
Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
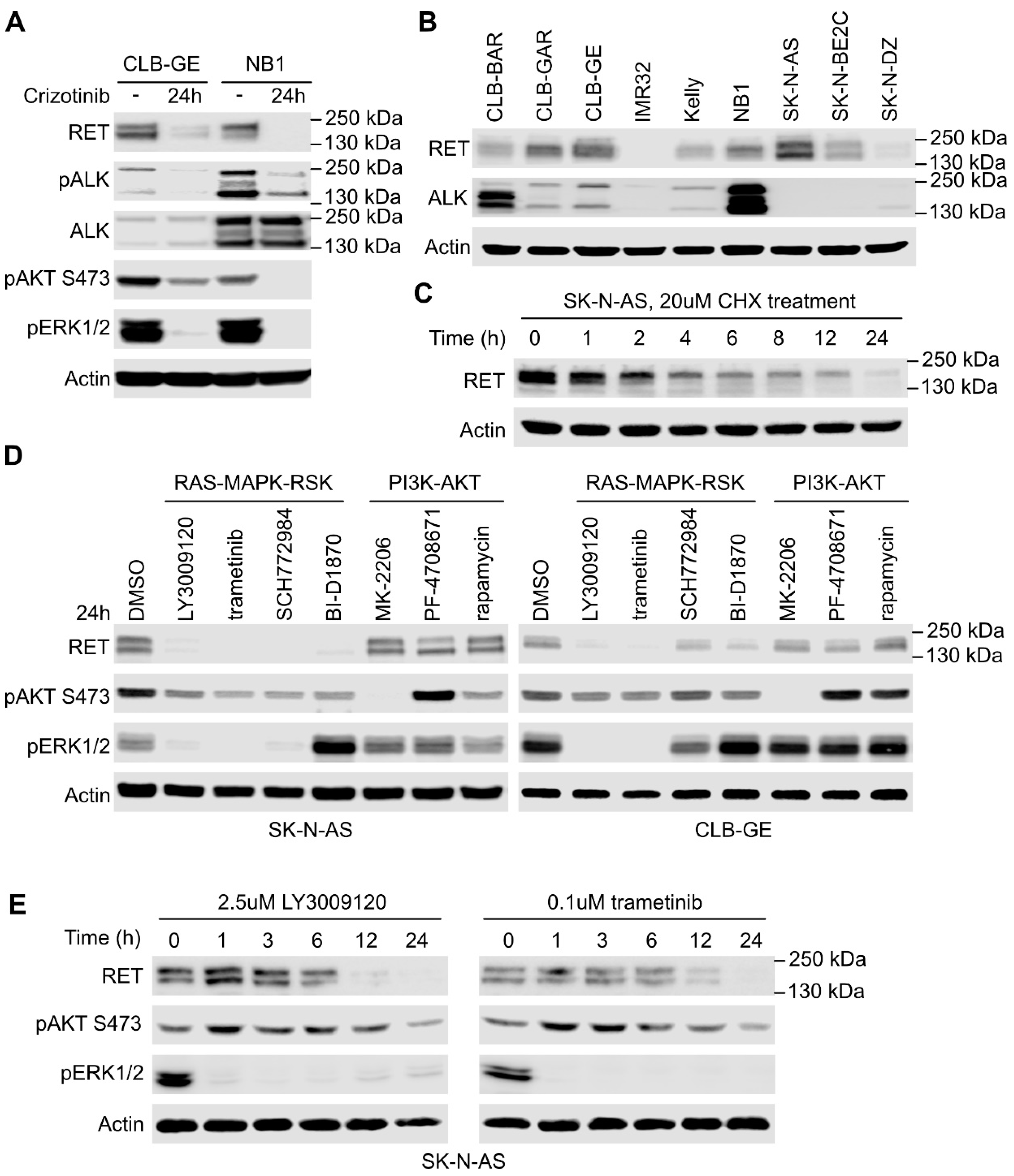
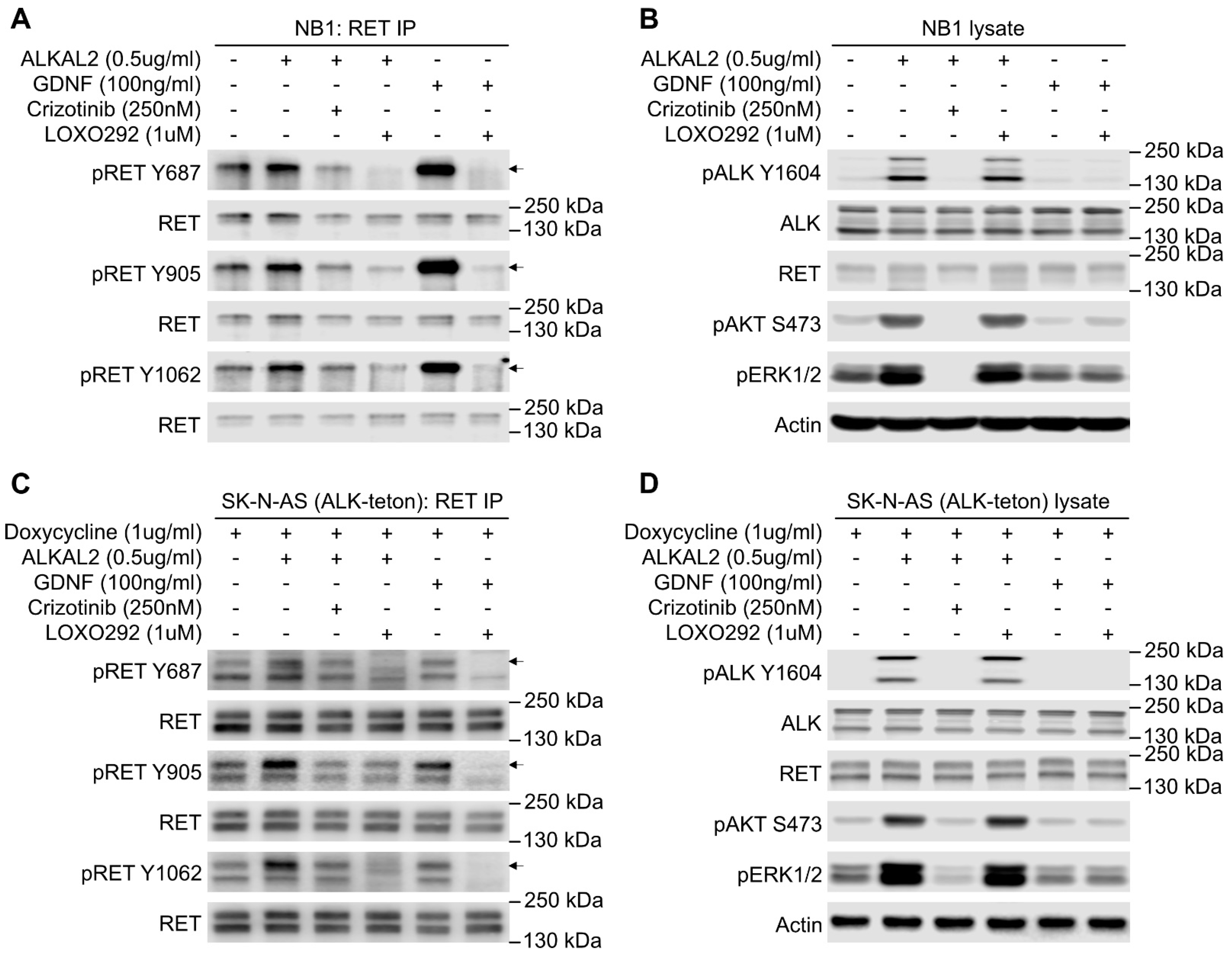
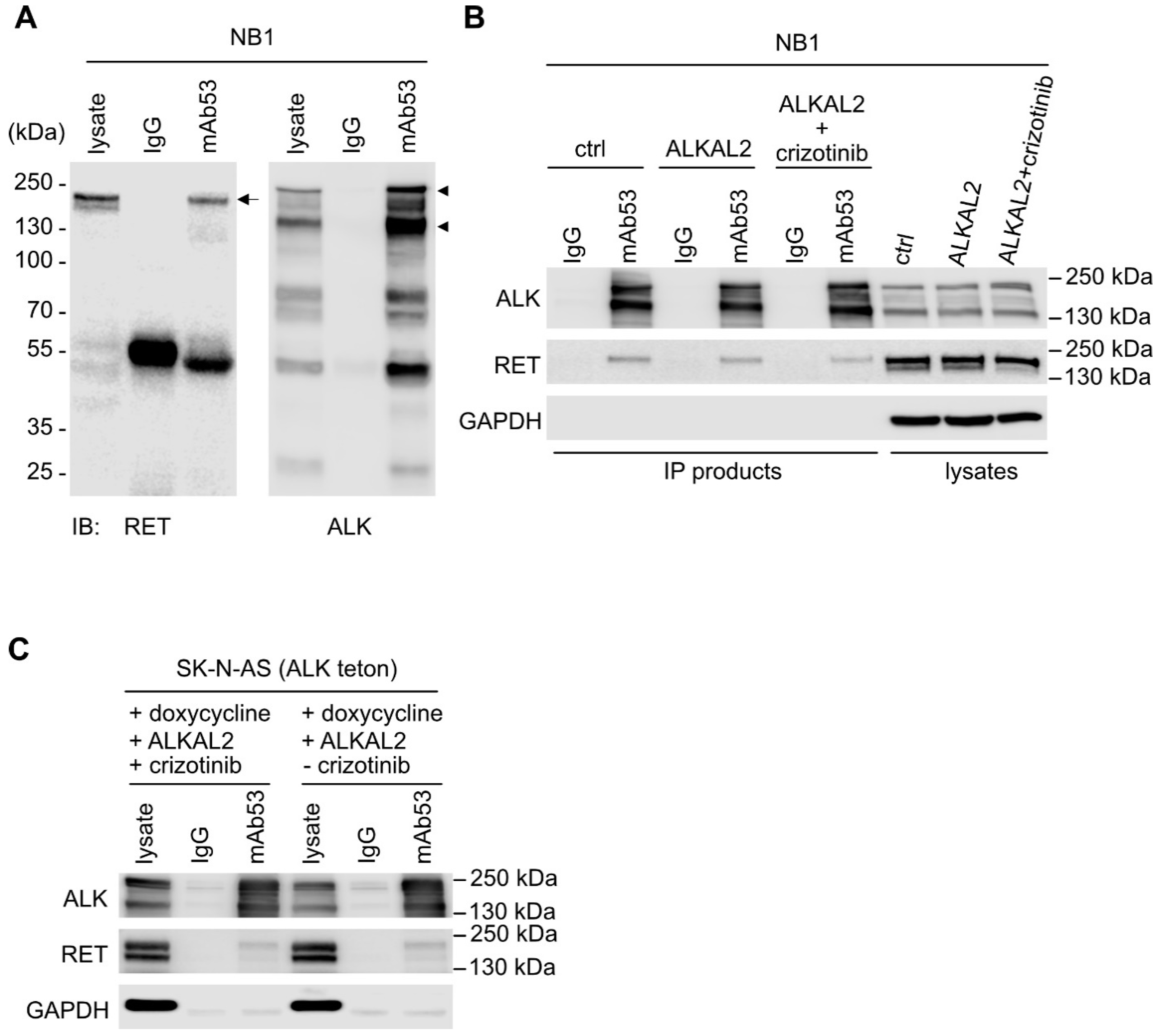
2.1. Stimulation of ALK in NB Cells Leads to RET Receptor Phosphorylation
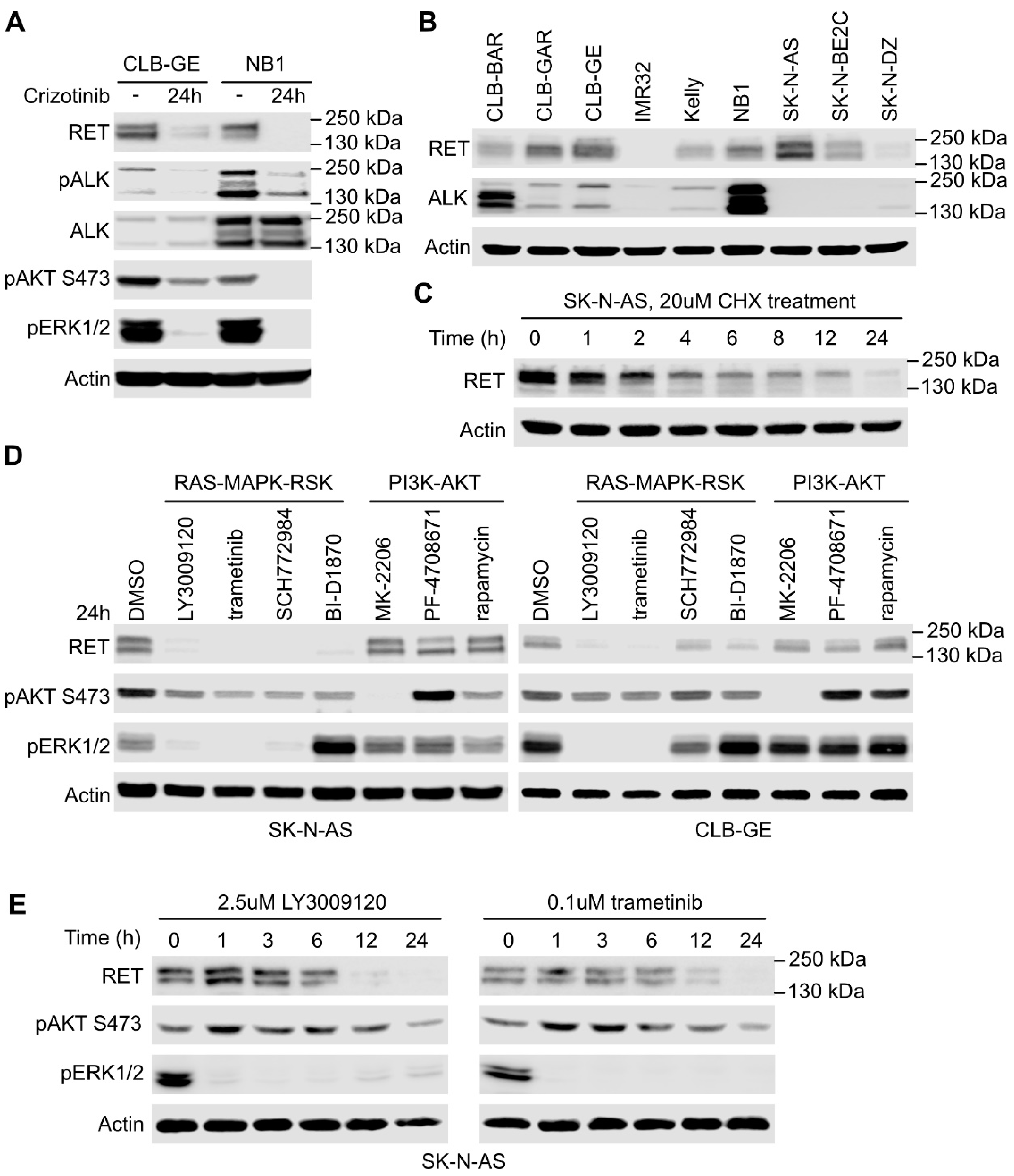
2.2. RAS/MAPK Signaling but Not PI3K/AKT Regulates RET Transcription
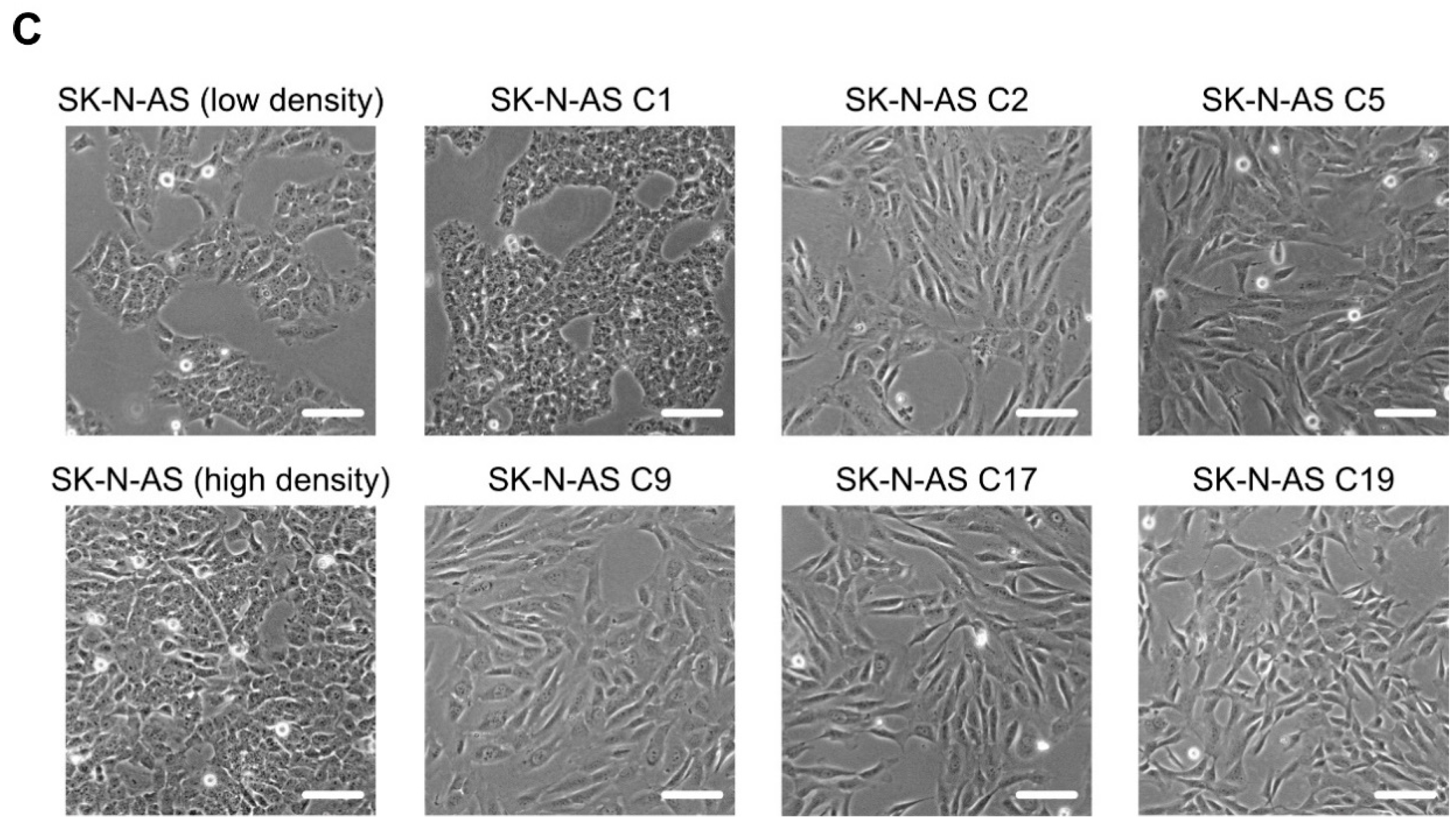
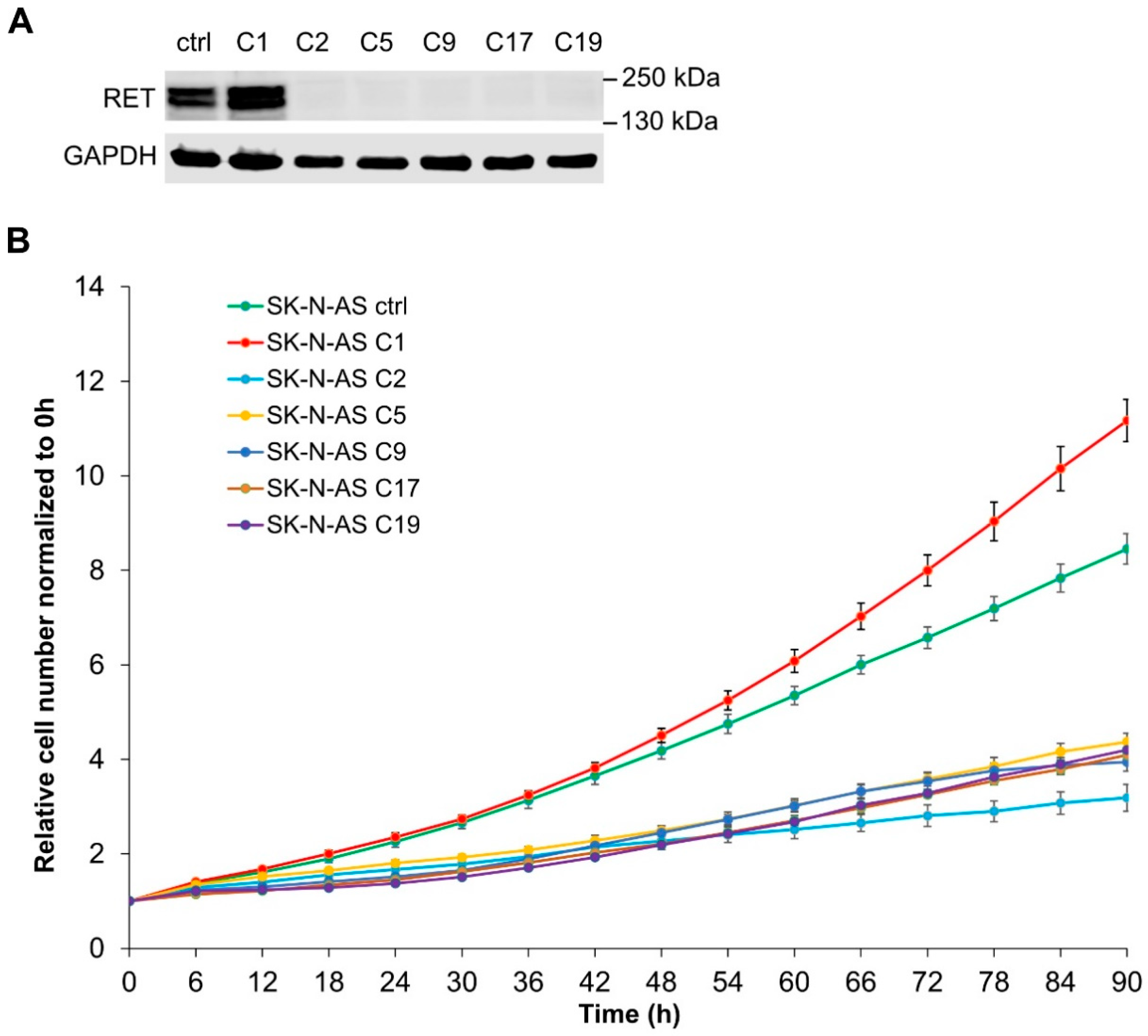
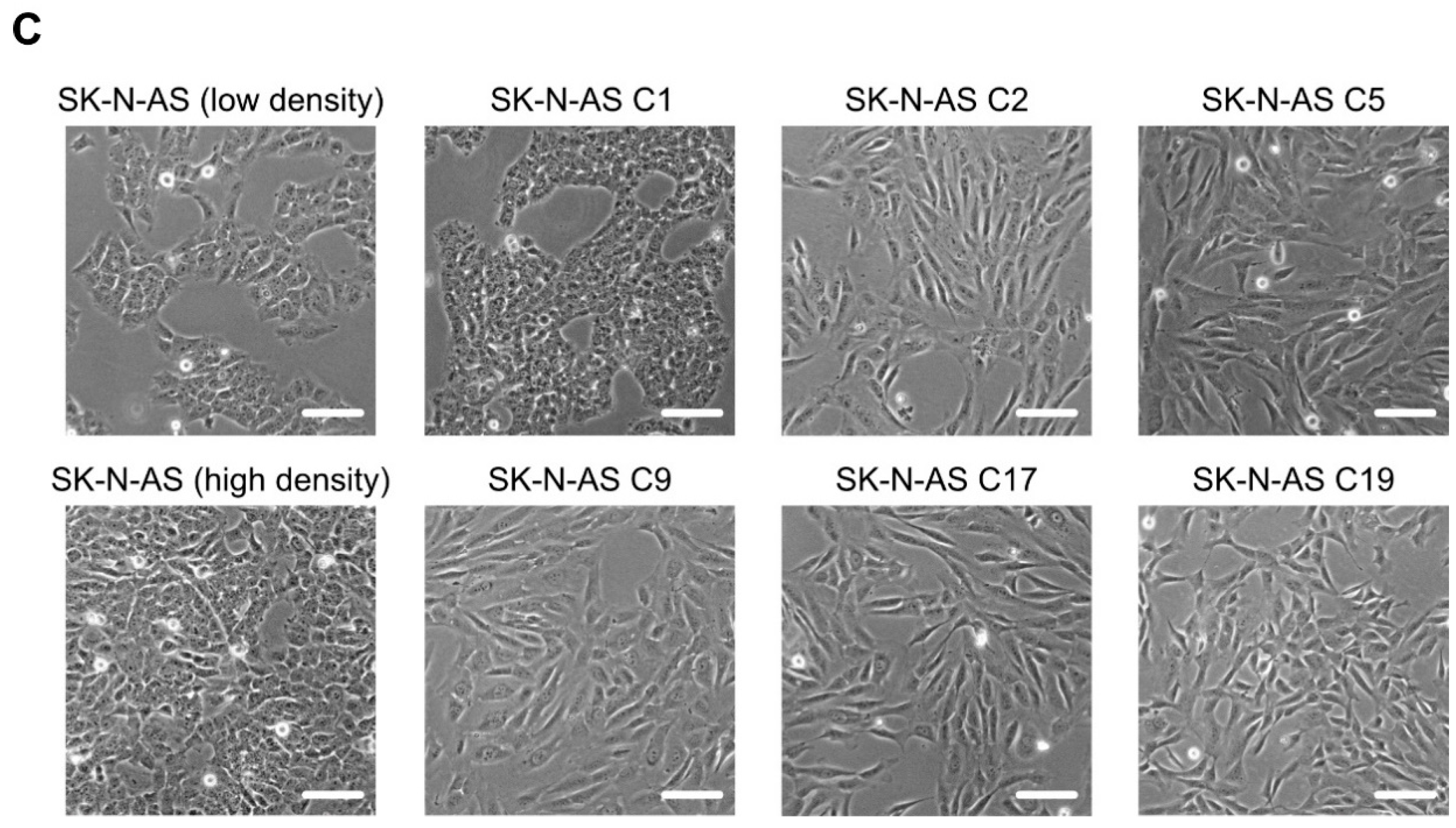
2.3. Loss of RET Leads to Phenotypic Changes and Reduced Proliferation
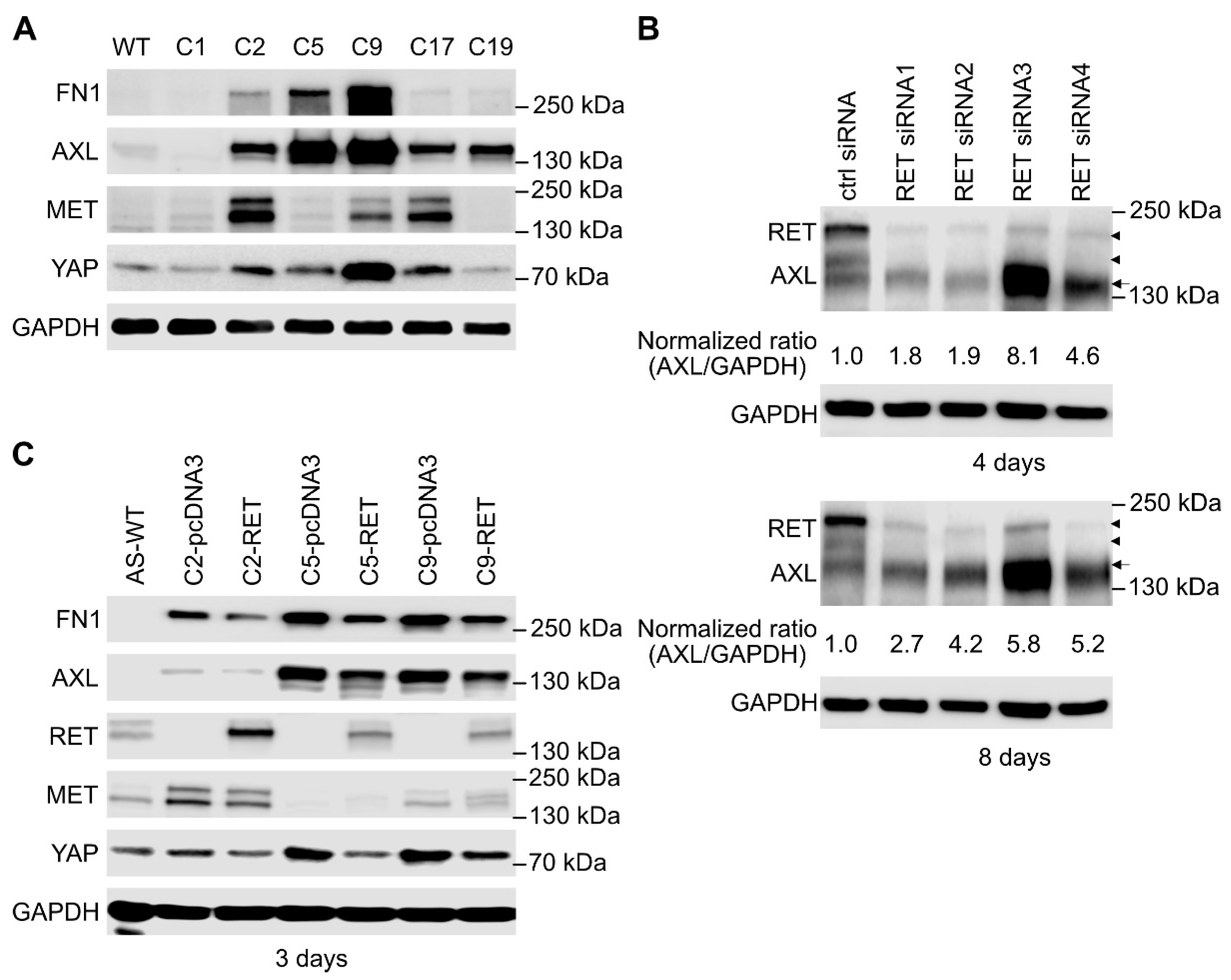
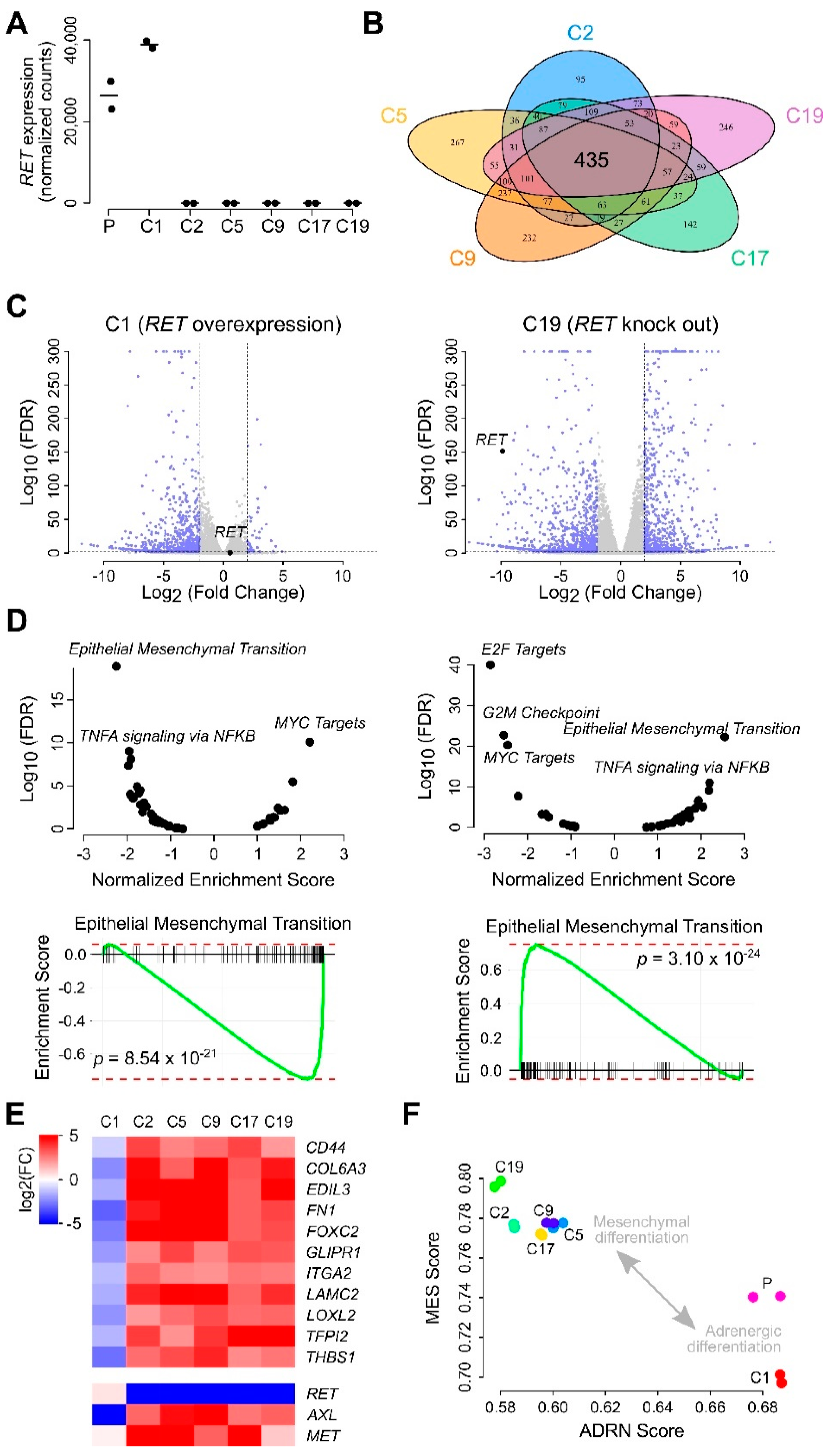
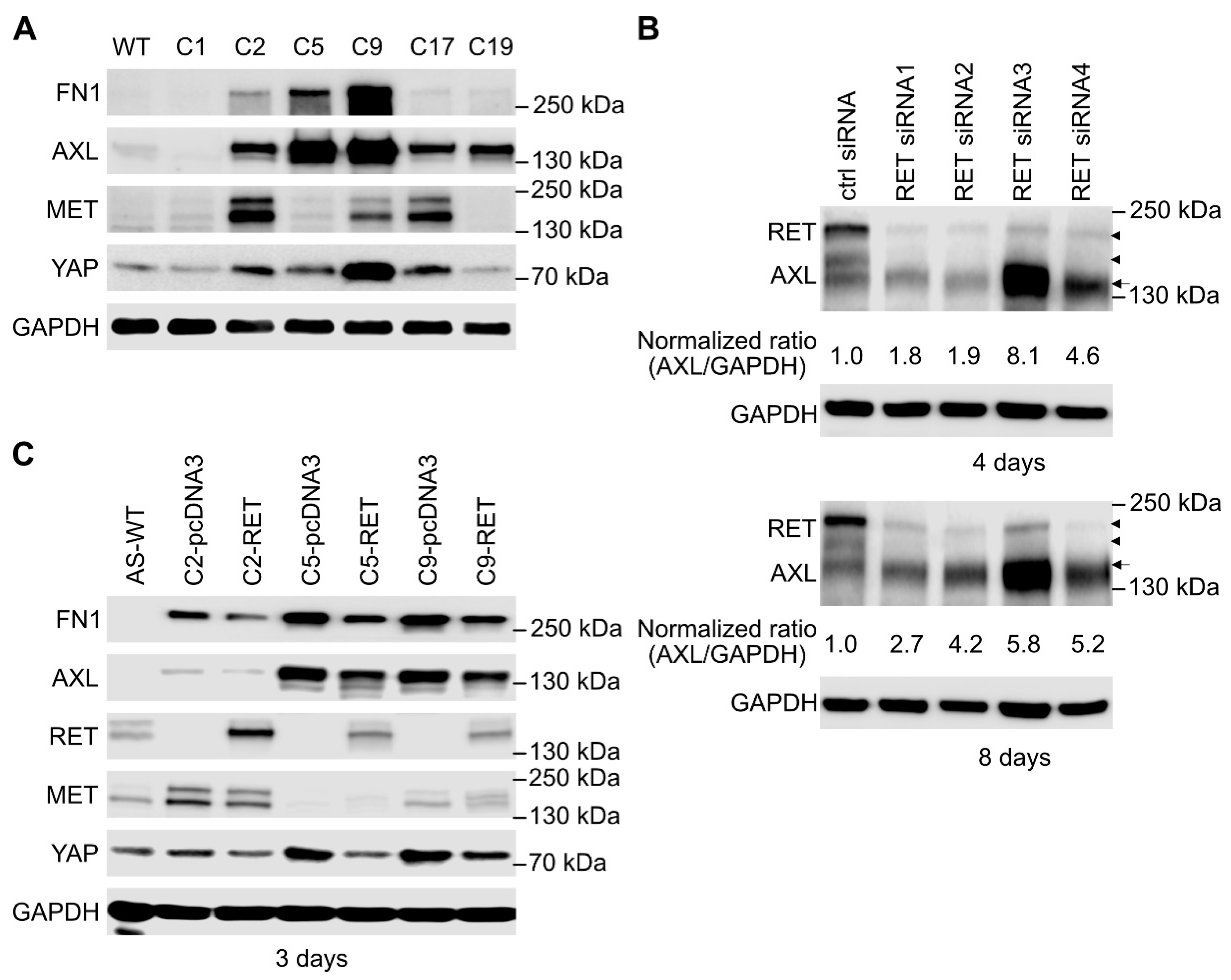
2.4. SK-N-AS RET KO NB Cells Display a Striking EMT Signature
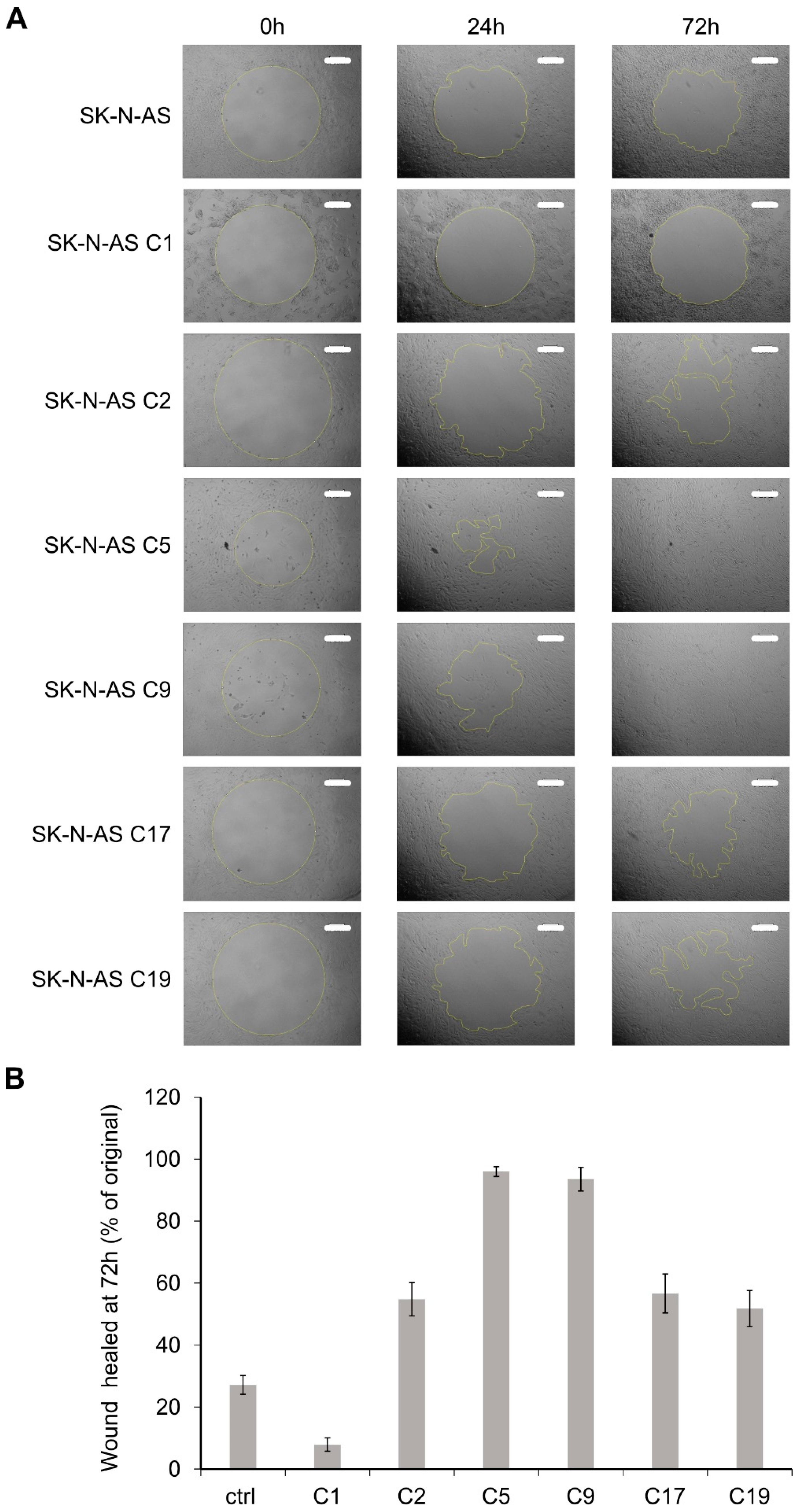
2.5. RET KO Cells Display Enhanced Cell Migration
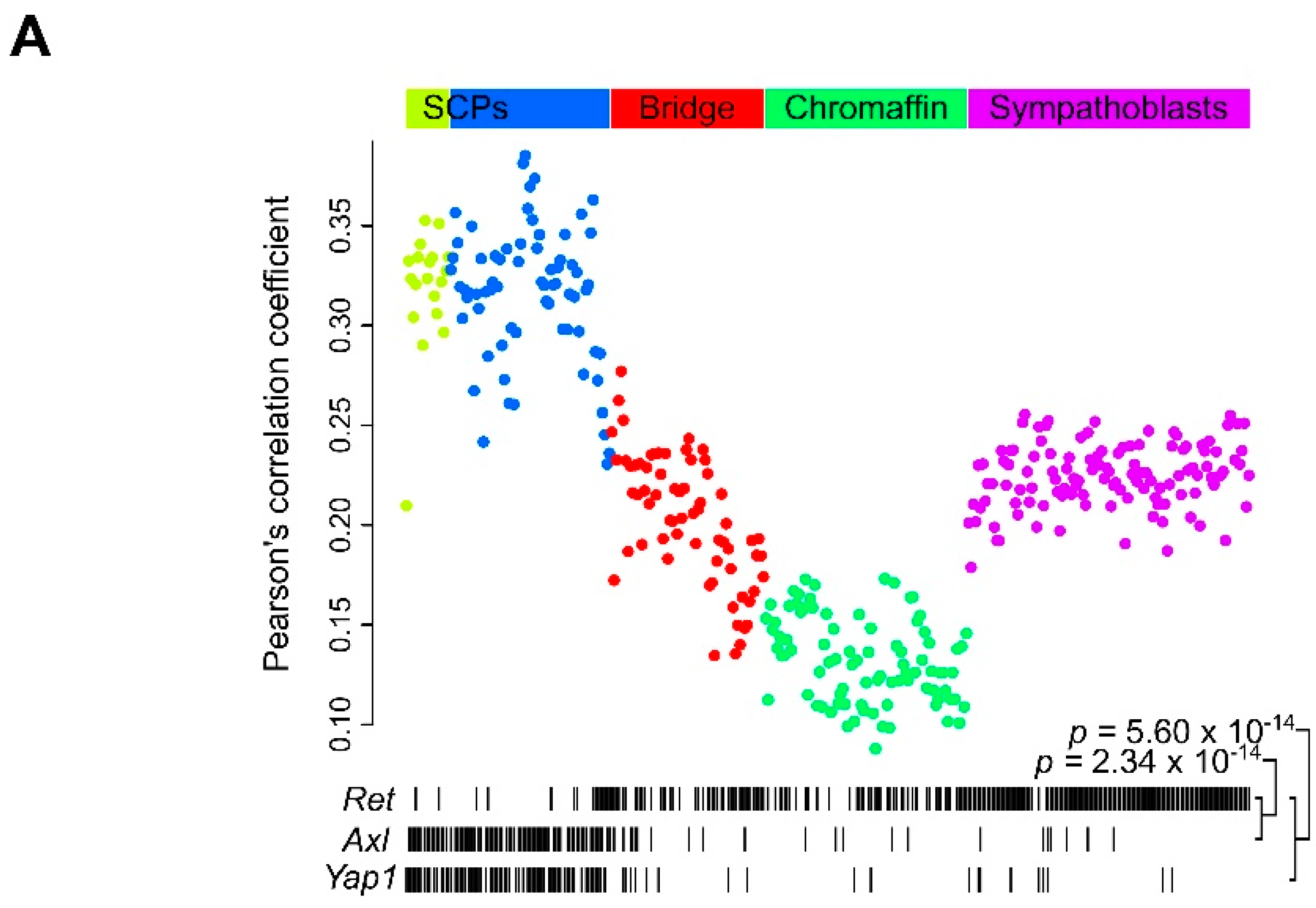
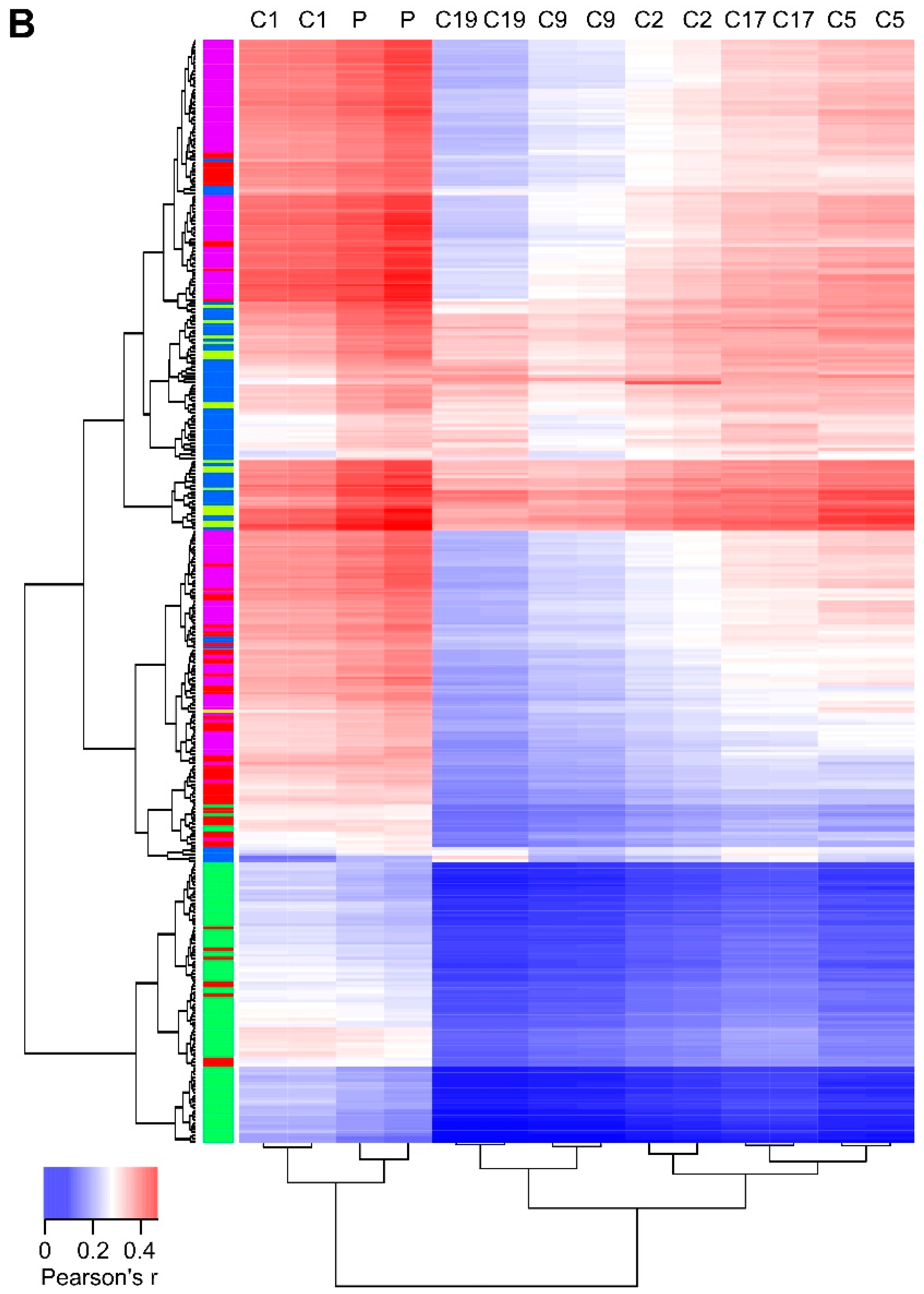
2.6. Ret Expression Correlates with Differentiation in the Developing Sympathaticosystem
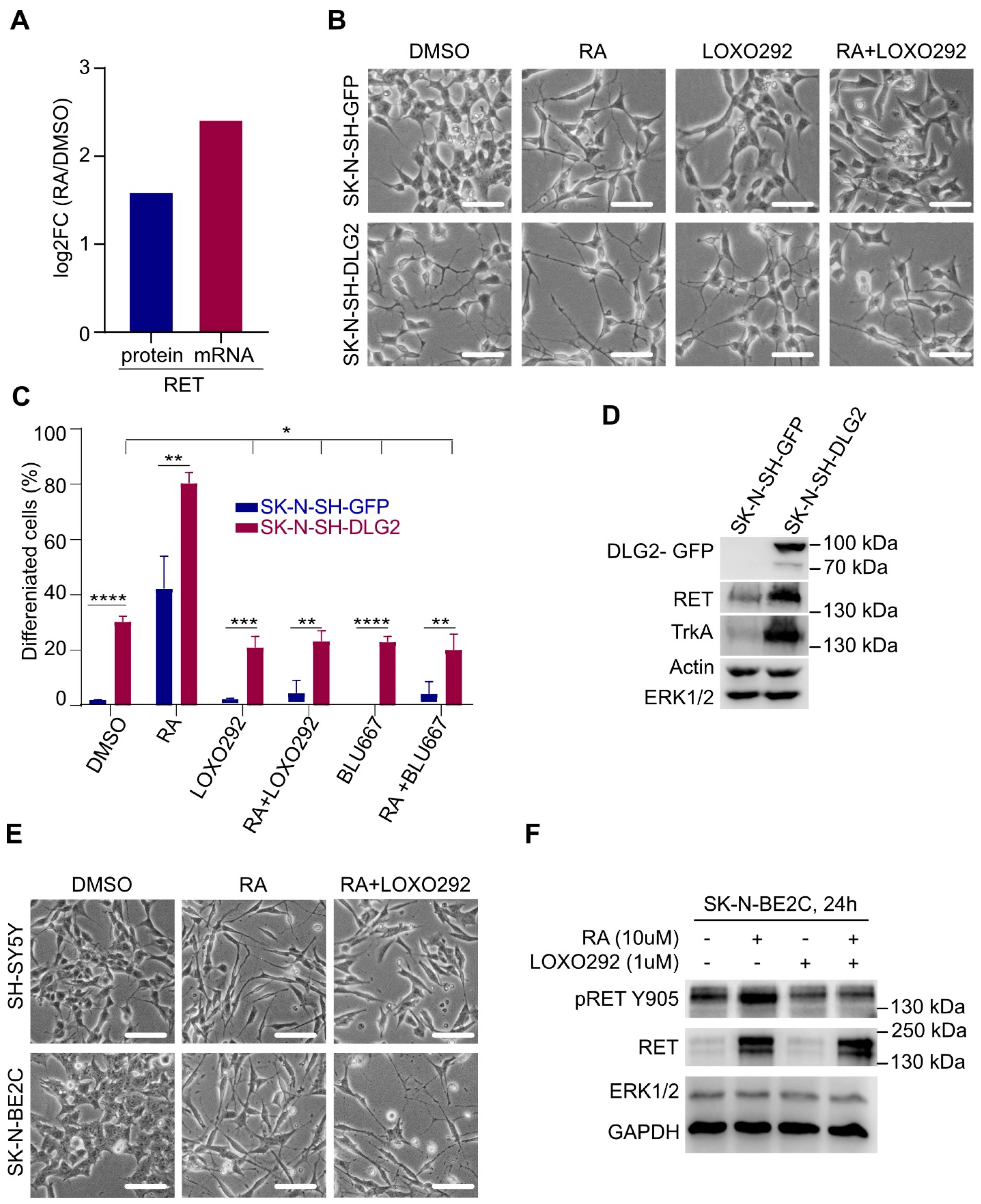
2.7. RET Is Required for Context-Dependent Differentiation of NB Cells
3. Discussion
4. Material and Methods
4.1. Antibodies and Inhibitors
4.2. Cell Culture and Cell Treatments
4.3. Plasmids, siRNA, and Cell Transfection
4.4. Cell Lysis, Immunoprecipitation, and Western Blot Analysis
4.5. Generation of CRISPR/Cas9 Engineered RET KO SK-N-AS Cells
4.6. Cell Proliferation Assay
4.7. Cell Migration Assay
4.8. Differentiation Assay
4.9. RNA-Seq Data Analysis
4.10. Proteomic Data Analysis
4.11. Gene Set Enrichment Analysis
