
Liquid Biopsy in Pancreatic Cancer: Are We Ready to Apply It in the Clinical Practice?


Abstract
Simple Summary
Abstract
1. Introduction
2. Methodologies for Liquid Biopsy
2.1. Cell Free DNA (cfDNA)
2.2. Exosomes
2.3. Circulating Tumor Cells (CTCs)
2.4. Cell-Free RNAs (cfRNAs)
2.5. Tumor Educated Platelets (TEPs)
3. Liquid Biopsy in Other Body Fluids for the Early Detection of PDAC
4. Early Detection of PDAC
4.1. ctDNA
4.2. Exosomes
4.3. CTCs
4.4. cfRNAs
4.5. Liquid Biopsy in the Diagnosis of PDAC
5. Detection of Recurrence
5.1. ctDNA
5.2. Exosomes
5.3. CTCs
5.4. miRNAs
6. Disease Monitoring
6.1. Neoadyuvant Treatment.
6.2. Metastatic Disease
7. Precision Medicine
8. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- NCCN Guidelines. Pancreatic Adenocarcinoma. Available online: https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf (accessed on 3 February 2021).
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Janssen, Q.P.; Buettner, S.; Suker, M.; Beumer, B.R.; Addeo, P.; Bachellier, P.; Bahary, N.; Bekaii-Saab, T.; Bali, M.A.; Besselink, M.G.; et al. Neoadjuvant FOLFIRINOX in Patients With Borderline Resectable Pancreatic Cancer: A Systematic Review and Patient-Level Meta-Analysis. J. Natl. Cancer Inst. 2019, 111, 782–794. [Google Scholar] [CrossRef]
- Tienhoven, G.V.; Versteijne, E.; Suker, M.; Groothuis, K.B.C.; Busch, O.R.; Bonsing, B.A.; Hingh, I.H.J.T.d.; Festen, S.; Patijn, G.A.; Vos-Geelen, J.d.; et al. Preoperative chemoradiotherapy versus immediate surgery for resectable and borderline resectable pancreatic cancer (PREOPANC-1): A randomized, controlled, multicenter phase III trial. J. Clin. Oncol. 2018, 36, LBA4002. [Google Scholar] [CrossRef]
- Sabater, L.; Munoz, E.; Rosello, S.; Dorcaratto, D.; Garces-Albir, M.; Huerta, M.; Roda, D.; Gomez-Mateo, M.C.; Ferrandez-Izquierdo, A.; Darder, A.; et al. Borderline resectable pancreatic cancer. Challenges and controversies. Cancer Treat. Rev. 2018, 68, 124–135. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Imamura, T.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Arita, T.; Konishi, H.; Shiozaki, A.; et al. Liquid biopsy in patients with pancreatic cancer: Circulating tumor cells and cell-free nucleic acids. World J. Gastroenterol. 2016, 22, 5627–5641. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- DiPardo, B.J.; Winograd, P.; Court, C.M.; Tomlinson, J.S. Pancreatic cancer circulating tumor cells: Applications for personalized oncology. Expert Rev. Mol. Diagn. 2018, 18, 809–820. [Google Scholar] [CrossRef]
- Kamyabi, N.; Bernard, V.; Maitra, A. Liquid biopsies in pancreatic cancer. Expert Rev. Anticancer Ther. 2019, 19, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Buscail, E.; Maulat, C.; Muscari, F.; Chiche, L.; Cordelier, P.; Dabernat, S.; Alix-Panabieres, C.; Buscail, L. Liquid Biopsy Approach for Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 852. [Google Scholar] [CrossRef]
- Junqueira-Neto, S.; Batista, I.A.; Costa, J.L.; Melo, S.A. Liquid Biopsy beyond Circulating Tumor Cells and Cell-Free DNA. Acta Cytol. 2019, 63, 479–488. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Park, S.S.; Lee, Y.K.; Norton, J.A.; Jeffrey, S.S. Liquid biopsy in pancreatic ductal adenocarcinoma: Current status of circulating tumor cells and circulating tumor DNA. Mol. Oncol. 2019, 13, 1623–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xu, J.W.; Cheng, Y.G.; Gao, J.Y.; Hu, S.Y.; Wang, L.; Zhan, H.X. Early detection of pancreatic cancer: Where are we now and where are we going? Int. J. Cancer 2017, 141, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136–168. [Google Scholar] [CrossRef]
- Rothe, F.; Laes, J.F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Stahlberg, A.; Krzyzanowski, P.M.; Egyud, M.; Filges, S.; Stein, L.; Godfrey, T.E. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat. Protoc. 2017, 12, 664–682. [Google Scholar] [CrossRef]
- Coumans, F.A.; van der Pol, E.; Boing, A.N.; Hajji, N.; Sturk, G.; van Leeuwen, T.G.; Nieuwland, R. Reproducible extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J. Extracell. Vesicles 2014, 3, 25922. [Google Scholar] [CrossRef]
- Woo, H.K.; Sunkara, V.; Park, J.; Kim, T.H.; Han, J.R.; Kim, C.J.; Choi, H.I.; Kim, Y.K.; Cho, Y.K. Exodisc for Rapid, Size-Selective, and Efficient Isolation and Analysis of Nanoscale Extracellular Vesicles from Biological Samples. ACS Nano 2017, 11, 1360–1370. [Google Scholar] [CrossRef]
- Liu, F.; Vermesh, O.; Mani, V.; Ge, T.J.; Madsen, S.J.; Sabour, A.; Hsu, E.C.; Gowrishankar, G.; Kanada, M.; Jokerst, J.V.; et al. The Exosome Total Isolation Chip. ACS Nano 2017, 11, 10712–10723. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.; Ansa-Addo, E.; Stratton, D.; Antwi-Baffour, S.; Jorfi, S.; Kholia, S.; Krige, L.; Lange, S.; Inal, J. A filtration-based protocol to isolate human plasma membrane-derived vesicles and exosomes from blood plasma. J. Immunol. Methods 2011, 371, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Zacharias, W.; Gercel-Taylor, C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol. Biol. 2011, 728, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Banko, P.; Lee, S.Y.; Nagygyorgy, V.; Zrinyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 48. [Google Scholar] [CrossRef]
- Miltenyi, S.; Muller, W.; Weichel, W.; Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 1990, 11, 231–238. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Powell, A.A.; Huber, D.E.; Berbee, J.G.; Roh, K.H.; Yu, W.; Xiao, W.; Davis, M.M.; Pease, R.F.; Mindrinos, M.N.; et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc. Natl. Acad. Sci. USA 2009, 106, 3970–3975. [Google Scholar] [CrossRef]
- Lu, N.N.; Xie, M.; Wang, J.; Lv, S.W.; Yi, J.S.; Dong, W.G.; Huang, W.H. Biotin-triggered decomposable immunomagnetic beads for capture and release of circulating tumor cells. Acs Appl. Mater. Interfaces 2015, 7, 8817–8826. [Google Scholar] [CrossRef]
