S.A. Geographic influences in the global rise of thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.R.; Dwyer, T.; McArdle, K.; Tucker, P.; Shugg, D. The changing incidence and spectrum of thyroid carcinoma in Tasmania (1978–1998) during a transition from iodine sufficiency to iodine deficiency. J. Clin. Endocrinol. Metab. 2000, 85, 1513–1517. [Google Scholar] [PubMed]
- Cameselle-Teijeiro, J.M.; Sobrinho-Simões, M. New WHO classification of thyroid tumors: A pragmatic categorization of thyroid gland neoplasms. Endocrinol. Diabetes Nutr. 2018, 65, 133–135. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs; IARC (International Agency for Research on Cancer): Lyon, France, 2017; ISBN 978-92-832-4493-6.
- Aschebrook-Kilfoy, B.; Ward, M.H.; Sabra, M.M.; Devesa, S.S. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011, 21, 125–134. [Google Scholar] [CrossRef]
- Coca-Pelaz, A.; Shah, J.P.; Hernandez-Prera, J.C.; Ghossein, R.A.; Rodrigo, J.P.; Hartl, D.M.; Olsen, K.D.; Shaha, A.R.; Zafereo, M.; Suarez, C.; et al. Papillary Thyroid Cancer—Aggressive Variants and Impact on Management: A Narrative Review. Adv. Ther. 2020, 37, 3112–3128. [Google Scholar] [CrossRef] [PubMed]
- Caria, P.; Vanni, R. Cytogenetic and molecular events in adenoma and well-differentiated thyroid follicular-cell neoplasia. Cancer Genet. Cytogenet. 2010, 203, 21–29. [Google Scholar] [CrossRef]
- Hundahl, S.A.; Fleming, I.D.; Fremgen, A.M.; Menck, H.R. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995. Cancer 1998, 83, 2638–2648. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Galetti, V. Iodine intake as a risk factor for thyroid cancer: A comprehensive review of animal and human studies. Thyroid Res. 2015, 8, 1–21. [Google Scholar] [CrossRef]
- Sobrinho-Simões, M.; Eloy, C.; Magalhães, J.; Lobo, C.; Amaro, T. Follicular thyroid carcinoma. Mod. Pathol. 2011, 24, S10–S18. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, Z.; Chen, C.; Zhao, B.; Cao, H.; Li, T.; Liu, X.; Wang, W.; Li, Y. Clinical characteristics and prognostic factors of Hurthle cell carcinoma: A population based study. BMC Cancer 2020, 20, 407. [Google Scholar] [CrossRef]
- Nunes da Silva, T.; Limbert, E.; Leite, V. Poorly Differentiated Thyroid Carcinoma Patients with Detectable Thyroglobulin Levels after Initial Treatment Show an Increase in Mortality and Disease Recurrence. ETJ 2018, 7, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly Differentiated Carcinoma of the Thyroid Gland: Current Status and Future Prospects. Thyroid 2019, 29, 311–321. [Google Scholar] [CrossRef]
- De Leo, S.; Trevisan, M.; Fugazzola, L. Recent advances in the management of anaplastic thyroid cancer. Thyroid Res. 2020, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.R.; Duffield, A.; Wilkinson, S.J.; Ware, R.; Greenaway, T.M.; Percival, J.; Hoffman, L. Two families with an autosomal dominant inheritance pattern for papillary carcinoma of the thyroid. J. Clin. Endocrinol. Metab. 1997, 82, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Kraimps, J.L.; Bouin-Pineau, M.H.; Amati, P.; Mothes, D.; Bonneau, D.; Maréchaud, R.; Barbier, J. Familial papillary carcinoma of the thyroid. Surgery 1997, 121, 715–718. [Google Scholar] [CrossRef]
- Malchoff, C.D.; Malchoff, D.M. Familial nonmedullary thyroid carcinoma. Cancer Control 2006, 13, 106–110. [Google Scholar] [CrossRef]