4.12. Correlation Analysis between RET KO Clones and Single-Cell Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into alk receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of alk as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in alk provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the alk kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the alk gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of alk kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed alk mutation spectrum in tumors with mycn amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [Green Version]
- Umapathy, G.; Mendoza-Garcia, P.; Hallberg, B.; Palmer, R.H. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019, 127, 288–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nusslein-Volhard, C.; Palmer, R.H. Alkals are in vivo ligands for alk family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, E.S.; Cheng, Q.N.; Reshetnyak, A.V.; Schlessinger, J.; Nicoli, S. Alk and ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase ltk. Proc. Natl. Acad. Sci. USA 2017, 114, 12027–12032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (fam150) are ligands of the receptor tyrosine kinases alk and ltk: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. Fam150a and fam150b are activating ligands for anaplastic lymphoma kinase. Elife 2015, 4, e09811. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic lymphoma kinase (alk) regulates initiation of transcription of mycn in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [Green Version]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of mycn causes neuroblastoma in transgenic mice. Embo J 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The alk(f1174l) mutation potentiates the oncogenic activity of mycn in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated alk collaborates with mycn in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Ono, S.; Saito, T.; Terui, K.; Yoshida, H.; Enomoto, H. Generation of conditional alk f1174l mutant mouse