- Tong, X.; Xiong, Y.; Zborowski, M.; Farag, S.S.; Chalmers, J.J. A novel high throughput immunomagnetic cell sorting system for potential clinical scale depletion of T cells for allogeneic stem cell transplantation. Exp. Hematol. 2007, 35, 1613–1622. [Google Scholar] [CrossRef][Green Version]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Adams, A.A.; Okagbare, P.I.; Feng, J.; Hupert, M.L.; Patterson, D.; Gottert, J.; McCarley, R.L.; Nikitopoulos, D.; Murphy, M.C.; Soper, S.A. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J. Am. Chem. Soc. 2008, 130, 8633–8641. [Google Scholar] [CrossRef]
- Gleghorn, J.P.; Pratt, E.D.; Denning, D.; Liu, H.; Bander, N.H.; Tagawa, S.T.; Nanus, D.M.; Giannakakou, P.A.; Kirby, B.J. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip 2010, 10, 27–29. [Google Scholar] [CrossRef]
- Gupta, V.; Jafferji, I.; Garza, M.; Melnikova, V.O.; Hasegawa, D.K.; Pethig, R.; Davis, D.W. ApoStream(), a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics 2012, 6, 24133. [Google Scholar] [CrossRef]
- Di Trapani, M.; Manaresi, N.; Medoro, G. DEPArray system: An automatic image-based sorter for isolation of pure circulating tumor cells. Cytom. Part A J. Int. Soc. Anal. Cytol. 2018, 93, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.I.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694–710. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.T.; Zhao, L.; Shen, Q.; Garcia, M.A.; Wu, D.; Hou, S.; Song, M.; Xu, X.; Ouyang, W.H.; Ouyang, W.W.; et al. NanoVelcro Chip for CTC enumeration in prostate cancer patients. Methods 2013, 64, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Campton, D.E.; Ramirez, A.B.; Nordberg, J.J.; Drovetto, N.; Clein, A.C.; Varshavskaya, P.; Friemel, B.H.; Quarre, S.; Breman, A.; Dorschner, M.; et al. High-recovery visual identification and single-cell retrieval of circulating tumor cells for genomic analysis using a dual-technology platform integrated with automated immunofluorescence staining. BMC Cancer 2015, 15, 360. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.; Gertler, R.; Friederichs, J.; Fuehrer, K.; Dahm, M.; Phelps, R.; Thorban, S.; Nekarda, H.; Siewert, J.R. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry 2002, 49, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Drula, R.; Ott, L.F.; Berindan-Neagoe, I.; Pantel, K.; Calin, G.A. MicroRNAs from Liquid Biopsy Derived Extracellular Vesicles: Recent Advances in Detection and Characterization Methods. Cancers 2020, 12, 2009. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Moran, S.; Martinez-Cardus, A.; Sayols, S.; Musulen, E.; Balana, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Bai, X.; Yadav, R.K.; Singh, A.; Li, G.; Ma, T.; Chen, W.; Liang, T. Liquid biopsy in pancreatic cancer: The beginning of a new era. Oncotarget 2018, 9, 26900–26933. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, O.; Gasparini, P.; Boeri, M.; Sozzi, G. Exo-miRNAs as a New Tool for Liquid Biopsy in Lung Cancer. Cancers 2019, 11, 888. [Google Scholar] [CrossRef]
- Shigeyasu, K.; Toden, S.; Zumwalt, T.J.; Okugawa, Y.; Goel, A. Emerging Role of MicroRNAs as Liquid Biopsy Biomarkers in Gastrointestinal Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2391–2399. [Google Scholar] [CrossRef]
- Szilagyi, M.; Pos, O.; Marton, E.; Buglyo, G.; Soltesz, B.; Keseru, J.; Penyige, A.; Szemes, T.; Nagy, B. Circulating Cell-Free Nucleic Acids: Main Characteristics and Clinical Application. Int. J. Mol. Sci. 2020, 21, 6827. [Google Scholar] [CrossRef]
- Wright, K.; de Silva, K.; Purdie, A.C.; Plain, K.M. Comparison of methods for miRNA isolation and quantification from ovine plasma. Sci. Rep. 2020, 10, 825. [Google Scholar] [CrossRef]
- Fortunato, O.; Boeri, M.; Verri, C.; Conte, D.; Mensah, M.; Suatoni, P.; Pastorino, U.; Sozzi, G. Assessment of circulating microRNAs in plasma of lung cancer patients. Molecules 2014, 19, 3038–3054. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Liao, Z.; Wang, Z.; Wang, Z.; Li, Y.; Qian, L.; Zhao, J.; Zong, H.; Kang, B.; et al. Plasma extracellular vesicle long RNA profiling identifies a diagnostic signature for the detection of pancreatic ductal adenocarcinoma. Gut 2020, 69, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.B.; Yin, H.S.; Wang, J.Y. Evaluating the diagnostic and prognostic value of long non-coding RNA SNHG15 in pancreatic ductal adenocarcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5892–5898. [Google Scholar] [CrossRef] [PubMed]
- Boriachek, K.; Umer, M.; Islam, M.N.; Gopalan, V.; Lam, A.K.; Nguyen, N.T.; Shiddiky, M.J.A. An amplification-free electrochemical detection of exosomal miRNA-21 in serum samples. Analyst 2018, 143, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, C.; Li, Y.; Ma, Y.; Deng, J.; Li, L.; Sun, J. Thermophoretic Detection of Exosomal microRNAs by Nanoflares. J. Am. Chem. Soc. 2020, 142, 4996–5001. [Google Scholar] [CrossRef]
- Leslie, M. Cell biology. Beyond clotting: The powers of platelets. Science 2010, 328, 562–564. [Google Scholar] [CrossRef]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Sol, N.; Wurdinger, T. Platelet RNA signatures for the detection of cancer. Cancer Metastasis Rev. 2017, 36, 263–272. [Google Scholar] [CrossRef]
- Best, M.G.; Wesseling, P.; Wurdinger, T. Tumor-Educated Platelets as a Noninvasive Biomarker Source for Cancer Detection and Progression Monitoring. Cancer Res. 2018, 78, 3407–3412. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef]
- Satoh, K. Molecular Approaches Using Body Fluid for the Early Detection of Pancreatic Cancer. Diagnostics 2021, 11, 375. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; Li, J.; Wang, F.; Chen, K.; Zheng, Y.; Wang, J.; Lu, W.; Zhou, Y.; Yin, Q.; et al. A meta-analysis of the diagnostic value of detecting K-ras mutation in pancreatic juice as a molecular marker for pancreatic cancer. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2016, 16, 605–614. [Google Scholar] [CrossRef]
