
Exploiting Clonal Evolution to Improve the Diagnosis and Treatment Efficacy Prediction in Pediatric AML


 , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Pediatric AML: Clinical Presentation
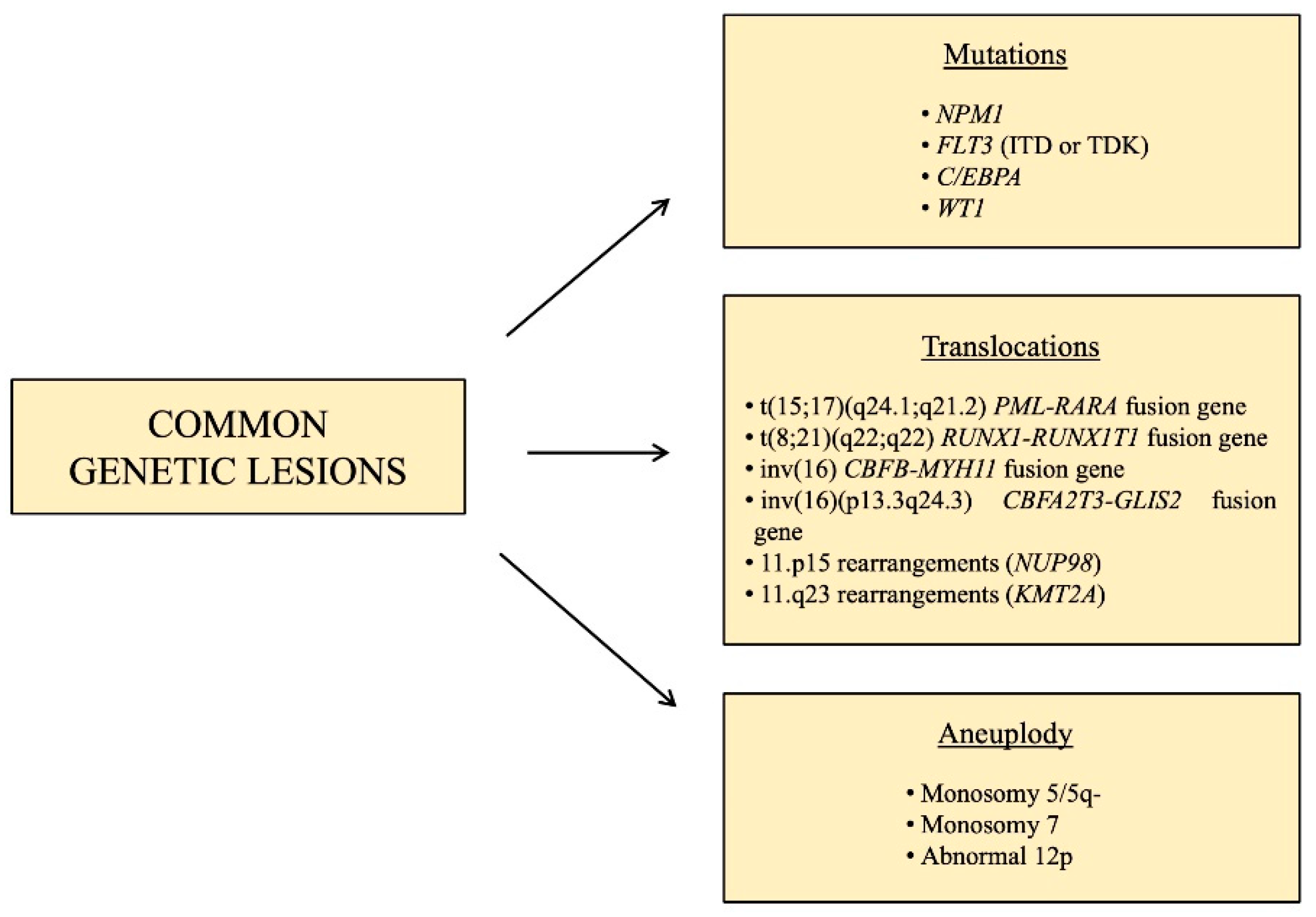
1.2. Pediatric AML: Common Genetic Lesions
1.3. Clonal Evolution in Pediatric AML: From Diagnosis to Relapse
- i.