- Xu, B.; Ghossein, R. Evolution of the Histologic Classification of Thyroid Neoplasms and its Impact on Clinical Management. Eur. J. Surg. Oncol. 2018, 44, 338–347. [Google Scholar] [CrossRef]
- Grossman, R.F.; Tu, S.H.; Duh, Q.Y.; Siperstein, A.E.; Novosolov, F.; Clark, O.H. Familial nonmedullary thyroid cancer. An emerging entity that warrants aggressive treatment. Arch. Surg. 1995, 130, 892–897, discussion 898–899. [Google Scholar]
- Loh, K.-C. Familial Nonmedullary Thyroid Carcinoma: A Meta-Review of Case Series. Thyroid 1997, 7, 107–113. [Google Scholar] [CrossRef]
- Alsanea, O.; Wada, N.; Ain, K.; Wong, M.; Taylor, K.; Ituarte, P.H.G.; Treseler, P.A.; Weier, H.-U.; Freimer, N.; Siperstein, A.E.; et al. Is familial non-medullary thyroid carcinoma more aggressive than sporadic thyroid cancer? A multicenter series. Surgery 2000, 128, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Noguchi, S.; Yamashita, H.; Yamashita, H.; Watanabe, S.; Ogawa, T.; Tsuno, A.; Murakami, A.; Miyauchi, A. Mutational analysis of the APC gene in cribriform-morula variant of papillary thyroid carcinoma. World J. Surg. 2006, 30, 775–779. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, W.; Li, J.; Su, A.; Wei, T.; Liu, F.; Zhu, J. Endocrine tumours: Familial nonmedullary thyroid carcinoma is a more aggressive disease: A systematic review and meta-analysis. Eur. J. Endocrinol. 2015, 172, R253–R262. [Google Scholar] [CrossRef]
- Murff, H.J.; Spigel, D.R.; Syngal, S. Does this Patient Have a Family History of Cancer: An Evidence-Based Analysis of the Accuracy of Family Cancer History; Centre for Reviews and Dissemination: Heslington, UK, 2004. [Google Scholar]
- Mazeh, H.; Sippel, R.S. Familial Nonmedullary Thyroid Carcinoma. Thyroid 2013, 23, 1049–1056. [Google Scholar] [CrossRef]
- Bonora, E.; Tallini, G.; Romeo, G. Genetic Predisposition to Familial Nonmedullary Thyroid Cancer: An Update of Molecular Findings and State-of-the-Art Studies. J. Oncol. 2010, 2010, 385206. [Google Scholar] [CrossRef] [PubMed]
- Peiling Yang, S.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292. [Google Scholar]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Courcoutsakis, N.A.; Abati, A.; Filie, A.; Doppman, J.L.; Carney, J.A.; Shawker, T. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J. Clin. Endocrinol. Metab. 1997, 82, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-E.; Oshima, J.; Fu, Y.-H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional Cloning of the Werner’s Syndrome Gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rio Frio, T.; Bahubeshi, A.; Kanellopoulou, C.; Hamel, N.; Niedziela, M.; Sabbaghian, N.; Pouchet, C.; Gilbert, L.; O’Brien, P.K.; Serfas, K.; et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA 2011, 305, 68–77. [Google Scholar] [CrossRef] [PubMed]
- van Os, N.J.H.; Roeleveld, N.; Weemaes, C.M.R.; Jongmans, M.C.J.; Janssens, G.O.; Taylor, A.M.R.; Hoogerbrugge, N.; Willemsen, M.A.A.P. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar]
- Carbone, M.; Arron, S.T.; Beutler, B.; Bononi, A.; Cavenee, W.; Cleaver, J.E.; Croce, C.M.; D’Andrea, A.; Foulkes, W.D.; Gaudino, G.; et al. Tumour predisposition and cancer syndromes as models to study gene-environment interactions. Nat. Rev. Cancer. 2020, 20, 533–549. [Google Scholar] [PubMed]