- Hata, T.; Ishida, M.; Motoi, F.; Yamaguchi, T.; Naitoh, T.; Katayose, Y.; Egawa, S.; Unno, M. Telomerase activity in pancreatic juice differentiates pancreatic cancer from chronic pancreatitis: A meta-analysis. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2016, 16, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Kim, Y.; Chia, D.; Spielmann, N.; Eibl, G.; Elashoff, D.; Wei, F.; Lin, Y.L.; Moro, A.; Grogan, T.; et al. Role of pancreatic cancer-derived exosomes in salivary biomarker development. J. Biol. Chem. 2013, 288, 26888–26897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Farrell, J.J.; Zhou, H.; Elashoff, D.; Akin, D.; Park, N.H.; Chia, D.; Wong, D.T. Salivary transcriptomic biomarkers for detection of resectable pancreatic cancer. Gastroenterology 2010, 138, 949–957.e7. [Google Scholar] [CrossRef]
- Xie, Z.; Yin, X.; Gong, B.; Nie, W.; Wu, B.; Zhang, X.; Huang, J.; Zhang, P.; Zhou, Z.; Li, Z. Salivary microRNAs show potential as a noninvasive biomarker for detecting resectable pancreatic cancer. Cancer Prev. Res. 2015, 8, 165–173. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, X.; Li, J.; Guo, Y.; Li, H.; Pan, X.; Jiang, J.; Liu, H.; Wu, B. Salivary HOTAIR and PVT1 as novel biomarkers for early pancreatic cancer. Oncotarget 2016, 7, 25408–25419. [Google Scholar] [CrossRef]
- Adachi, J.; Kumar, C.; Zhang, Y.; Olsen, J.V.; Mann, M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 2006, 7, R80. [Google Scholar] [CrossRef]
- Napoli, C.; Sperandio, N.; Lawlor, R.T.; Scarpa, A.; Molinari, H.; Assfalg, M. Urine metabolic signature of pancreatic ductal adenocarcinoma by (1)h nuclear magnetic resonance: Identification, mapping, and evolution. J. Proteome Res. 2012, 11, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Davis, V.W.; Schiller, D.E.; Eurich, D.; Bathe, O.F.; Sawyer, M.B. Pancreatic ductal adenocarcinoma is associated with a distinct urinary metabolomic signature. Ann. Surg. Oncol. 2013, 20 (Suppl. S3), S415–S423. [Google Scholar] [CrossRef]
- Lusczek, E.R.; Paulo, J.A.; Saltzman, J.R.; Kadiyala, V.; Banks, P.A.; Beilman, G.; Conwell, D.L. Urinary 1H-NMR metabolomics can distinguish pancreatitis patients from healthy controls. JOP J. Pancreas 2013, 14, 161–170. [Google Scholar] [CrossRef]
- Mayerle, J.; Kalthoff, H.; Reszka, R.; Kamlage, B.; Peter, E.; Schniewind, B.; Gonzalez Maldonado, S.; Pilarsky, C.; Heidecke, C.D.; Schatz, P.; et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut 2018, 67, 128–137. [Google Scholar] [CrossRef]
- Roy, R.; Zurakowski, D.; Wischhusen, J.; Frauenhoffer, C.; Hooshmand, S.; Kulke, M.; Moses, M.A. Urinary TIMP-1 and MMP-2 levels detect the presence of pancreatic malignancies. Br. J. Cancer 2014, 111, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Hogendorf, P.; Durczynski, A.; Skulimowski, A.; Kumor, A.; Poznanska, G.; Strzelczyk, J. Neutrophil Gelatinase-Associated Lipocalin (NGAL) concentration in urine is superior to CA19-9 and Ca 125 in differentiation of pancreatic mass: Preliminary report. Cancer Biomark. Sect. A Dis. Markers 2016, 16, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Shu, X.O.; Li, H.L.; Yang, G.; Wen, W.; Gao, Y.T.; Cai, Q.; Rothman, N.; Yin, H.Y.; Lan, Q.; et al. Prospective study of urinary prostaglandin E2 metabolite and pancreatic cancer risk. Int. J. Cancer 2017, 141, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Soufi, M.; Carr, R.A.; Flick, K.F.; Wu, H.; Colgate, C.L.; Schmidt, C.M. Performance of candidate urinary biomarkers for pancreatic cancer—Correlation with pancreatic cyst malignant progression? Am. J. Surg. 2020, 219, 492–495. [Google Scholar] [CrossRef]
- Debernardi, S.; O’Brien, H.; Algahmdi, A.S.; Malats, N.; Stewart, G.D.; Pljesa-Ercegovac, M.; Costello, E.; Greenhalf, W.; Saad, A.; Roberts, R.; et al. A combination of urinary biomarker panel and PancRISK score for earlier detection of pancreatic cancer: A case-control study. PLoS Med. 2020, 17, e1003489. [Google Scholar] [CrossRef]
- Terasawa, H.; Kinugasa, H.; Ako, S.; Hirai, M.; Matsushita, H.; Uchida, D.; Tomoda, T.; Matsumoto, K.; Horiguchi, S.; Kato, H.; et al. Utility of liquid biopsy using urine in patients with pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2019, 20, 1348–1353. [Google Scholar] [CrossRef]
- Debernardi, S.; Massat, N.J.; Radon, T.P.; Sangaralingam, A.; Banissi, A.; Ennis, D.P.; Dowe, T.; Chelala, C.; Pereira, S.P.; Kocher, H.M.; et al. Noninvasive urinary miRNA biomarkers for early detection of pancreatic adenocarcinoma. Am. J. Cancer Res. 2015, 5, 3455–3466. [Google Scholar]
- Yoshizawa, N.; Sugimoto, K.; Tameda, M.; Inagaki, Y.; Ikejiri, M.; Inoue, H.; Usui, M.; Ito, M.; Takei, Y. miR-3940-5p/miR-8069 ratio in urine exosomes is a novel diagnostic biomarker for pancreatic ductal adenocarcinoma. Oncol. Lett. 2020, 19, 2677–2684. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- US Preventive Services Task Force; Owens, D.K.; Davidson, K.W.; Krist, A.H.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Curry, S.J.; Doubeni, C.A.; Epling, J.W., Jr.; et al. Screening for Pancreatic Cancer: US Preventive Services Task Force Reaffirmation Recommendation Statement. JAMA 2019, 322, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Sho, M.; Akahori, T.; Nakagawa, K.; Nakamura, K. Application of liquid biopsy for surgical management of pancreatic cancer. Ann. Gastroenterol. Surg. 2020, 4, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Sefrioui, D.; Blanchard, F.; Toure, E.; Basile, P.; Beaussire, L.; Dolfus, C.; Perdrix, A.; Paresy, M.; Antonietti, M.; Iwanicki-Caron, I.; et al. Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br. J. Cancer 2017, 117, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Lipton, L.; Cohen, J.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Burge, M.; Cooray, P.; Nagrial, A.; et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Ding, X.Q.; Zhu, H.; Wang, R.X.; Pan, X.R.; Tong, J.H. KRAS Mutant Allele Fraction in Circulating Cell-Free DNA Correlates With Clinical Stage in Pancreatic Cancer Patients. Front. Oncol. 2019, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated with Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108–118.e4. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef]