- Cohesin complex gene mutations;
- ii.
- Transcription factors and epigenetic regulators mutations;
- iii.
- Signaling molecules mutations.
2. Identifying Genetic Heterogeneity and Subclones Populations Exploiting High Throughput Techniques
3. iPSC Model as a Promising Tool to Study the Clonal Evolution of AML
4. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.J.M.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.S.; et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef]
- Masetti, R.; Castelli, I.; Astolfi, A.; Bertuccio, S.N.; Indio, V.; Togni, M.; Belotti, T.; Serravalle, S.; Tarantino, G.; Zecca, M.; et al. Genomic complexity and dynamics of clonal evolution in childhood acute myeloid leukemia studied with whole-exome sequencing. Oncotarget 2016, 7, 56746–56757. [Google Scholar] [CrossRef]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Pession, A.; Locatelli, F. Acute myeloid leukemia in infants: Biology and treatment. Front. Pediatr. 2015, 3. [Google Scholar] [CrossRef]
- Pession, A.; Masetti, R.; Rizzari, C.; Putti, M.C.; Casale, F.; Fagioli, F.; Luciani, M.; Nigro, L.L.; Menna, G.; Micalizzi, C.; et al. Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 2013, 122, 170–178. [Google Scholar] [CrossRef]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.-P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.-H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwaan, C.; van den Heuvel-Eibrink, M. Pediatric AML: From biology to clinical management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.T.; Bruel, A.V.D.; Bankhead, C.; Mitchell, C.D.; Phillips, B.; Thompson, M.J. Clinical presentation of childhood leukaemia: A systematic review and meta-analysis. Arch. Dis. Child. 2016, 101, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Masetti, R.; Rondelli, R.; Zecca, M.; Fagioli, F.; Rovelli, A.; Messina, C.; Lanino, E.; Bertaina, A.; Favre, C.; et al. Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transpl. 2015, 50, 2. [Google Scholar]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Gasperini, P.; Pession, A. All-trans retinoic acid in the treatment of pediatric acute promyelocytic leukemia. Expert Rev. Anticancer Ther. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Testi, A.M.; Pession, A.; Diverio, D.; Grimwade, D.; Gibson, B.; De Azevedo, A.C.; Moran, L.; Leverger, G.; Elitzur, S.; Hasle, H.; et al. Risk-adapted treatment of acute promyelocytic leukemia: Results from the International consortium for childhood APL. Blood 2018, 132, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hills, R.K.; Moorman, A.V.; Grimwade, D.J.; Hann, I.; Webb, D.K.H.; Wheatley, K.; De Graaf, S.S.N.; Berg, E.V.D.; Burnett, A.K.; et al. Cytogenetics of Childhood acute myeloid leukemia: United Kingdom medical research council treatment trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016, 122. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.E.; Rau, R.E. The genomics of acute myeloid leukemia in children. Cancer Metastasis Rev. 2020, 39, 189–209. [Google Scholar] [CrossRef]
- Lonetti, A.; Indio, V.; Laginestra, M.A.; Tarantino, G.; Chiarini, F.; Astolfi, A.; Bertuccio, S.N.; Martelli, A.M.; Locatelli, F.; Pession, A.; et al. Inhibition of methyltransferase DOT1L sensitizes to sorafenib treatment AML cells irrespective of MLL-rearrangements: A novel therapeutic strategy for pediatric AML. Cancers 2020, 12, 1972. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Bertuccio, S.N.; Astolfi, A.; Chiarini, F.; Lonetti, A.; Indio, V.; De Luca, M.; Bandini, J.; Serravalle, S.; Franzoni, M.; et al. Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J. Hematol. Oncol. 2017, 10, 26. [Google Scholar] [CrossRef]