models for the study of neuroblastoma pathogenesis. Genesis 2019, 57, e23323. [Google Scholar] [CrossRef]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugene, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated alk triggers prolonged neurogenesis and ret upregulation providing a therapeutic target in alk-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated alk induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra191. [Google Scholar] [CrossRef]
- Mulligan, L.M. Ret revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, I.; Kumps, C.; Claeys, S.; Lindner, S.; Beckers, A.; Janssens, E.; Carter, D.R.; Cazes, A.; Cheung, B.B.; De Mariano, M.; et al. Upregulation of mapk negative feedback regulators and ret in mutant alk neuroblastoma: Implications for targeted treatment. Clin. Cancer Res. 2015, 21, 3327–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui-Pierchala, B.A.; Milbrandt, J.; Johnson, E.M., Jr. Ngf utilizes c-ret via a novel gfl-independent, inter-rtk signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron 2002, 33, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Menacho, I.; Barnouin, K.; Goodman, K.; Martinez-Torres, R.J.; Borg, A.; Murray-Rust, J.; Mouilleron, S.; Knowles, P.; McDonald, N.Q. Oncogenic ret kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol. Cell 2014, 53, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.C.; Vyse, S.; Huang, P.H. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov. Today 2017, 22, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, C.L.; D’Alessio, A.; de Franciscis, V.; Cerchia, L. A cross-talk between trkb and ret tyrosine kinases receptors mediates neuroblastoma cells differentiation. PLoS ONE 2008, 3, e1643. [Google Scholar] [CrossRef]