- Okada, T.; Mizukami, Y.; Ono, Y.; Sato, H.; Hayashi, A.; Kawabata, H.; Koizumi, K.; Masuda, S.; Teshima, S.; Takahashi, K.; et al. Digital PCR-based plasma cell-free DNA mutation analysis for early-stage pancreatic tumor diagnosis and surveillance. J. Gastroenterol. 2020, 55, 1183–1193. [Google Scholar] [CrossRef]
- Maire, F.; Micard, S.; Hammel, P.; Voitot, H.; Levy, P.; Cugnenc, P.H.; Ruszniewski, P.; Puig, P.L. Differential diagnosis between chronic pancreatitis and pancreatic cancer: Value of the detection of KRAS2 mutations in circulating DNA. Br. J. Cancer 2002, 87, 551–554. [Google Scholar] [CrossRef]
- Cao, F.; Wei, A.; Hu, X.; He, Y.; Zhang, J.; Xia, L.; Tu, K.; Yuan, J.; Guo, Z.; Liu, H.; et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin. Epigenet. 2020, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Thege, F.I.; Santana, S.M.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Maggs, L.R.; Kochman, M.L.; Ginsberg, G.G.; Lieb, J.G.; et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014, 146, 647–651. [Google Scholar] [CrossRef]
- Liu, H.; Sun, B.; Wang, S.; Liu, C.; Lu, Y.; Li, D.; Liu, X. Circulating Tumor Cells as a Biomarker in Pancreatic Ductal Adenocarcinoma. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Tien, Y.W.; Kuo, H.C.; Ho, B.I.; Chang, M.C.; Chang, Y.T.; Cheng, M.F.; Chen, H.L.; Liang, T.Y.; Wang, C.F.; Huang, C.Y.; et al. A High Circulating Tumor Cell Count in Portal Vein Predicts Liver Metastasis From Periampullary or Pancreatic Cancer: A High Portal Venous CTC Count Predicts Liver Metastases. Medicine 2016, 95, e3407. [Google Scholar] [CrossRef] [PubMed]
- Buscail, E.; Alix-Panabieres, C.; Quincy, P.; Cauvin, T.; Chauvet, A.; Degrandi, O.; Caumont, C.; Verdon, S.; Lamrissi, I.; Moranvillier, I.; et al. High Clinical Value of Liquid Biopsy to Detect Circulating Tumor Cells and Tumor Exosomes in Pancreatic Ductal Adenocarcinoma Patients Eligible for Up-Front Surgery. Cancers 2019, 11, 1656. [Google Scholar] [CrossRef] [PubMed]
- Allenson, K.; Castillo, J.; San Lucas, F.A.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2017, 28, 741–747. [Google Scholar] [CrossRef]
- Lai, X.; Wang, M.; McElyea, S.D.; Sherman, S.; House, M.; Korc, M. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Cancer Lett. 2017, 393, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Que, R.; Ding, G.; Chen, J.; Cao, L. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J. Surg. Oncol. 2013, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sheng, Y.; Kwak, K.J.; Shi, J.; Yu, B.; Lee, L.J. A signal-amplifiable biochip quantifies extracellular vesicle-associated RNAs for early cancer detection. Nat. Commun. 2017, 8, 1683. [Google Scholar] [CrossRef]
- Madhavan, B.; Yue, S.; Galli, U.; Rana, S.; Gross, W.; Muller, M.; Giese, N.A.; Kalthoff, H.; Becker, T.; Buchler, M.W.; et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int. J. Cancer 2015, 136, 2616–2627. [Google Scholar] [CrossRef]
- Deng, T.; Yuan, Y.; Zhang, C.; Zhang, C.; Yao, W.; Wang, C.; Liu, R.; Ba, Y. Identification of Circulating MiR-25 as a Potential Biomarker for Pancreatic Cancer Diagnosis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, J.; Gao, W.; Huang, L.; Liu, Y.; Li, X.; Li, Z.; Yu, X. Meta-analysis of the Diagnostic Performance of Circulating MicroRNAs for Pancreatic Cancer. Int. J. Med Sci. 2021, 18, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, H.; Chen, N.; Hao, J.; Jin, H.; Ma, X. Diagnostic value of various liquid biopsy methods for pancreatic cancer: A systematic review and meta-analysis. Medicine 2020, 99, e18581. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, H.; Nouso, K.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Tsutsumi, K.; Kato, H.; Matsubara, T.; Okada, H.; Yamamoto, K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015, 121, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Dabritz, J.; Preston, R.; Hanfler, J.; Oettle, H. Follow-up study of K-ras mutations in the plasma of patients with pancreatic cancer: Correlation with clinical features and carbohydrate antigen 19-9. Pancreas 2009, 38, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Krimmel, J.D.; Schmitt, M.W.; Harrell, M.I.; Agnew, K.J.; Kennedy, S.R.; Emond, M.J.; Loeb, L.A.; Swisher, E.M.; Risques, R.A. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 6005–6010. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, Q.; Li, X.; Su, W.; Li, G.; Ma, T.; Gao, S.; Lou, J.; Que, R.; Zheng, L.; et al. Monitoring Tumor Burden in Response to FOLFIRINOX Chemotherapy Via Profiling Circulating Cell-Free DNA in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 196–203. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Ning, N.; Chen, Q.; Yang, Z.; Guo, Y.; Xu, D.; Zhang, D.; Zhan, T.; Cui, W. Patterns of circulating tumor cells identified by CEP8, CK and CD45 in pancreatic cancer. Int. J. Cancer 2015, 136, 1228–1233. [Google Scholar] [CrossRef]
- Kaczor-Urbanowicz, K.E.; Cheng, J.; King, J.C.; Sedarat, A.; Pandol, S.J.; Farrell, J.J.; Wong, D.T.W.; Kim, Y. Reviews on Current Liquid Biopsy for Detection and Management of Pancreatic Cancers. Pancreas 2020, 49, 1141–1152. [Google Scholar] [CrossRef]
- Ren, C.; Han, C.; Zhang, J.; He, P.; Wang, D.; Wang, B.; Zhao, P.; Zhao, X. Detection of apoptotic circulating tumor cells in advanced pancreatic cancer following 5-fluorouracil chemotherapy. Cancer Biol. Ther. 2011, 12, 700–706. [Google Scholar] [CrossRef]
- Ankeny, J.S.; Court, C.M.; Hou, S.; Li, Q.; Song, M.; Wu, D.; Chen, J.F.; Lee, T.; Lin, M.; Sho, S.; et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br. J. Cancer 2016, 114, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Cauley, C.E.; Pitman, M.B.; Zhou, J.; Perkins, J.; Kuleman, B.; Liss, A.S.; Fernandez-Del Castillo, C.; Warshaw, A.L.; Lillemoe, K.D.; Thayer, S.P. Circulating Epithelial Cells in Patients with Pancreatic Lesions: Clinical and Pathologic Findings. J. Am. Coll. Surg. 2015, 221, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Chapman, C.G.; Xu, P.; Koons, A.; Konda, V.J.; Siddiqui, U.D.; Waxman, I. Acquisition of Portal Venous Circulating Tumor Cells From Patients With Pancreaticobiliary Cancers by Endoscopic Ultrasound. Gastroenterology 2015, 149, 1794–1803.e94. [Google Scholar] [CrossRef]
