
Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects


 , , ,
, , ,  , ,
, ,  ,
,  and
and
Simple Summary
Abstract
1. Introduction
2. Results
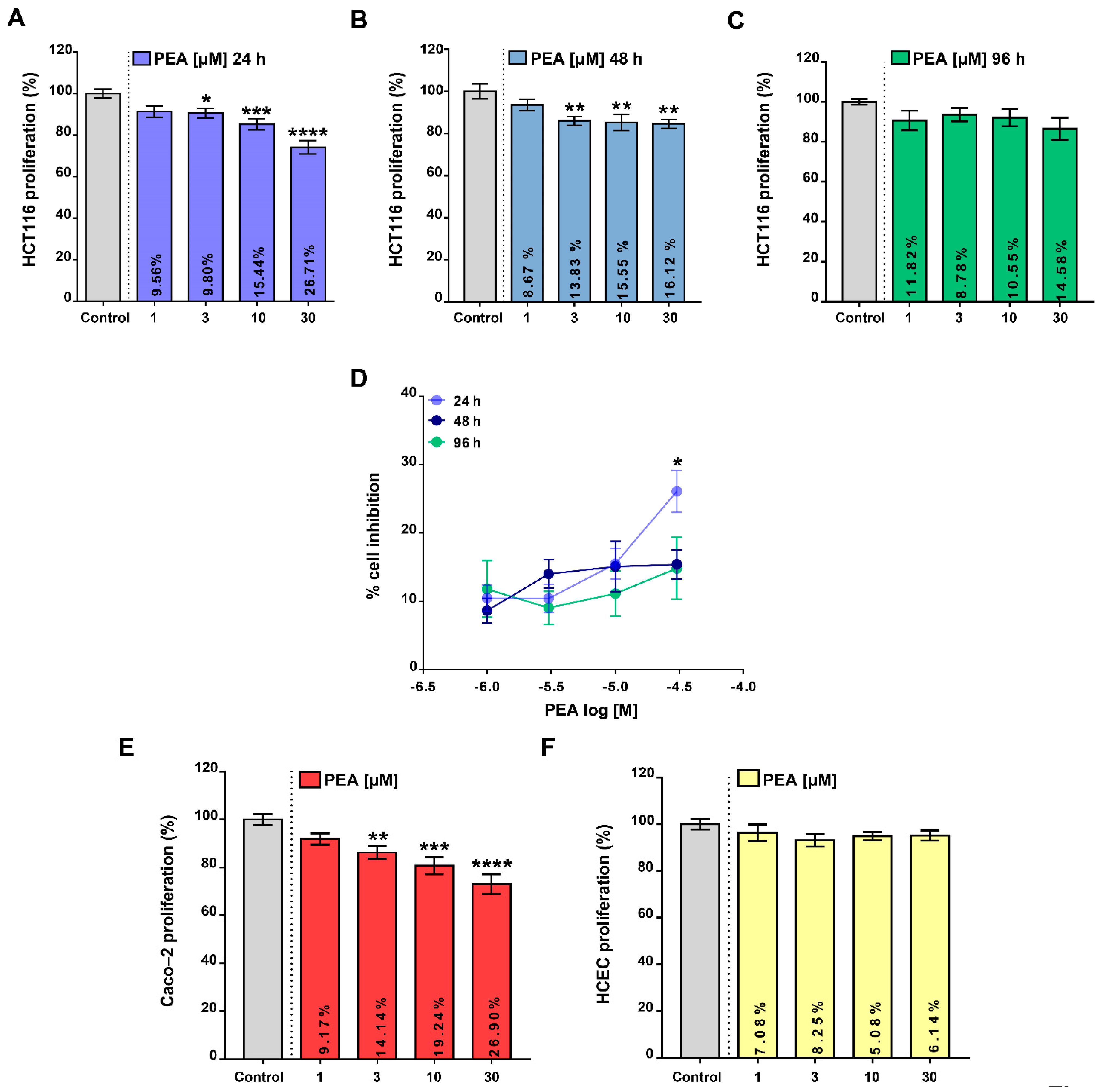
2.1. Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation without Affecting the Proliferation of Healthy Colonic Epithelial Cells
2.2. Palmitoylethanolamide Reduces CRC Cell Proliferation via PPAR-α and GPR55
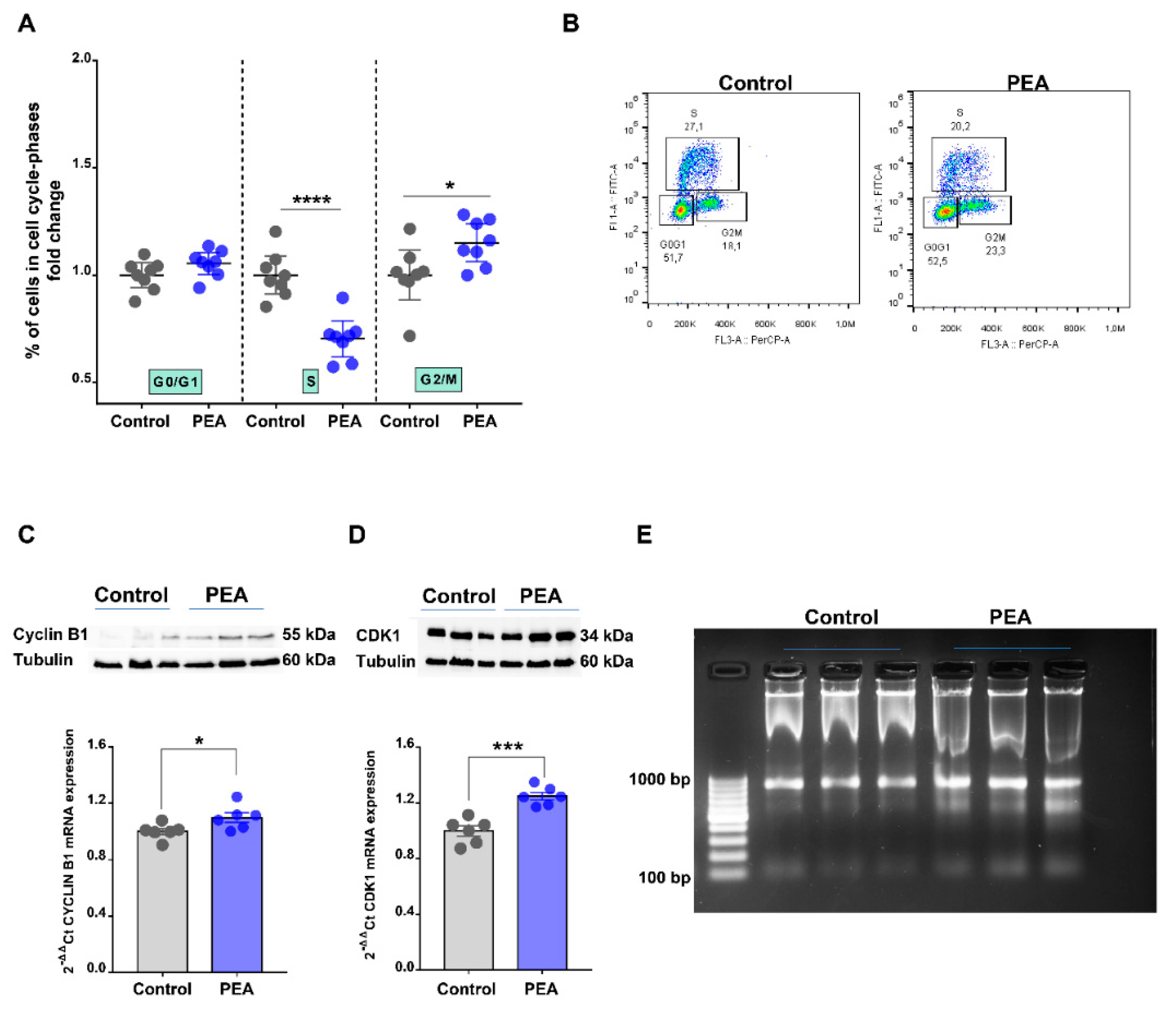
2.3. Palmitoylethanolamide Promotes Cell Cycle Arrest in the G2/M Phase in CRC Cells by Promoting DNA Damage
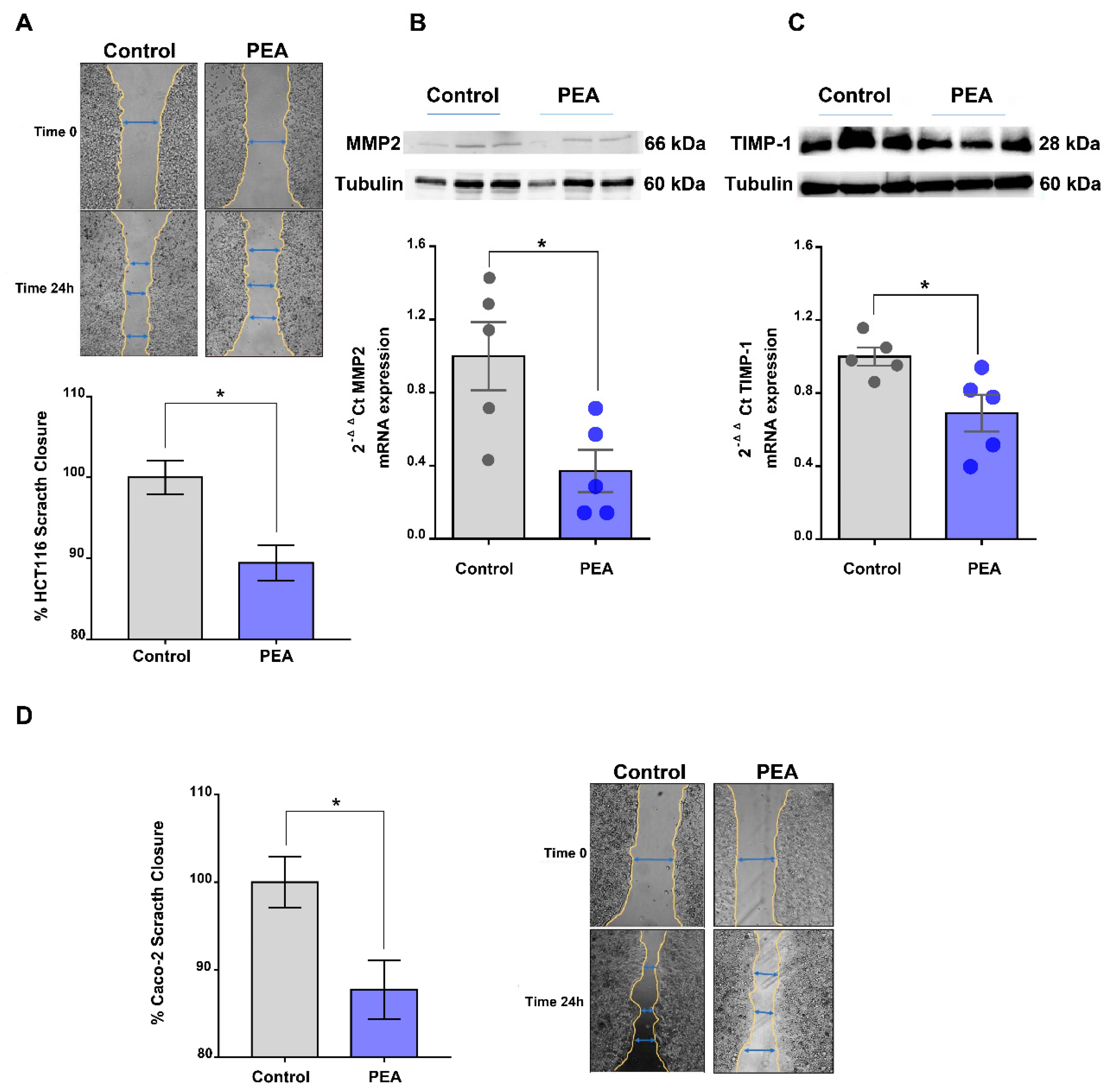
2.4. Palmitoylethanolamide Reduces Tumor Cell Migration
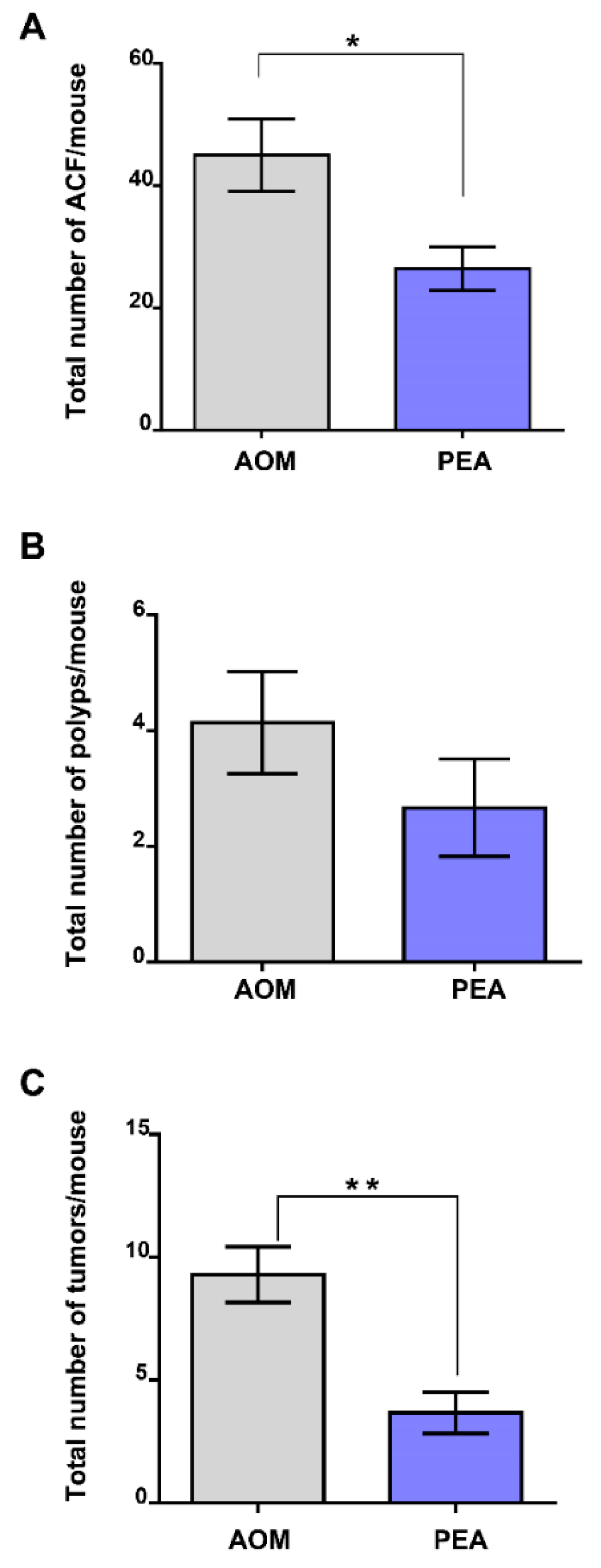
2.5. Palmitoylethanolamide Prevents Tumor Development in a Murine Model of Colon Cancer
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines
4.3. BrdU Incorporation
4.4. Scratch Assay
4.5. Cell Cycle Analysis
4.6. Gene Expression Analysis by Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
4.7. Western Blot Analysis
4.8. DNA Fragmentation Assay
4.9. Mice
4.10. Azoxymethane (AOM) Murine Model of Colon Cancer
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2018 with focus on colorectal cancer. Ann. Oncol. 2018, 29, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef]