- Charkes, N.D. On the prevalence of familial nonmedullary thyroid cancer in multiply affected kindreds. Thyroid 2006, 16, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, F.; Stark, M.; Tocco, T.; Ayadi, H.; Delisle, M.; Goldgar, D.; Schlumberger, M.; Romeo, G.; Canzian, F. Genetic Heterogeneity in Familial Nonmedullary Thyroid Carcinoma: Exclusion of Linkage to RET, MNG1, and TCO in 56 Families. J. Clin. Endocrinol. Metab. 1999, 84, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Moses, W.; Weng, J.; Kebebew, E. Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid 2011, 21, 367–371. [Google Scholar] [PubMed]
- Guilmette, J.; Nosé, V. Hereditary and familial thyroid tumours. Histopathology 2018, 72, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Navas-Carrillo, D.; Ríos, A.; Rodríguez, J.M.; Parrilla, P.; Orenes-Piñero, E. Familial nonmedullary thyroid cancer: Screening, clinical, molecular and genetic findings. Biochim. Biophys. Acta 2014, 1846, 468–476. [Google Scholar] [CrossRef]
- Malchoff, C.D.; Sarfarazi, M.; Tendler, B.; Forouhar, F.; Whalen, G.; Joshi, V.; Arnold, A.; Malchoff, D.M. Papillary thyroid carcinoma associated with papillary renal neoplasia: Genetic linkage analysis of a distinct heritable tumor syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 1758–1764. [Google Scholar]
- McKay, J.D.; Lesueur, F.; Jonard, L.; Pastore, A.; Williamson, J.; Hoffman, L.; Burgess, J.; Duffield, A.; Papotti, M.; Stark, M.; et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am. J. Hum. Genet. 2001, 69, 440–446. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Bronisz, A.; Liyanarachchi, S.; Nagy, R.; Li, W.; Huang, Y.; Akagi, K.; Saji, M.; Kula, D.; Wojcicka, A.; et al. SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J. Clin. Endocrinol. Metab. 2013, 98, E973–E980. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cdc42--the centre of polarity. J. Cell. Sci. 2004, 117, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Canzian, F.; Shayeghi, M.; Stark, M.; Shugart, Y.Y.; Biggs, P.; Mangion, J.; Hamoudi, R.; Rosenblatt, J.; Buu, P.; et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am. J. Hum. Genet. 1997, 61, 1123–1130. [Google Scholar] [CrossRef]
- Khan, N.E.; Bauer, A.J.; Schultz, K.A.P.; Doros, L.; Decastro, R.M.; Ling, A.; Lodish, M.B.; Harney, L.A.; Kase, R.G.; Carr, A.G.; et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Macrae, I.J.; Zhou, K.; Li, F.; Repic, A.; Brooks, A.N.; Cande, W.Z.; Adams, P.D.; Doudna, J.A. Structural basis for double-stranded RNA processing by Dicer. Science 2006, 311, 195–198. [Google Scholar] [CrossRef]
- Rutter, M.M.; Jha, P.; Schultz, K.A.P.; Sheil, A.; Harris, A.K.; Bauer, A.J.; Field, A.L.; Geller, J.; Hill, D.A. DICER1 Mutations and Differentiated Thyroid Carcinoma: Evidence of a Direct Association. J. Clin. Endocrinol. Metab. 2016, 101, 1–5. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhang, L.; Holloway, A.K.; Wu, X.; Su, L.; Kebebew, E. MiR-886-3p regulates cell proliferation and migration, and is dysregulated in familial non-medullary thyroid cancer. PLoS ONE 2011, 6, e24717. [Google Scholar] [CrossRef]
- Yuan, Z.; Yang, Z.; Zheng, Q. Deregulation of microRNA expression in thyroid tumors. J. Zhejiang Univ. Sci. B 2014, 15, 212–224. [Google Scholar] [CrossRef]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; de la Chapelle, A. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 2015, 5, 10566. [Google Scholar] [CrossRef] [PubMed]