- Li, X.; Gao, P.; Wang, Y.; Wang, X. Blood-Derived microRNAs for Pancreatic Cancer Diagnosis: A Narrative Review and Meta-Analysis. Front. Physiol. 2018, 9, 685. [Google Scholar] [CrossRef]
- Yan, Q.; Hu, D.; Li, M.; Chen, Y.; Wu, X.; Ye, Q.; Wang, Z.; He, L.; Zhu, J. The Serum MicroRNA Signatures for Pancreatic Cancer Detection and Operability Evaluation. Front. Bioeng. Biotechnol. 2020, 8, 379. [Google Scholar] [CrossRef]
- Komatsu, S.; Ichikawa, D.; Miyamae, M.; Kawaguchi, T.; Morimura, R.; Hirajima, S.; Okajima, W.; Ohashi, T.; Imamura, T.; Konishi, H.; et al. Malignant potential in pancreatic neoplasm; new insights provided by circulating miR-223 in plasma. Expert Opin. Biol. Ther. 2015, 15, 773–785. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Gercke, N.; Strauch, K.; Wieboldt, R.; Matthai, E.; Wagner, V.; Rospleszcz, S.; Schafer, A.; Franke, F.S.; Mintziras, I.; et al. The Combination of MiRNA-196b, LCN2, and TIMP1 is a Potential Set of Circulating Biomarkers for Screening Individuals at Risk for Familial Pancreatic Cancer. J. Clin. Med. 2018, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- Brychta, N.; Krahn, T.; von Ahsen, O. Detection of KRAS Mutations in Circulating Tumor DNA by Digital PCR in Early Stages of Pancreatic Cancer. Clin. Chem. 2016, 62, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, S.; Liao, W.; Chen, E.; Wang, X.; Song, Y.; Duan, F.; Deng, W.; Li, S. LINC00460 promotes pancreatic cancer progression by sponging miR-491-5p. J. Gene Med. 2021, 2021, e3333. [Google Scholar] [CrossRef]
- Metzenmacher, M.; Varaljai, R.; Hegedus, B.; Cima, I.; Forster, J.; Schramm, A.; Scheffler, B.; Horn, P.A.; Klein, C.A.; Szarvas, T.; et al. Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer. Cancers 2020, 12, 353. [Google Scholar] [CrossRef]
- Takahashi, K.; Ota, Y.; Kogure, T.; Suzuki, Y.; Iwamoto, H.; Yamakita, K.; Kitano, Y.; Fujii, S.; Haneda, M.; Patel, T.; et al. Circulating extracellular vesicle-encapsulated HULC is a potential biomarker for human pancreatic cancer. Cancer Sci. 2020, 111, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Groot, V.P.; Mosier, S.; Javed, A.A.; Teinor, J.A.; Gemenetzis, G.; Ding, D.; Haley, L.M.; Yu, J.; Burkhart, R.A.; Hasanain, A.; et al. Circulating Tumor DNA as a Clinical Test in Resected Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4973–4984. [Google Scholar] [CrossRef]
- Daamen, L.A.; Groot, V.P.; Goense, L.; Wessels, F.J.; Borel Rinkes, I.H.; Intven, M.P.W.; van Santvoort, H.C.; Molenaar, I.Q. The diagnostic performance of CT versus FDG PET-CT for the detection of recurrent pancreatic cancer: A systematic review and meta-analysis. Eur. J. Radiol. 2018, 106, 128–136. [Google Scholar] [CrossRef]
- Daamen, L.A.; Groot, V.P.; Heerkens, H.D.; Intven, M.P.W.; van Santvoort, H.C.; Molenaar, I.Q. Systematic review on the role of serum tumor markers in the detection of recurrent pancreatic cancer. HPB Off. J. Int. Hepato Pancreato Biliary Assoc. 2018, 20, 297–304. [Google Scholar] [CrossRef]
- Azizian, A.; Ruhlmann, F.; Krause, T.; Bernhardt, M.; Jo, P.; Konig, A.; Kleiss, M.; Leha, A.; Ghadimi, M.; Gaedcke, J. CA19-9 for detecting recurrence of pancreatic cancer. Sci. Rep. 2020, 10, 1332. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Hadano, N.; Murakami, Y.; Uemura, K.; Hashimoto, Y.; Kondo, N.; Nakagawa, N.; Sueda, T.; Hiyama, E. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br. J. Cancer 2016, 115, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Iinuma, H.; Wada, K.; Minezaki, S.; Kawamura, S.; Kainuma, M.; Ikeda, Y.; Shibuya, M.; Miura, F.; Sano, K. Usefulness of exosome-encapsulated microRNA-451a as a minimally invasive biomarker for prediction of recurrence and prognosis in pancreatic ductal adenocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2018, 25, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Iinuma, H.; Wada, K.; Takahashi, K.; Minezaki, S.; Kainuma, M.; Shibuya, M.; Miura, F.; Sano, K. Exosome-encapsulated microRNA-4525, microRNA-451a and microRNA-21 in portal vein blood is a high-sensitive liquid biomarker for the selection of high-risk pancreatic ductal adenocarcinoma patients. J. Hepato-Biliary-Pancreat. Sci. 2019, 26, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hugenschmidt, H.; Labori, K.J.; Borgen, E.; Brunborg, C.; Schirmer, C.B.; Seeberg, L.T.; Naume, B.; Wiedswang, G. Preoperative CTC-Detection by CellSearch((R)) Is Associated with Early Distant Metastasis and Impaired Survival in Resected Pancreatic Cancer. Cancers 2021, 13, 485. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzis, G.; Groot, V.P.; Yu, J.; Ding, D.; Teinor, J.A.; Javed, A.A.; Wood, L.D.; Burkhart, R.A.; Cameron, J.L.; Makary, M.A.; et al. Circulating Tumor Cells Dynamics in Pancreatic Adenocarcinoma Correlate With Disease Status: Results of the Prospective CLUSTER Study. Ann. Surg. 2018, 268, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Blackford, A.L.; Weiss, M.J.; Cameron, J.L.; He, J.; Goggins, M.; Rasheed, Z.A.; Wolfgang, C.L.; Wood, L.D. Circulating Tumor Cells Expressing Markers of Tumor-Initiating Cells Predict Poor Survival and Cancer Recurrence in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.C.Y.; Po, J.W.; Becker, T.M.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Circulating tumour cells in pancreatic cancer: A systematic review and meta-analysis of clinicopathological implications. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2021, 21, 103–114. [Google Scholar] [CrossRef]
- Poruk, K.E.; Valero, V., 3rd; Saunders, T.; Blackford, A.L.; Griffin, J.F.; Poling, J.; Hruban, R.H.; Anders, R.A.; Herman, J.; Zheng, L.; et al. Circulating Tumor Cell Phenotype Predicts Recurrence and Survival in Pancreatic Adenocarcinoma. Ann. Surg. 2016, 264, 1073–1081. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Piva, F.; Marinelli, O.; Maggi, F.; Bianchi, F.; Bittoni, A.; Berardi, R.; Giampieri, R.; et al. Expression Profiling of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma Patients: Biomarkers Predicting Overall Survival. Front. Oncol. 2019, 9, 874. [Google Scholar] [CrossRef]