- Witek, B.; El Wakil, A.; Nord, C.; Ahlgren, U.; Eriksson, M.; Vernersson-Lindahl, E.; Helland, A.; Alexeyev, O.A.; Hallberg, B.; Palmer, R.H. Targeted disruption of alk reveals a potential role in hypogonadotropic hypogonadism. PLoS ONE 2015, 10, e0123542. [Google Scholar] [CrossRef]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharm. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Ret-deficient mice: An animal model for hirschsprung’s disease and renal agenesis. J. Intern. Med. 1995, 238, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Uesaka, T.; Nagashimada, M.; Yonemura, S.; Enomoto, H. Diminished ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J. Clin. Investig. 2008, 118, 1890–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofstra, R.M.; Cheng, N.C.; Hansen, C.; Stulp, R.P.; Stelwagen, T.; Clausen, N.; Tommerup, N.; Caron, H.; Westerveld, A.; Versteeg, R.; et al. No mutations found by ret mutation scanning in sporadic and hereditary neuroblastoma. Hum. Genet. 1996, 97, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Peaston, A.E.; Camacho, M.L.; Norris, M.D.; Haber, M.; Marsh, D.J.; Robinson, B.G.; Hyland, V.J.; Marshall, G.M. Absence of men2a- or 2b-type ret mutations in primary neuroblastoma tumour tissue. Mol. Cell Probes. 1998, 12, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynden, J.; Umapathy, G.; Ashouri, A.; Cervantes-Madrid, D.; Szydzik, J.; Ruuth, K.; Koster, J.; Larsson, E.; Guan, J.; Palmer, R.H.; et al. Phosphoproteome and gene expression profiling of alk inhibition in neuroblastoma cell lines reveals conserved oncogenic pathways. Sci. Signal. 2018, 11, 557. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Delisle, L.; Pierre-Eugene, C.; Louis-Brennetot, C.; Surdez, D.; Raynal, V.; Baulande, S.; Boeva, V.; Grossetete-Lalami, S.; Combaret, V.; Peuchmaur, M.; et al. Activated alk signals through the erk-etv5-ret pathway to drive neuroblastoma oncogenesis. Oncogene 2018, 37, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Ishizaka, Y.; Nagao, M.; Hara, M.; Ishikawa, T. Expression of the ret proto-oncogene product in human normal and neoplastic tissues of neural crest origin. J. Pathol. 1994, 172, 255–260. [Google Scholar] [CrossRef]
- Tahira, T.; Ishizaka, Y.; Itoh, F.; Nakayasu, M.; Sugimura, T.; Nagao, M. Expression of the ret proto-oncogene in human neuroblastoma cell lines and its increase during neuronal differentiation induced by retinoic acid. Oncogene 1991, 6, 2333–2338. [Google Scholar]
- D’Alessio, A.; De Vita, G.; Cali, G.; Nitsch, L.; Fusco, A.; Vecchio, G.; Santelli, G.; Santoro, M.; de Franciscis, V. Expression of the ret oncogene induces differentiation of sk-n-be neuroblastoma cells. Cell Growth Differ 1995, 6, 1387–1394. [Google Scholar]
- Hishiki, T.; Nimura, Y.; Isogai, E.; Kondo, K.; Ichimiya, S.; Nakamura, Y.; Ozaki, T.; Sakiyama, S.; Hirose, M.; Seki, N.; et al. Glial cell line-derived neurotrophic factor/neurturin-induced differentiation and its enhancement by retinoic acid in primary human neuroblastomas expressing c-ret, gfr alpha-1, and gfr alpha-2. Cancer Res. 1998, 58, 2158–2165. [Google Scholar]
- Peterson, S.; Bogenmann, E. The ret and trka pathways collaborate to regulate neuroblastoma differentiation. Oncogene 2004, 23, 213–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siaw, J.T.; Javanmardi, N.; Van den Eynden, J.; Lind, D.E.; Fransson, S.; Martinez-Monleon, A.; Djos, A.; Sjoberg, R.M.; Ostensson, M.; Caren, H.; et al. 11q deletion or alk activity curbs dlg2 expression to maintain an undifferentiated state in neuroblastoma. Cell Rep. 2020, 32, 108171. [Google Scholar] [CrossRef] [PubMed]