- Masetti, R.; Bertuccio, S.N.; Guidi, V.; Cerasi, S.; Lonetti, A.; Pession, A. Uncommon cytogenetic abnormalities identifying high-risk acute myeloid leukemia in children. Futur. Oncol. 2020, 16, 2747–2762. [Google Scholar] [CrossRef]
- Masetti, R.; Bertuccio, S.N.; Pession, A.; Locatelli, F. CBFA2T3-GLIS2-positive acute myeloid leukaemia. A peculiar paediatric entity. Br. J. Haematol. 2018, 184, 337–347. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Yamato, G.; Ohki, K.; Tabuchi, K.; Sotomatsu, M.; Tomizawa, D.; Kinoshita, A.; Arakawa, H.; Saito, A.M.; et al. Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br. J. Haematol. 2020, 188, 528–539. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.D.E.; Branstetter, C.; Ma, J.; Li, Y.; Walsh, M.P.; Cheng, J.; Obulkasim, A.; Dang, J.; Easton, J.; Verboon, L.J.; et al. Pediatric non–down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 2017, 49, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Malinge, S.; Izraeli, S.; Crispino, J.D. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood 2009, 113, 2619–2628. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Sandahl, J.D.; Coenen, E.A.; Forestier, E.; Harbott, J.; Johansson, B.; Kerndrup, G.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; Cayuela, J.-M.; et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: An international study of 62 patients. Haematologica 2014, 99, 865–872. [Google Scholar] [CrossRef]
- Cazzaniga, G.; Dell’Oro, M.G.; Mecucci, C.; Giarin, E.; Masetti, R.; Rossi, V.; Locatelli, F.; Martelli, M.F.; Basso, G.; Pession, A.; et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 2005, 106, 1419–1422. [Google Scholar] [CrossRef][Green Version]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512.e9. [Google Scholar] [CrossRef]
- Leroy, H.; Roumier, C.; Huyghe, P.; Biggio, V.; Fenaux, P.; Preudhomme, C. CEBPA point mutations in hematological malignancies. Leukemia 2005, 19, 329–334. [Google Scholar] [CrossRef]
- Sexauer, A.N.; Tasian, S.K. Targeting FLT3 signaling in childhood acute myeloid leukemia. Front. Pediatr. 2017, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Von Neuhoff, C.; Dworzak, M.; Bourquin, J.-P.; Bradtke, J.; Göhring, G.; Escherich, G.; Fleischhack, G.; Graf, N.; Gruhn, B.; et al. Genotype-outcome correlations in pediatric AML: The impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 2017, 31, 2807–2814. [Google Scholar] [CrossRef]
- Aparicio, S.; Caldas, C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Schuback, H.L.; Ries, R.E.; Wai, D.; Hampton, O.A.; Trevino, L.R.; Alonzo, T.A.; Auvil, J.M.G.; Davidsen, T.M.; Gesuwan, P.; et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res. 2016, 76, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Shiba, N.; Yoshida, K.; Shiraishi, Y.; Okuno, Y.; Yamato, G.; Hara, Y.; Nagata, Y.; Chiba, K.; Tanaka, H.; Terui, K.; et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br. J. Haematol. 2016, 175, 476–489. [Google Scholar] [CrossRef]
- Salman, H.; Shuai, X.; Nguyen-Lefebvre, A.T.; Giri, B.; Ren, M.; Rauchman, M.; Robbins, L.; Hou, W.; Korkaya, H.; Ma, Y. SALL1 expression in acute myeloid leukemia. Oncotarget 2018, 9, 7442–7452. [Google Scholar] [CrossRef]
- Walter, C.; Schneider, M.; Von Neuhoff, N.; Hanenberg, H.; Reinhardt, D.; Rasche, M. Mutational landscape of pediatric acute myeloid leukemia: A report of the AML-BFM study group with a targeted NGS approach in 525 patients integrating De Novo, relapsed and secondary AML. Blood 2019, 134, 1398. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.; Hovestadt, V.; Ii, M.H.W.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Story, J.L.; et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef]