- Miyamae, M.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Hirajima, S.; Okajima, W.; Ohashi, T.; Imamura, T.; Konishi, H.; Shiozaki, A.; et al. Plasma microRNA profiles: Identification of miR-744 as a novel diagnostic and prognostic biomarker in pancreatic cancer. Br. J. Cancer 2015, 113, 1467–1476. [Google Scholar] [CrossRef]
- Karasek, P.; Gablo, N.; Hlavsa, J.; Kiss, I.; Vychytilova-Faltejskova, P.; Hermanova, M.; Kala, Z.; Slaby, O.; Prochazka, V. Pre-operative Plasma miR-21-5p Is a Sensitive Biomarker and Independent Prognostic Factor in Patients with Pancreatic Ductal Adenocarcinoma Undergoing Surgical Resection. Cancer Genom. Proteom. 2018, 15, 321–327. [Google Scholar] [CrossRef]
- Slater, E.P.; Strauch, K.; Rospleszcz, S.; Ramaswamy, A.; Esposito, I.; Kloppel, G.; Matthai, E.; Heeger, K.; Fendrich, V.; Langer, P.; et al. MicroRNA-196a and -196b as Potential Biomarkers for the Early Detection of Familial Pancreatic Cancer. Transl. Oncol. 2014, 7, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369. [Google Scholar] [CrossRef]
- Pietrasz, D.; Pecuchet, N.; Garlan, F.; Didelot, A.; Dubreuil, O.; Doat, S.; Imbert-Bismut, F.; Karoui, M.; Vaillant, J.C.; Taly, V.; et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Ayub, M.; Rothwell, D.G.; Gulati, S.; Kilerci, B.; Hollebecque, A.; Sun Leong, H.; Smith, N.K.; Sahoo, S.; Descamps, T.; et al. Analysis of circulating cell-free DNA identifies KRAS copy number gain and mutation as a novel prognostic marker in Pancreatic cancer. Sci. Rep. 2019, 9, 11610. [Google Scholar] [CrossRef] [PubMed]
- Toledano-Fonseca, M.; Cano, M.T.; Inga, E.; Rodriguez-Alonso, R.; Gomez-Espana, M.A.; Guil-Luna, S.; Mena-Osuna, R.; de la Haba-Rodriguez, J.R.; Rodriguez-Ariza, A.; Aranda, E. Circulating Cell-Free DNA-Based Liquid Biopsy Markers for the Non-Invasive Prognosis and Monitoring of Metastatic Pancreatic Cancer. Cancers 2020, 12, 1754. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Woo, S.M.; Park, B.; Yoon, K.A.; Kim, Y.H.; Joo, J.; Lee, W.J.; Han, S.S.; Park, S.J.; Kong, S.Y. Prognostic Implications of Multiplex Detection of KRAS Mutations in Cell-Free DNA from Patients with Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2018, 64, 726–734. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Bachet, J.B.; Blons, H.; Hammel, P.; Hariry, I.E.; Portales, F.; Mineur, L.; Metges, J.P.; Mulot, C.; Bourreau, C.; Cain, J.; et al. Circulating Tumor DNA is Prognostic and Potentially Predictive of Eryaspase Efficacy in Second-line in Patients with Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5208–5216. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, C.; Jiang, J.; Luo, G.; Lu, Y.; Jin, K.; Guo, M.; Zhang, Z.; Xu, J.; Liu, L.; et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int. J. Cancer 2017, 140, 2344–2350. [Google Scholar] [CrossRef]
- He, J.; Blair, A.B.; Groot, V.P.; Javed, A.A.; Burkhart, R.A.; Gemenetzis, G.; Hruban, R.H.; Waters, K.M.; Poling, J.; Zheng, L.; et al. Is a Pathological Complete Response Following Neoadjuvant Chemoradiation Associated With Prolonged Survival in Patients With Pancreatic Cancer? Ann. Surg. 2018, 268, 1–8. [Google Scholar] [CrossRef]
- Yin, L.; Pu, N.; Thompson, E.; Miao, Y.; Wolfgang, C.; Yu, J. Improved Assessment of Response Status in Patients with Pancreatic Cancer Treated with Neoadjuvant Therapy using Somatic Mutations and Liquid Biopsy Analysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Sugimori, M.; Sugimori, K.; Tsuchiya, H.; Suzuki, Y.; Tsuyuki, S.; Kaneta, Y.; Hirotani, A.; Sanga, K.; Tozuka, Y.; Komiyama, S.; et al. Quantitative monitoring of circulating tumor DNA in patients with advanced pancreatic cancer undergoing chemotherapy. Cancer Sci. 2020, 111, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Rashid, S.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical significance of promoter methylation status of tumor suppressor genes in circulating DNA of pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Huguet, F.; Louvet, C.; Mineur, L.; Bouche, O.; Chibaudel, B.; Artru, P.; Desseigne, F.; Bachet, J.B.; Mathiot, C.; et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: The ancillary CirCe 07 study to the LAP 07 trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Court, C.M.; Ankeny, J.S.; Sho, S.; Winograd, P.; Hou, S.; Song, M.; Wainberg, Z.A.; Girgis, M.D.; Graeber, T.G.; Agopian, V.G.; et al. Circulating Tumor Cells Predict Occult Metastatic Disease and Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2018, 25, 1000–1008. [Google Scholar] [CrossRef]
- Han, L.; Chen, W.; Zhao, Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Okubo, K.; Uenosono, Y.; Arigami, T.; Mataki, Y.; Matsushita, D.; Yanagita, S.; Kurahara, H.; Sakoda, M.; Kijima, Y.; Maemura, K.; et al. Clinical impact of circulating tumor cells and therapy response in pancreatic cancer. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2017, 43, 1050–1055. [Google Scholar] [CrossRef]
- Xu, Y.; Qin, T.; Li, J.; Wang, X.; Gao, C.; Xu, C.; Hao, J.; Liu, J.; Gao, S.; Ren, H. Detection of Circulating Tumor Cells Using Negative Enrichment Immunofluorescence and an In Situ Hybridization System in Pancreatic Cancer. Int. J. Mol. Sci. 2017, 18, 622. [Google Scholar] [CrossRef]
- Yu, K.H.; Ricigliano, M.; Hidalgo, M.; Abou-Alfa, G.K.; Lowery, M.A.; Saltz, L.B.; Crotty, J.F.; Gary, K.; Cooper, B.; Lapidus, R.; et al. Pharmacogenomic modeling of circulating tumor and invasive cells for prediction of chemotherapy response and resistance in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5281–5289. [Google Scholar] [CrossRef]
- Dotan, E.; Alpaugh, R.K.; Ruth, K.; Negin, B.P.; Denlinger, C.S.; Hall, M.J.; Astsaturov, I.; McAleer, C.; Fittipaldi, P.; Thrash-Bingham, C.; et al. Prognostic Significance of MUC-1 in Circulating Tumor Cells in Patients With Metastatic Pancreatic Adenocarcinoma. Pancreas 2016, 45, 1131–1135. [Google Scholar] [CrossRef]
- Kulemann, B.; Rosch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef]