- Tetri, L.H.; Kolla, V.; Golden, R.L.; Iyer, R.; Croucher, J.L.; Choi, J.H.; Macfarland, S.P.; Naraparaju, K.; Guan, P.; Nguyen, F.; et al. Ret receptor expression and interaction with trk receptors in neuroblastomas. Oncol. Rep. 2020, 44, 263–272. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative wnt signaling activates yap/taz. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szemes, M.; Greenhough, A.; Malik, K. Wnt signaling is a major determinant of neuroblastoma cell lineages. Front. Mol. Neurosci. 2019, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Borenas, M.; Umapathy, G.; Lai, W.Y.; Lind, D.E.; Witek, B.; Guan, J.; Mendoza-Garcia, P.; Masudi, T.; Claeys, A.; Chuang, T.P.; et al. Alk ligand alkal2 potentiates mycn-driven neuroblastoma in the absence of alk mutation. Embo J. 2021, 40, e105784. [Google Scholar] [CrossRef] [PubMed]
- Mus, L.M.; Lambertz, I.; Claeys, S.; Kumps, C.; Van Loocke, W.; Van Neste, C.; Umapathy, G.; Vaapil, M.; Bartenhagen, C.; Laureys, G.; et al. The ets transcription factor etv5 is a target of activated alk in neuroblastoma contributing to increased tumour aggressiveness. Sci. Rep. 2020, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr. Opin. Cell Biol. 2018, 55, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Bronner, M.E. Neural crest lineage analysis: From past to future trajectory. Development 2020, 147, 20. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. Taz promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [Green Version]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357, 6346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, J.; Huang, R.Y. Axl-driven emt state as a targetable conduit in cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [Green Version]