- Couch, D.G.; Tasker, C.; Theophilidou, E.; Lund, J.N.; O’Sullivan, S.E. Cannabidiol and palmitoylethanolamide are anti-inflammatory in the acutely inflamed human colon. Clin. Sci. 2017, 131, 2611–2626. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.; Wend, P.; Purfurst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef]
- Maccarrone, M.; Attina, M.; Cartoni, A.; Bari, M.; Finazzi-Agro, A. Gas chromatography-mass spectrometry analysis of endogenous cannabinoids in healthy and tumoral human brain and human cells in culture. J. Neurochem. 2001, 76, 594–601. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Bisogno, T.; Ligresti, A.; Bifulco, M.; Melck, D.; Di Marzo, V. Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam. Clin. Pharmacol. 2002, 16, 297–302. [Google Scholar] [CrossRef]
- Fraguas-Sanchez, A.I.; Martin-Sabroso, C.; Torres-Suarez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 175, 2566–2580. [Google Scholar] [CrossRef] [PubMed]
- Sarnelli, G.; Gigli, S.; Capoccia, E.; Iuvone, T.; Cirillo, C.; Seguella, L.; Nobile, N.; D’Alessandro, A.; Pesce, M.; Steardo, L.; et al. Palmitoylethanolamide Exerts Antiproliferative Effect and Downregulates VEGF Signaling in Caco-2 Human Colon Carcinoma Cell Line Through a Selective PPAR-alpha-Dependent Inhibition of Akt/mTOR Pathway. Phytother. Res. 2016, 30, 963–970. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. Br. J. Pharmacol. 2019, 176 (Suppl. 1), S297–S396. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Gil-Henn, H.; Samson, A.O. Invasion and metastasis: The elusive hallmark of cancer. Oncogene 2020, 39, 2024–2026. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2, 1998–2004. [Google Scholar] [CrossRef]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Hansoulle, L.; Dauguet, N.; Muccioli, G.G.; Gallez, B.; Lambert, D.M. Increasing antiproliferative properties of endocannabinoids in N1E-115 neuroblastoma cells through inhibition of their metabolism. PLoS ONE 2011, 6, e26823. [Google Scholar] [CrossRef]
- Granberg, M.; Fowler, C.J.; Jacobsson, S.O. Effects of the cannabimimetic fatty acid derivatives 2-arachidonoylglycerol, anandamide, palmitoylethanolamide and methanandamide upon IgE-dependent antigen-induced beta-hexosaminidase, serotonin and TNF alpha release from rat RBL-2H3 basophilic leukaemic cells. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 364, 66–73. [Google Scholar] [CrossRef]
- Laezza, C.; Pisanti, S.; Crescenzi, E.; Bifulco, M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Kaz, A.M.; Brentnall, T.A. Genetic testing for colon cancer. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef]
- Capasso, R.; Izzo, A.A.; Fezza, F.; Pinto, A.; Capasso, F.; Mascolo, N.; Di Marzo, V. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br. J. Pharmacol. 2001, 134, 945–950. [Google Scholar] [CrossRef]
- Capasso, R.; Orlando, P.; Pagano, E.; Aveta, T.; Buono, L.; Borrelli, F.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide normalizes intestinal motility in a model of post-inflammatory accelerated transit: Involvement of CB(1) receptors and TRPV1 channels. Br. J. Pharmacol. 2014, 171, 4026–4037. [Google Scholar] [CrossRef]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeted Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| CDK1 | GGATGTGCTTATGCAGGATTCC | CATGTACTGACCAGGAGGGATAG |
| CYCLIN B1 | AACTTTCGCCTGAGCCTATTTT | TTGGTCTGACTGCTTGCTCTT |
| E-CADHERIN | ATTTTTCCCTCGACACCCGAT | TCCCAGGCGTAGACCAAGA |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
| MMP2 | CCCACTGCGGTTTTCTCGAAT | CAAAGGGGTATCCATCGCCAT |
| SLUG | TGTGACAAGGAATATGTGAGCC | TGAGCCCTCAGATTTGACCTG |
| SNAI1 | ACTGCAACAAGGAATACCTCAG | GCACTGGTACTTCTTGACATCTG |
| TGF-β | CTAATGGTGGAAACCCACAACG | TATCGCCAGGAATTGTTGCTG |
| TIMP1 | CTTCTGCAATTCCGACCTCGT | ACGCTGGTATAAGGTGGTCTG |
