
Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
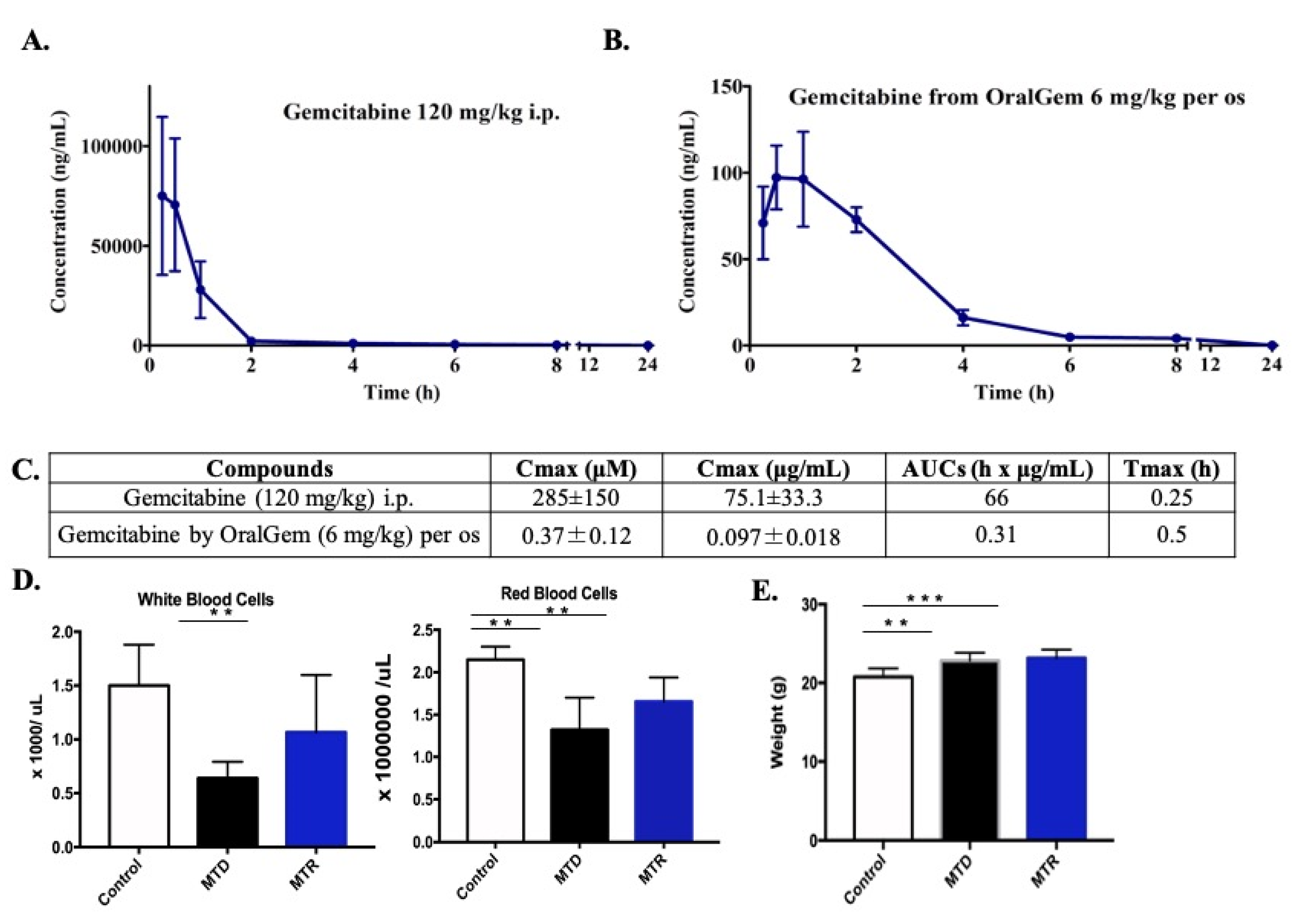
2.1. MTD Gemcitabine Dosing Generates High Circulating Drug Concentrations Compared to MTR OralGem Administration