- Fernandez-Lazaro, D.; Hernandez, J.L.G.; Garcia, A.C.; Castillo, A.C.D.; Hueso, M.V.; Cruz-Hernandez, J.J. Clinical Perspective and Translational Oncology of Liquid Biopsy. Diagnostics 2020, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Vietsch, E.E.; Graham, G.T.; McCutcheon, J.N.; Javaid, A.; Giaccone, G.; Marshall, J.L.; Wellstein, A. Circulating cell-free DNA mutation patterns in early and late stage colon and pancreatic cancer. Cancer Genet. 2017, 218–219, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Veronese, N.; Nottegar, A.; Cappelletti, V.; Daidone, M.G.; Smith, L.; Parris, C.; Brosens, L.A.A.; Caruso, M.G.; Cheng, L.; et al. Liquid Biopsy as Surrogate for Tissue for Molecular Profiling in Pancreatic Cancer: A Meta-Analysis Towards Precision Medicine. Cancers 2019, 11, 1152. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet. Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci. Rep. 2015, 5, 18425. [Google Scholar] [CrossRef] [PubMed]
- Botrus, G.; Kosirorek, H.; Sonbol, M.B.; Kusne, Y.; Uson Junior, P.L.S.; Borad, M.J.; Ahn, D.H.; Kasi, P.M.; Drusbosky, L.M.; Dada, H.; et al. Circulating Tumor DNA-Based Testing and Actionable Findings in Patients with Advanced and Metastatic Pancreatic Adenocarcinoma. Oncologist 2021. [Google Scholar] [CrossRef]
- Lee, A.C.; Svedlund, J.; Darai, E.; Lee, Y.; Lee, D.; Lee, H.B.; Kim, S.M.; Kim, O.; Bae, H.J.; Choi, A.; et al. OPENchip: An on-chip in situ molecular profiling platform for gene expression analysis and oncogenic mutation detection in single circulating tumour cells. Lab Chip 2020, 20, 912–922. [Google Scholar] [CrossRef] [PubMed]



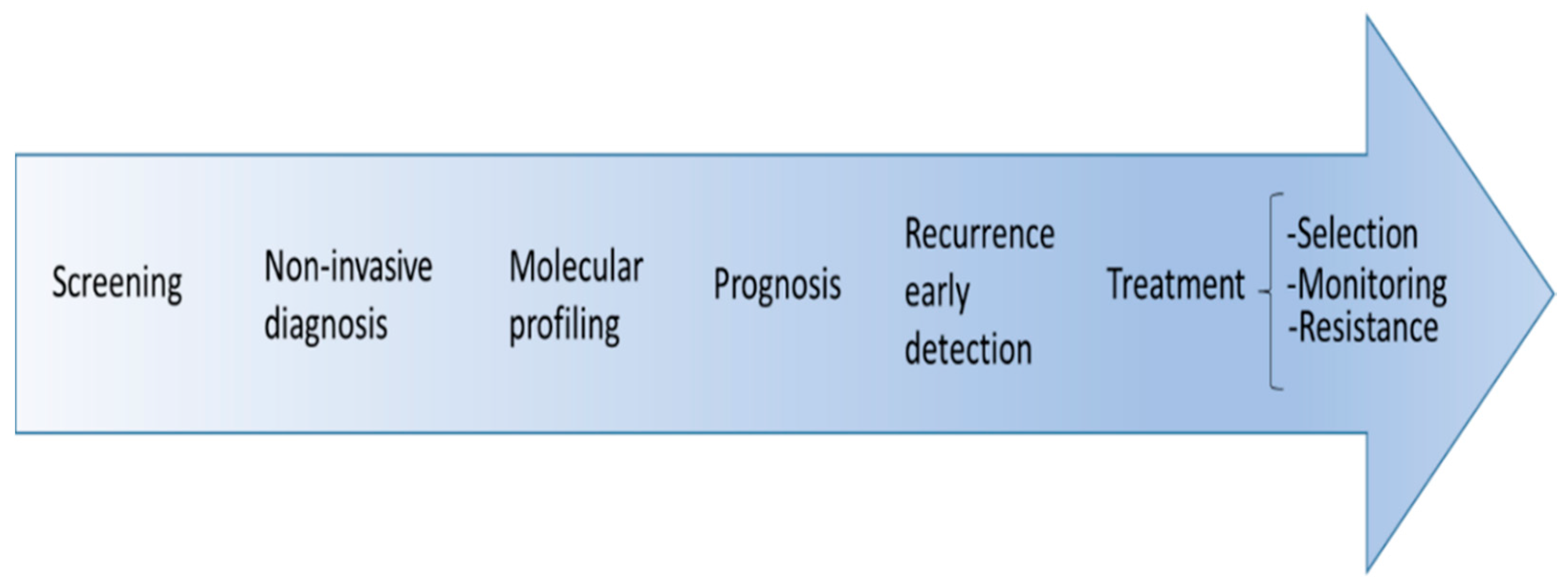{kind=link}
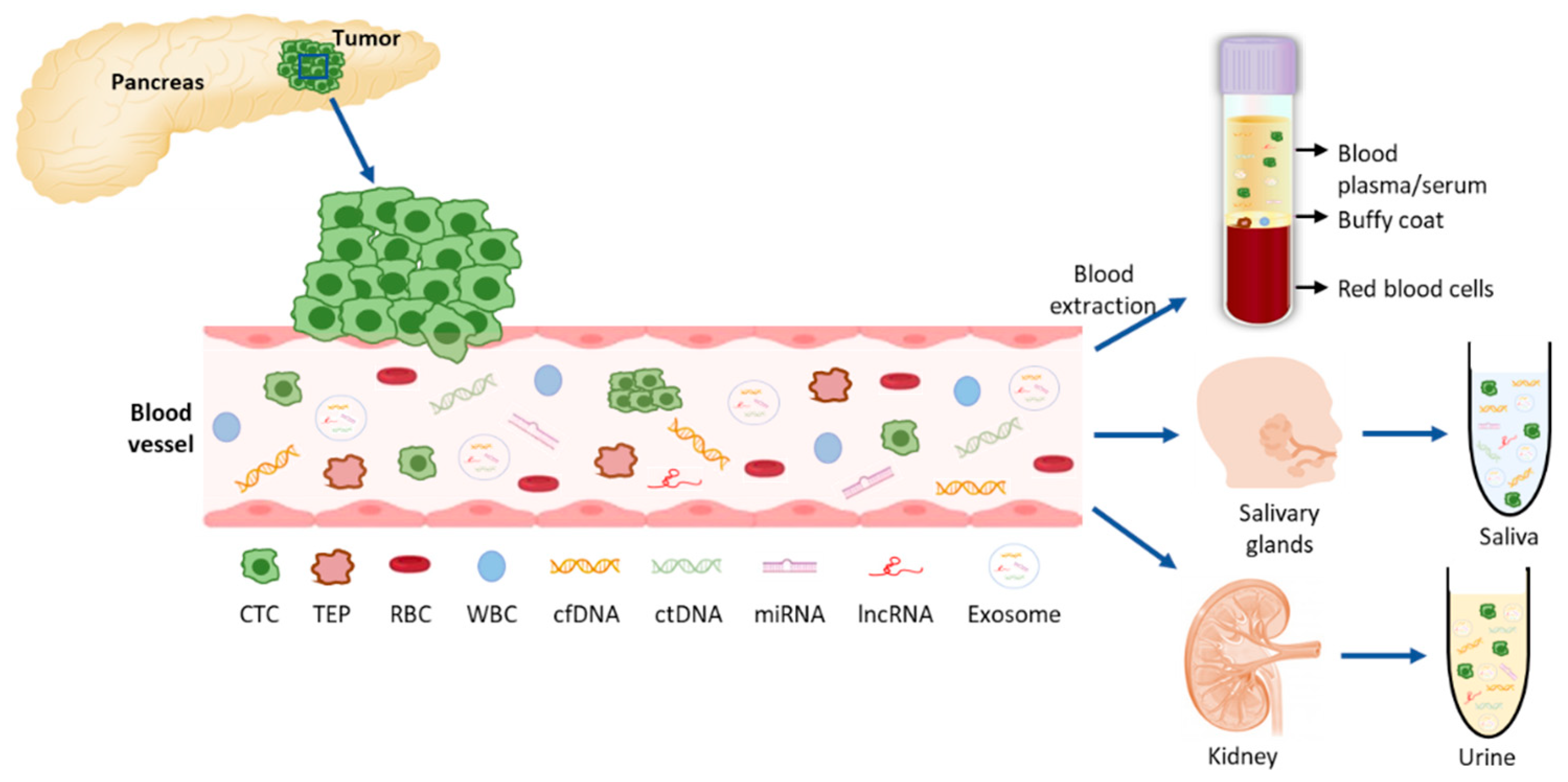{kind=link}
| LB Component | Technique | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|
| cfDNA | qPCR | Fast & Inexpensive High specificity | Lower sensitivity (0.1%) Detects only point mutations | [24,25,26,27,28,29,30] |
| dPCR* (ddPCR, BEAMING) | High sensitivity (0.01%) & specificity | Detects only point mutations Expensive | ||
| NGS | High DNA input permits high throughput analysis and screen for unknown variants (WGS &WES) Can identify structural variants and copy number variations | Variable sensitivity (0.1% aprox.) Expensive | ||
| Exosomes | Density-based isolation* (centrifugation) | Inexpensive Independent of marker expression | Time consuming High volume sample required Can damage exosomes Contaminated sample | [20,21,22,31,32,33,34,35] |
| Size-based isolation | Fast & Inexpensive Independent of marker expression | Contaminated sample | ||
| Affinity-based isolation | High purity and specificity | Low sample yield | ||
| Commercial kits | Fast & Simple | Expensive | ||
| CTCs | Immunoaffinity enrichment* | Positive enrichment: - Very specific - High capture efficiency & purity Negative enrichment: - Label-free CTCs obtained | Only one subpopulation captured Lower purity | [20,24,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50] |
| Physical methods (size & density) | Represent tumor heterogeneity Fast & Simple Less expensive Label-free CTCs obtained | Must be followed with immuno-labelling techniques to distinguish CTCs | ||
| cfRNAs | RT-qPCR | Fast & Inexpensive High specificity | Low sensitivity in samples with low abundance cfRNA | [51] |
| ddPCR* | Higher sensitivity & accuracy Lower sample volume required More reproducible than qPCR | Tedious assay optimization |
| Study Technique | Specific/Relevant Molecular Findings | Stage | Nº Patients PDAC/Control | Sensitivity (%) | Specificity (%) | Ref. | |
|---|---|---|---|---|---|---|---|
| ctDNA | ctDNA | KRAS mutant allele fraction > 5% | Early PDAC Metastatic | 90/37 104/37 | 34 54 | NR NR | [97] |
| Mutations at codons 12, 13 and 61 of KRAS | Early PDAC | 112 | 62 | NR | [95] | ||
| Mutations KRAS exon 2 (codons 12 and 13) | All stages | 52/10 | 65 | 75 | [94] | ||
| KRAS MAFs | All stages | 110/52 | 47 | NR | [96] | ||
| Mutations at codons 12, 13 of KRAS | Early PDAC | 221/182 | 30 | NR | [98] | ||
| Mutations at codons 12, 13 and 61 of KRAS | Early PDAC | 112/76 | 71 | NR | [99] | ||
| ctDNA combined with other serum tumor markers | ctDNA+ CA19.9 | Mutations KRAS exon 2 | All stages | 47/31 | 85-98 | 77-81 | [100] |
| ctDNA+ CA19.9 | KRAS MAFs | All stages | 110/52 | 47 | [96] | ||
| KRAS ctDNA + CEA + CA19.9 + HGF + osteopontin | Mutations at codons 12, 13 of KRAS | Early PDAC | 221/182 | 64 | 99 | [98] | |
| cfDNA methylome | 5-methylcytosine and 5-hydroxymethylcytosine | Global DNA hypomethylation | All stages | 72/136 | 94 | 95 | [101] |
| CTCs | CellSearch® | > 3 CTCs/mL | IPMN PDAC | 21/19 8/19 | 33 73 | NR | [102] |
| CD45/CEP8/DAPI staining-FISH | CTC detection | PDAC | 95/48 | 76 | 68 | [103] | |
| Anti-EpCAM Portal-vein blood | Number of CTCs in 2 mL portal venous blood (if > 112 indicate hepatic metastasis) | Metastatic Resectable | 17 43 | 100 58 | NR | [104] | |
| Combined analysis CTC + exosomes | CTC detection and GPC1-positive-exosome detection | Early PDAC | 22/28 | 100 | 80 | [105] | |
| Exosomes | KRAS mutations in exoDNA | Presence mutant KRAS in circulating exosome-derived DNA | Early PDAC Locally advanced Metastatic | 33/54 15/54 20/54 | 67 80 85 | NR NR NR | [106] |
| GPC1-exosomes | CTC detection and GPC1-positive-exosome detection | Early PDAC | 22/28 | 50 | 90 | [105] | |
| miRNAs of exosomes | miRNAs in exosomes | All stages | Reported increased expression in PDAC | NR | [107,108,109,110] | ||
| miRNAs | miR-21, miR-25 | miR-21, miR-25 | All stages | 303/760 | 75 | 93 | [111] |
| Meta-analysis | Presence of different miRNAs | All stages | 4,326/4,277 | 79 | 74 | [112] |
| Trial | Trial Design | Trial Purpose | Study Population | n | Primary Endpoint | Technique | Ref. |
|---|---|---|---|---|---|---|---|
| DYNAMIC-Pancreas: ctDNA Analysis Informing Adjuvant Chemotherapy in Early Stage PDAC: A Multicenter Randomized Study | Phase II/III | Prognostic | PDAC locally advance treated with neoadjuvant chemotherapy and surgery | 408 | DFS | ctDNA | ACTRN12618 000335291 |