- Bunone, G.; Borrello, M.G.; Picetti, R.; Bongarzone, I.; Peverali, F.A.; de Franciscis, V.; Della Valle, G.; Pierotti, M.A. Induction of ret proto-oncogene expression in neuroblastoma cells precedes neuronal differentiation and is not mediated by protein synthesis. Exp. Cell Res. 1995, 217, 92–99. [Google Scholar] [CrossRef]
- Oppenheimer, O.; Cheung, N.K.; Gerald, W.L. The ret oncogene is a critical component of transcriptional programs associated with retinoic acid-induced differentiation in neuroblastoma. Mol. Cancer Ther. 2007, 6, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.M.; Huang, P.H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. Alk inhibitor resistance in alk(f1174l)-driven neuroblastoma is associated with axl activation and induction of emt. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.W.; Borenas, M.; Lind, D.E.; Cervantes-Madrid, D.; Umapathy, G.; Palmer, R.H.; Hallberg, B. Alectinib, an anaplastic lymphoma kinase inhibitor, abolishes alk activity and growth in alk-positive neuroblastoma cells. Front. Oncol. 2019, 9, 579. [Google Scholar] [CrossRef]
- Kodama, T.; Tsukaguchi, T.; Satoh, Y.; Yoshida, M.; Watanabe, Y.; Kondoh, O.; Sakamoto, H. Alectinib shows potent antitumor activity against ret-rearranged non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2910–2918. [Google Scholar] [CrossRef] [Green Version]
- Smith-Hicks, C.L.; Sizer, K.C.; Powers, J.F.; Tischler, A.S.; Costantini, F. C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2b. Embo J. 2000, 19, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Yoshioka, Y.; Maeda, K.; Bito, Y.; Fukumoto, T.; Uesaka, T.; Enomoto, H. Mice conditionally expressing ret(c618f) mutation display c cell hyperplasia and hyperganglionosis of the enteric nervous system. Genesis 2019, 57, e23292. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Kalcheim, C.; Unsicker, K. The development of the chromaffin cell lineage from the neural crest. Auton. Neurosci. 2009, 151, 10–16. [Google Scholar] [CrossRef]