| TWIST | GTCCGCAGTCTTACGAGGAG | GCTTGAGGGTCTGAATCTTGCT |
| VIMENTIN | AGTCCACTGAGTACCGGAGAC | CATTTCACGCATCTGGCGTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, E.; Venneri, T.; Lucariello, G.; Cicia, D.; Brancaleone, V.; Nanì, M.F.; Cacciola, N.A.; Capasso, R.; Izzo, A.A.; Borrelli, F.; et al. Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects. Cancers 2021, 13, 1923. https://doi.org/10.3390/cancers13081923
Pagano E, Venneri T, Lucariello G, Cicia D, Brancaleone V, Nanì MF, Cacciola NA, Capasso R, Izzo AA, Borrelli F, et al. Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects. Cancers. 2021; 13(8):1923. https://doi.org/10.3390/cancers13081923
Chicago/Turabian StylePagano, Ester, Tommaso Venneri, Giuseppe Lucariello, Donatella Cicia, Vincenzo Brancaleone, M. Francesca Nanì, Nunzio A. Cacciola, Raffaele Capasso, Angelo A. Izzo, Francesca Borrelli, and et al. 2021. "Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects" Cancers 13, no. 8: 1923. https://doi.org/10.3390/cancers13081923
APA StylePagano, E., Venneri, T., Lucariello, G., Cicia, D., Brancaleone, V., Nanì, M. F., Cacciola, N. A., Capasso, R., Izzo, A. A., Borrelli, F., & Romano, B. (2021). Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects. Cancers, 13(8), 1923. https://doi.org/10.3390/cancers13081923