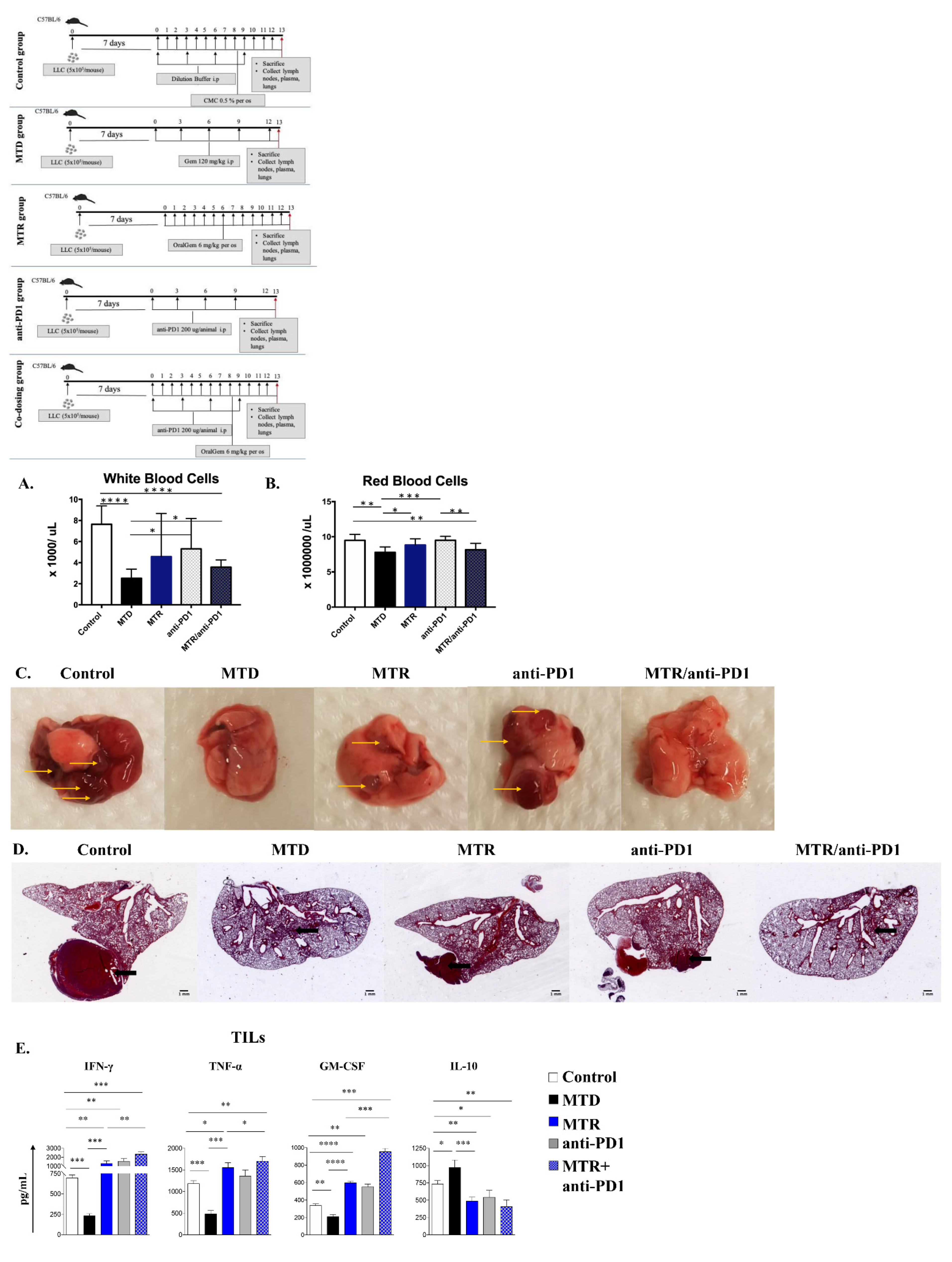
2.2. Decreased White Blood Cell Toxicity upon OralGem MTR Compared to MTD Administration
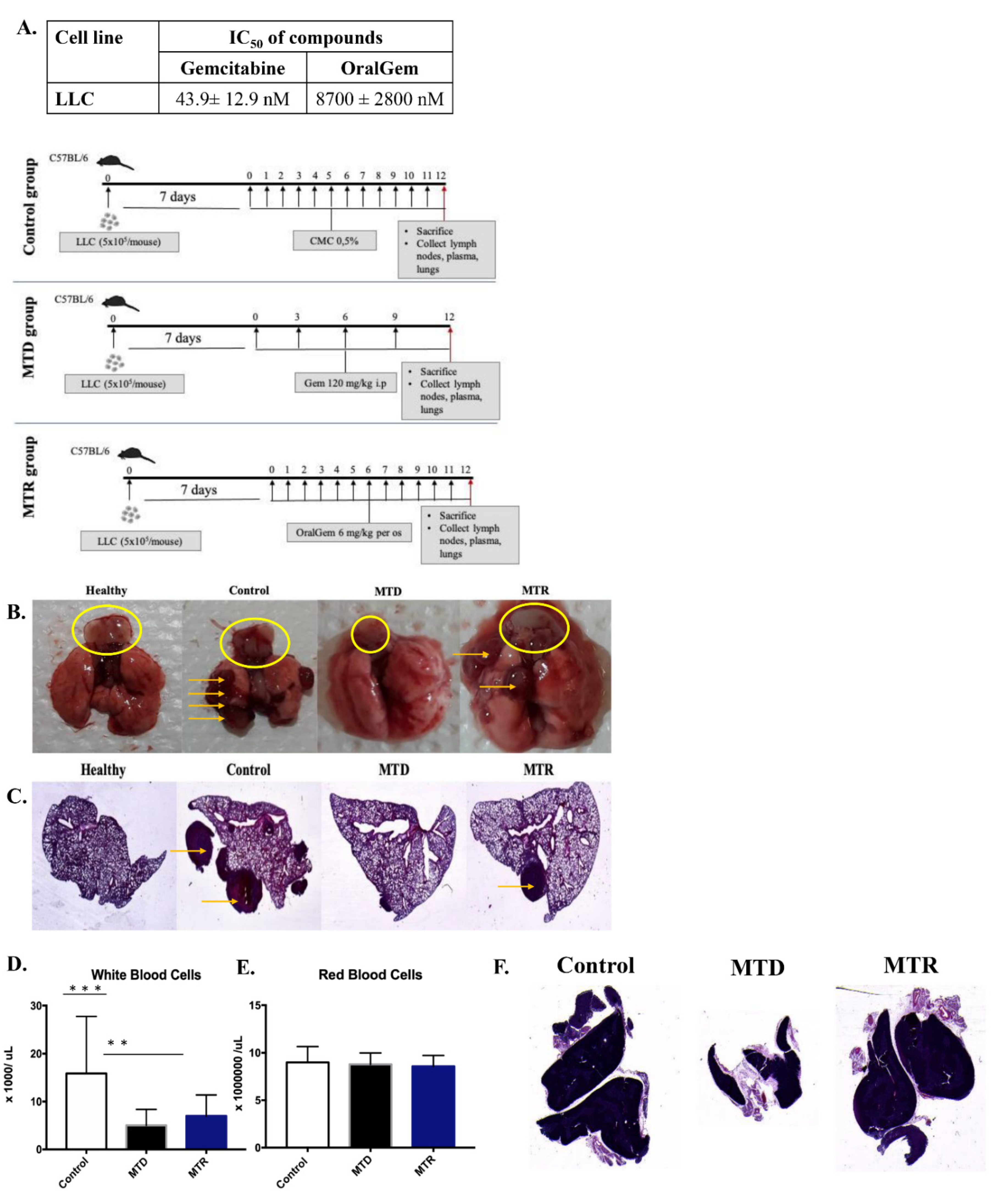