| Mutation of KRAS, CDKN2A, SMAD4 and TP53 in PDAC | Role of Liquid Biopsy in Preoperative Diagnosis | Diagnostic | Non metastatic PDAC without any systemic metastatic spread at preoperative imaging | 50 | 1-Presence of venous and/or arterial invasion 2-Early recurrence [<12 months from resection], local or systemic recurrence after resection | KRAS, CDKN2A, SMAD4 and TP53 mutation on circulating cfDNA | NCT03524677 |
| Prognostic Role of ctDNA in Resectable PDAC (PROJECTION) | Comparison of DFS of patients with preoperative presence of ctDNA (Group A) and absence of ctDNA (Group B) | Diagnostic Prognostic | Resectable PDAC | 200 | To determine the stage, the remission or the progression of PDAC | Collected prior of surgery and within 14 days before start of adjuvant chemotherapy. | NCT04246203 |
| Detection of High Expression Levels of EMT-Transcription Factor mRNAs in Patients with PDAC and Their Diagnostic Potential | Case control | Diagnostic | Case: Cases Subjects affected by PDAC Control: Healthy Subject enrolled following colon cancer screening via colonoscopy | 850 | DFS | Detection and quantification of EMT-transcription factor mRNA levels in blood | NCT04323917 |
| Verification of Predictive Biomarkers for PDAC Treatment Using Multicenter Liquid Biopsy | Observational | Diagnostic | Subjects affected by PDAC | 662 | Clinical applicability | 1-Quantification and monitoring of KRAS mutations Using ddPCR in ctDNA 2-Discovery of biomarkers through ctDNA panel | NCT04241367 |
| Circulating Extracellular Exosomal Small RNA as Potential Biomarker for Human PDAC | Cohort-prospective | Diagnostic Early-detection | PDAC and other pancreatic lesions | 102 | Sensitivity and specificity of exo-sRNA analysis | exo-sRNA | NCT04636788 |
| Diagnostic Accuracy of CTCs and Onco-exosome Quantification in the Diagnosis of PDAC-PANC-CTC (PANC-CTC) | Cohort-prospective | Diagnostic | PDAC | 52 | Sensitivity and diagnostic application of CTC detection | CTC | NCT03032913 |
| PRIMUS002: Looking at 2 Neo-adjuvant Treatment Regimens for Resectable and Borderline Resectable PDAC | Phase II non-randomized | Prognostic | -FOLFOX-A (FOLFOX + Nab-paclitaxel) -AG: Nabpaclitaxel-Gemcitabie | 278 | Time to progression | ctDNA | NCT04176952 |
| Tumor Markers, Liquid Biopsies, and Patient Reported Outcomes in Metastatic Colorectal, Pancreas, Biliary, and Esophagogastric Cancers | Observational multicohort | Prognostic | Gastrointestinal Cancer | 600 | RECIST response | CEA, CA19.9 and ctDNA | NCT04776837 |
| PLATON-Platform for Analyzing Targetable Tumor Mutations (Pilot-study) | Observational multicohort | Diagnostic | Gastrointestinal Cancer | 200 | Relative frequency of targetable mutations | FoundationOne®CDx and FoundationOne®Liquid | NCT04484636 |
| ctDNA in Pancreatic Cancer | Prospective observational | Diagnostic | Resectable pancreatic cancer | 100 | Analysis of Factors Related to PDAC Recurrence Using ctDNA | ctDNA | NCT02934984 |
| A Study of Blood Based Biomarkers for Pancreas Adenocarcinoma | Prospective observational | Diagnostic Early-detection | PDAC and benign pancreatic disease | 750 | Sensitivity for early diagnosis | Proteins and proteases, functional DNA repair assays, exosomes, stromal elements, cRNAs and ctDNA | NCT 03334708 |
| Blood Markers of Early Pancreas Cancer | Prospective observational | Early-detection | New onset diabetes, high risk pre-diabetes Pancreatic cystic neoplasms and pancreatitis Familial risk | 1250 | Sensitivity for early diagnosis | cfDNA | NCT03568630 |
| Nalirinox Neo-pancreas RAS Mut ctDNA Study | Phase II | Prognostic | Patients with Resectable PDAC Treated with Neoadjuvant NALIRINOX | 20 | Monitoring response | KRAS ctDNA | NCT04010552 |
| Tumor Stage | Nº Patients Total ctDNA + ctDNA- | Results | Ref. |
|---|---|---|---|
| Resectable | 59 29 30 | RFS 8mo if pre-surgery ctDNA + vs. 19mo if ctDNA- (p < 0.01) | [135] |
| Resectable | 37 23 14 | RFS 10.3mo if pre-surgery ctDNA + vs. RFS not reached (p = 0.002) | [95] |
| Resectable | 34 14 * 20 * | ExoDNA KRAS MAF peak of ≥1% after treatment is associated with tumor progression | [97] |
| Advanced/metastatic | 104 50 54 | OS 6.5mo if ctDNA + vs. 19mo if ctDNA- (p < 0.001) | [154] |
| Advanced/metastatic | 55 42 13 | OS 2.5mo if ctDNA + with copy number gain vs. 5.5mo without copy number gain vs. 10.6mo if ctDNA- (p < 0.001) | [155] |
| Metastatic | 61 47 14 | OS 5.6mo if ctDNA + vs. 12.4mo if ctDNA- (p < 0.001) | [156] |
| Metastatic | 102 70 32 | OS 8.6mo if ctDNA + vs. 14.6mo if ctDNA- (p < 0.02) PFS 3.5mo if ctDNA + vs. 10.7mo if ctDNA- (p < 0.02) | [97] |
| Any stage | 77 60 17 | KRAS MAF peak of <0.415% is associated with longer PFS and OS | [157] |
| Advanced/metastatic | 54 36 18 | Decrease in KRAS ctDNA levels during chemotherapy (d14) is an early indicator of response to treatment | [158] |
| Metastatic | 113 77 36 | Early change in ctDNA levels (d28) was correlated with ORR, PFS and OS | [159] |
| Advanced/metastatic | 38 17 21 | The dynamics of total cfDNA concentration correlated with tumor burden following chemotherapy | [117] |
| Metastatic | 188 65 123 | OS 4.7mo if ctDNA + vs. 6mo if ctDNA- (p = 0.015) | [160] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heredia-Soto, V.; Rodríguez-Salas, N.; Feliu, J. Liquid Biopsy in Pancreatic Cancer: Are We Ready to Apply It in the Clinical Practice? Cancers 2021, 13, 1986. https://doi.org/10.3390/cancers13081986
Heredia-Soto V, Rodríguez-Salas N, Feliu J. Liquid Biopsy in Pancreatic Cancer: Are We Ready to Apply It in the Clinical Practice? Cancers. 2021; 13(8):1986. https://doi.org/10.3390/cancers13081986
Chicago/Turabian StyleHeredia-Soto, Victoria, Nuria Rodríguez-Salas, and Jaime Feliu. 2021. "Liquid Biopsy in Pancreatic Cancer: Are We Ready to Apply It in the Clinical Practice?" Cancers 13, no. 8: 1986. https://doi.org/10.3390/cancers13081986
APA StyleHeredia-Soto, V., Rodríguez-Salas, N., & Feliu, J. (2021). Liquid Biopsy in Pancreatic Cancer: Are We Ready to Apply It in the Clinical Practice? Cancers, 13(8), 1986. https://doi.org/10.3390/cancers13081986