- Canzian, F.; Amati, P.; Harach, H.R.; Kraimps, J.L.; Lesueur, F.; Barbier, J.; Levillain, P.; Romeo, G.; Bonneau, D. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am. J. Hum. Genet. 1998, 63, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Pasini, B.; Ceccherini, I.; Romeo, G. RET mutations in human disease. Trends Genet. 1996, 12, 138–144. [Google Scholar] [CrossRef]
- Diquigiovanni, C.; Bergamini, C.; Evangelisti, C.; Isidori, F.; Vettori, A.; Tiso, N.; Argenton, F.; Costanzini, A.; Iommarini, L.; Anbunathan, H.; et al. Mutant MYO1F alters the mitochondrial network and induces tumor proliferation in thyroid cancer. Int. J. Cancer 2018, 143, 1706–1719. [Google Scholar] [CrossRef]
- He, H.; Nagy, R.; Liyanarachchi, S.; Jiao, H.; Li, W.; Suster, S.; Kere, J.; de la Chapelle, A. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. 2009, 69, 625–631. [Google Scholar] [CrossRef]
- Cavaco, B.M.; Batista, P.F.; Sobrinho, L.G.; Leite, V. Mapping a New Familial Thyroid Epithelial Neoplasia Susceptibility Locus to Chromosome 8p23.1-p22 by High-Density Single-Nucleotide Polymorphism Genome-Wide Linkage Analysis. J. Clin. Endocrinol. Metab. 2008, 93, 4426–4430. [Google Scholar] [CrossRef]
- Ye, F.; Gao, H.; Xiao, L.; Zuo, Z.; Liu, Y.; Zhao, Q.; Chen, H.; Feng, W.; Fu, B.; Sun, L.; et al. Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int. J. Cancer 2019, 144, 1321–1330. [Google Scholar] [CrossRef]
- Orois, A.; Gara, S.K.; Mora, M.; Halperin, I.; Martínez, S.; Alfayate, R.; Kebebew, E.; Oriola, J. NOP53 as A Candidate Modifier Locus for Familial Non-Medullary Thyroid Cancer. Genes 2019, 10, 899. [Google Scholar] [CrossRef]
- Lee, S.; Ahn, Y.-M.; Kim, J.-Y.; Cho, Y.-E.; Park, J.-H. Downregulation of NOP53 Ribosome Biogenesis Factor Leads to Abnormal Nuclear Division and Chromosomal Instability in Human Cervical Cancer Cells. Pathol. Oncol. Res. 2020, 26, 453–459. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Jonasson, J.G.; Sigurdsson, A.; Bergthorsson, J.T.; He, H.; Blondal, T.; Geller, F.; Jakobsdottir, M.; et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 2009, 41, 460–464. [Google Scholar] [CrossRef]
- De Felice, M.; Ovitt, C.; Biffali, E.; Rodriguez-Mallon, A.; Arra, C.; Anastassiadis, K.; Macchia, P.E.; Mattei, M.G.; Mariano, A.; Schöler, H.; et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat. Genet. 1998, 19, 395–398. [Google Scholar] [CrossRef]
- Bonora, E.; Rizzato, C.; Diquigiovanni, C.; Oudot-Mellakh, T.; Campa, D.; Vargiolu, M.; Guedj, M.; The NMTC Consortium; McKay, J.D.; Romeo, G.; et al. The FOXE1 locus is a major genetic determinant for familial nonmedullary thyroid carcinoma: FOXE1 and familial nonmedullary thyroid carcinoma. Int. J. Cancer 2014, 134, 2098–2107. [Google Scholar] [CrossRef]
- Tomaz, R.A.; Sousa, I.; Silva, J.G.; Santos, C.; Teixeira, M.R.; Leite, V.; Cavaco, B.M. FOXE1 polymorphisms are associated with familial and sporadic nonmedullary thyroid cancer susceptibility. Clin. Endocrinol. 2012, 77, 926–933. [Google Scholar] [CrossRef]
- Pereira, J.S.; da Silva, J.G.; Tomaz, R.A.; Pinto, A.E.; Bugalho, M.J.; Leite, V.; Cavaco, B.M. Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine 2015, 49, 204–214. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, W.; Liyanarachchi, S.; Jendrzejewski, J.; Srinivas, M.; Davuluri, R.V.; Nagy, R.; de la Chapelle, A. Genetic Predisposition to Papillary Thyroid Carcinoma: Involvement of FOXE1, TSHR, and a Novel lincRNA Gene, PTCSC2. J. Clin. Endocrinol. Metab. 2015, 100, E164–E172. [Google Scholar] [CrossRef]