- Saito, D.; Takase, Y.; Murai, H.; Takahashi, Y. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science 2012, 336, 1578–1581. [Google Scholar] [CrossRef]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Creminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. Gencode: The reference human genome annotation for the encode project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Suomi, T.; Seyednasrollah, F.; Jaakkola, M.K.; Faux, T.; Elo, L.L. Rots: An r package for reproducibility-optimized statistical testing. PLoS Comput. Biol. 2017, 13, e1005562. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siaw, J.T.; Gabre, J.L.; Uçkun, E.; Vigny, M.; Zhang, W.; Van den Eynden, J.; Hallberg, B.; Palmer, R.H.; Guan, J. Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells. Cancers 2021, 13, 1909. https://doi.org/10.3390/cancers13081909
Siaw JT, Gabre JL, Uçkun E, Vigny M, Zhang W, Van den Eynden J, Hallberg B, Palmer RH, Guan J. Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells. Cancers. 2021; 13(8):1909. https://doi.org/10.3390/cancers13081909
Chicago/Turabian StyleSiaw, Joachim T., Jonatan L. Gabre, Ezgi Uçkun, Marc Vigny, Wancun Zhang, Jimmy Van den Eynden, Bengt Hallberg, Ruth H. Palmer, and Jikui Guan. 2021. "Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells" Cancers 13, no. 8: 1909. https://doi.org/10.3390/cancers13081909
APA StyleSiaw, J. T., Gabre, J. L., Uçkun, E., Vigny, M., Zhang, W., Van den Eynden, J., Hallberg, B., Palmer, R. H., & Guan, J. (2021). Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells. Cancers, 13(8), 1909. https://doi.org/10.3390/cancers13081909