- Leick, M.B.; Levis, M.J. The future of targeting FLT3 activation in AML. Curr. Hematol. Malign. Rep. 2017, 12, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.-L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nat. Cell Biol. 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Pereira, W.O.; Correia, R.P.; Hamerschlak, N.; Bacal, N.S.; Campregher, P.V. immunophenotypic evolution of blast populations in pediatric acute myeloid leukemia. Einstein 2016, 14, 288–289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Cohen, N.M.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nat. Cell Biol. 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Jeong, D.; Lee, D.S.; Kim, N.; Choi, S.; Kim, K.; Kim, S.M.; Im, K.; Park, H.S.; Yun, J.; Lim, K.M.; et al. Prevalence of germline predisposition gene mutations in pediatric acute myeloid leukemia: Genetic background of pediatric AML. Leuk. Res. 2019, 85, 106210. [Google Scholar] [CrossRef]
- Porter, C.C. Germ line mutations associated with leukemias. Hematology 2016, 2016, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Yun, W.; Lee, S.-T.; Choi, J.R.; Yoo, K.H.; Koo, H.H.; Jung, C.W.; Kim, S.H. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci. Rep. 2020, 10, 14297. [Google Scholar] [CrossRef]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell 2017, 20, 329–344.e7. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable Leukemia. Cell Stem Cell 2017, 20, 315–328.e7. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, S.N.; Boudia, F.; Cambot, M.; Lopez, C.K.; Lordier, L.; Donada, A.; Robert, E.; Thirant, C.; Aid, Z.; Serravalle, S.; et al. The pediatric acute leukemia fusion oncogene ETO2-GLIS2 increases self-renewal and alters differentiation in a human induced pluripotent stem cells-derived model. HemaSphere 2020, 4, e319. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Group of Mutations | Genes | Timing |
|---|---|---|
| Cohesin complex genes | RAD21, SMC3, STAG2 | Diagnosis |
| ASXL1, ASXL2 | Diagnosis | |
| ASXL3 | Relapse | |
| BCOR, BCORL1, EZH2 | Diagnosis | |
| Signaling molecules | NRAS, CREBBP, KIT, FLT3-ITD | Relapse |
| PTPN11 | Diagnosis/relapse | |
| Others | SETD2, TYK2 | Diagnosis/relapse |
| SALL1 | Relapse | |
| C/EBPA | Diagnosis | |
| WT1 | Diagnosis/relapse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuccio, S.N.; Anselmi, L.; Masetti, R.; Lonetti, A.; Cerasi, S.; Polidori, S.; Serravalle, S.; Pession, A. Exploiting Clonal Evolution to Improve the Diagnosis and Treatment Efficacy Prediction in Pediatric AML. Cancers 2021, 13, 1995. https://doi.org/10.3390/cancers13091995
Bertuccio SN, Anselmi L, Masetti R, Lonetti A, Cerasi S, Polidori S, Serravalle S, Pession A. Exploiting Clonal Evolution to Improve the Diagnosis and Treatment Efficacy Prediction in Pediatric AML. Cancers. 2021; 13(9):1995. https://doi.org/10.3390/cancers13091995
Chicago/Turabian StyleBertuccio, Salvatore Nicola, Laura Anselmi, Riccardo Masetti, Annalisa Lonetti, Sara Cerasi, Sara Polidori, Salvatore Serravalle, and Andrea Pession. 2021. "Exploiting Clonal Evolution to Improve the Diagnosis and Treatment Efficacy Prediction in Pediatric AML" Cancers 13, no. 9: 1995. https://doi.org/10.3390/cancers13091995
APA StyleBertuccio, S. N., Anselmi, L., Masetti, R., Lonetti, A., Cerasi, S., Polidori, S., Serravalle, S., & Pession, A. (2021). Exploiting Clonal Evolution to Improve the Diagnosis and Treatment Efficacy Prediction in Pediatric AML. Cancers, 13(9), 1995. https://doi.org/10.3390/cancers13091995