- Wang, Y.; He, H.; Li, W.; Phay, J.; Shen, R.; Yu, L.; Hancioglu, B.; de la Chapelle, A. MYH9 binds to lncRNA gene PTCSC2 and regulates FOXE1 in the 9q22 thyroid cancer risk locus. Proc. Natl. Acad. Sci. USA 2017, 114, 474–479. [Google Scholar] [CrossRef]
- Sellers, J.R. Myosins: A diverse superfamily. Biochim. Biophys. Acta Mol. Cell Res. 2000, 1496, 3–22. [Google Scholar] [CrossRef]
- Guazzi, S.; Price, M.; De Felice, M.; Damante, G.; Mattei, M.G.; Di Lauro, R. Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and displays a novel DNA binding specificity. EMBO J. 1990, 9, 3631–3639. [Google Scholar] [CrossRef]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; Chapelle, A. de la The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.W.; Lang, B.H.H.; Liu, T.; Shum, C.K.Y.; So, M.-T.; Lau, D.K.C.; Leon, T.Y.Y.; Cherny, S.S.; Tsai, S.Y.; Lo, C.-Y.; et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J. Natl. Cancer Inst. 2009, 101, 162–175. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Jonasson, J.G.; Masson, G.; He, H.; Jonasdottir, A.; Sigurdsson, A.; Stacey, S.N.; Johannsdottir, H.; et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat. Genet. 2012, 44, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Thorleifsson, G.; Sigurdsson, J.K.; Stefansdottir, L.; Jonasson, J.G.; Gudjonsson, S.A.; Gudbjartsson, D.F.; Masson, G.; Johannsdottir, H.; Halldorsson, G.H.; et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 2017, 8, 14517. [Google Scholar] [CrossRef] [PubMed]
- Son, H.-Y.; Hwangbo, Y.; Yoo, S.-K.; Im, S.-W.; Yang, S.D.; Kwak, S.-J.; Park, M.S.; Kwak, S.H.; Cho, S.W.; Ryu, J.S.; et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat. Commun. 2017, 8, 15966. [Google Scholar] [CrossRef]
- Mahdieh, N.; Rabbani, B. An overview of mutation detection methods in genetic disorders. Iran. J. Pediatr. 2013, 23, 375–388. [Google Scholar]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Försti, A. Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef]
- Siołek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Giangiobbe, S.; Skopelitou, D.; Miao, B.; Paramasivam, N.; Diquigiovanni, C.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. Whole Genome Sequencing Prioritizes CHEK2, EWSR1, and TIAM1 as Possible Predisposition Genes for Familial Non-Medullary Thyroid Cancer. Front. Endocrinol. 2021, 12, 600682. [Google Scholar] [CrossRef]
- Wójcicka, A.; Czetwertyńska, M.; Świerniak, M.; Długosińska, J.; Maciąg, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Płoski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.-P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect—Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef]
- Yuan, X.; Liu, T.; Xu, D. Telomerase reverse transcriptase promoter mutations in thyroid carcinomas: Implications in precision oncology—A narrative review. Ann. Transl. Med. 2020, 8, 1244. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte Telomere Length and Risk of Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metab. 2008, 93, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Capuano, S.; Capezzone, M.; Benigni, M.; Pisu, M.; Marchisotta, S.; Pacini, F. Lack of mutations of the telomerase RNA component in familial papillary thyroid cancer with short telomeres. Thyroid 2012, 22, 363–368. [Google Scholar] [CrossRef]
- He, M.; Bian, B.; Gesuwan, K.; Gulati, N.; Zhang, L.; Nilubol, N.; Kebebew, E. Telomere Length is Shorter in Affected Members of Families with Familial Nonmedullary Thyroid Cancer. Thyroid 2013, 23, 301–307. [Google Scholar] [CrossRef]
- He, H.; Li, W.; Comiskey, D.F.; Liyanarachchi, S.; Nieminen, T.T.; Wang, Y.; DeLap, K.E.; Brock, P.; de la Chapelle, A. A Truncating Germline Mutation of TINF2 in Individuals with Thyroid Cancer or Melanoma Results in Longer Telomeres. Thyroid 2020, 30, 204–213. [Google Scholar] [CrossRef]
- Srivastava, A.; Miao, B.; Skopelitou, D.; Kumar, V.; Kumar, A.; Paramasivam, N.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer. Cancers 2020, 12, 1441. [Google Scholar] [CrossRef]
- Wilson, T.L.-S.; Hattangady, N.; Lerario, A.M.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Cha, K.B.; Else, T. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam. Cancer 2017, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.A.; Lupo, P.J.; Morton, L.M.; Yasui, Y.A.; Sapkota, Y.A.; Arnold, M.A.; Aubert, G.; Neglia, J.P.; Turcotte, L.M.; Leisenring, W.M.; et al. Genetic variation in POT1 and risk of thyroid subsequent malignant neoplasm: A report from the Childhood Cancer Survivor Study. PLoS ONE 2020, 15, e0228887. [Google Scholar]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Civitareale, D.; Lonigro, R.; Sinclair, A.J.; Di Lauro, R. A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO J. 1989, 8, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.P.; López-Márquez, A.; Santisteban, P. Thyroid transcription factors in development, differentiation and disease. Nat. Rev. Endocrinol. 2015, 11, 29–42. [Google Scholar] [CrossRef]
- Lazzaro, D.; Price, M.; de Felice, M.; Di Lauro, R. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development 1991, 113, 1093–1104. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Robenshtok, E.; Cantara, S.; Castagna, M.G. Familial non-medullary thyroid cancer: A critical review. J. Endocrinol. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Chromosomal Position | Locus Name | Candidate Gene | Reference | Technique |
|---|---|---|---|---|
| 1q21 | fPTC1/PRN1 | - | [42] | Linkage Analysis/WES |
| 2q21 | NMTC1 | - | [43] | |
| 12p14 | - | SRGAP1 | [44] | |
| 14q32 | MNG1 | DICER1 | [34,46] | |
| - | miR-886-3p and miR-20a | [51] | ||
| 16p13.3 | - | SRRM2 | [53] | |
| 19p13.2 | TCO | MYO1F | [54,56] | |
| 8q24 | - | AK023948 | [57] | |
| 8p23.1-p22 | FTEN | - | [58] | |
| 15q23 | - | MAP2K5 | [59] | |
| 19q13.33 | - | NOP53 | [60] | |
| 9q22.33 | - | FOXE1 | [62] | GWAS |
| 9q22.33 | - | PTCS2 | [67] | |
| 9q22.33 | - | MYH9 | [68] | |
| 14q13.3 | - | NKX2-1 | [62,72] | |
| 2q35 | - | DIRC3 | [73,74,75] | |
| 22q12.1 | - | CHEK2 | [79] | WGS |
| 14q12 | - | TINF2 | [91] | |
| 7q31.33 | - | POT1 | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diquigiovanni, C.; Bonora, E. Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC). Cancers 2021, 13, 2178. https://doi.org/10.3390/cancers13092178
Diquigiovanni C, Bonora E. Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC). Cancers. 2021; 13(9):2178. https://doi.org/10.3390/cancers13092178
Chicago/Turabian StyleDiquigiovanni, Chiara, and Elena Bonora. 2021. "Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC)" Cancers 13, no. 9: 2178. https://doi.org/10.3390/cancers13092178
APA StyleDiquigiovanni, C., & Bonora, E. (2021). Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC). Cancers, 13(9), 2178. https://doi.org/10.3390/cancers13092178