2.3. Enhanced Antitumor Efficacy, Associated with Reduced Angiogenesis upon MTR OralGem Administration
2.4. MTR OralGem-Treated Mice Exhibit Decreased Tumor Metastasis in the Lung
2.5. MTR OralGem Treatment Is Efficacious in a Syngeneic Model of Lung Cancer
2.6. MTR OralGem Chemotherapy Is Characterized by Reduced Toxicity in the Syngeneic Lung Cancer Model
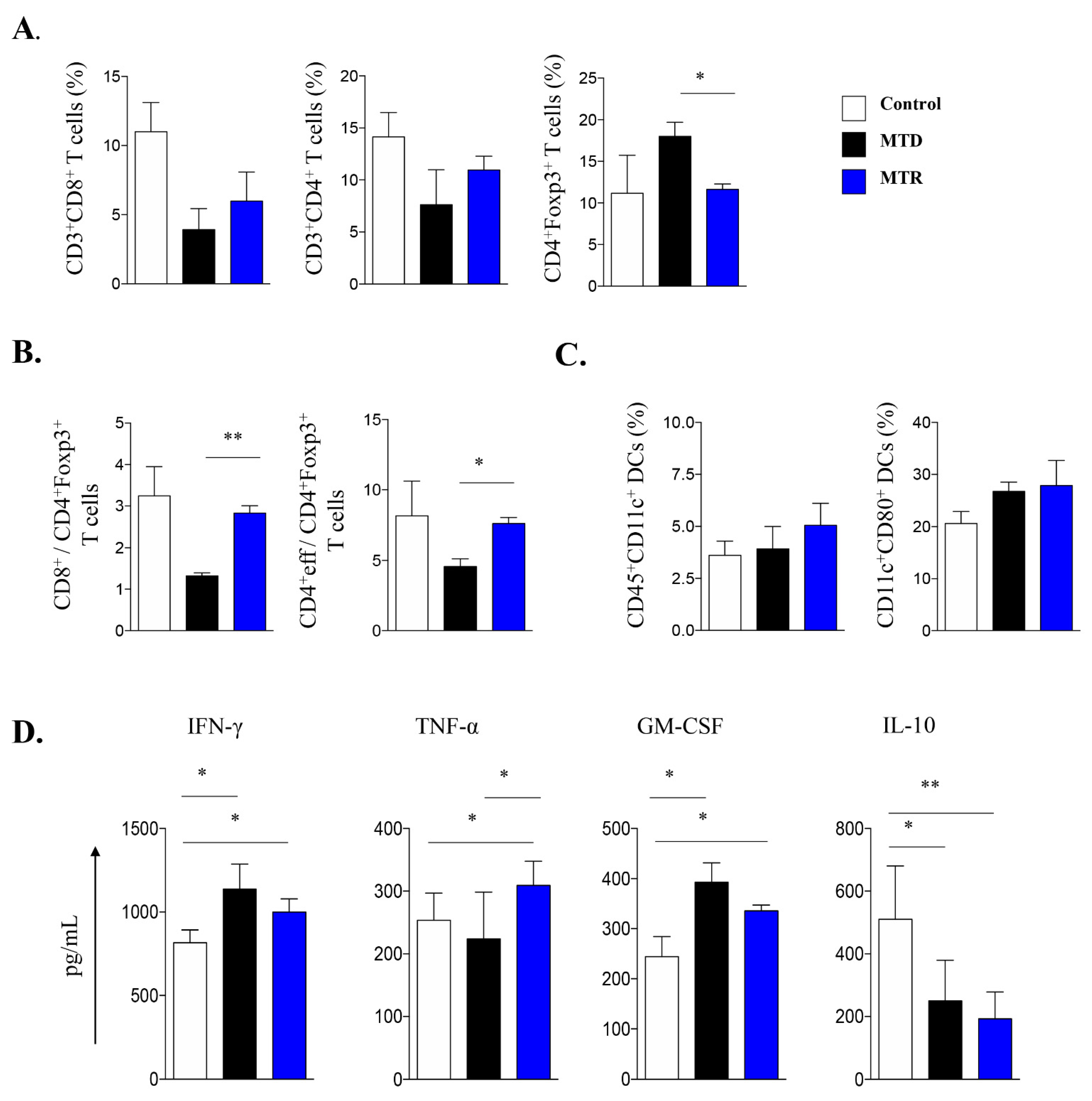
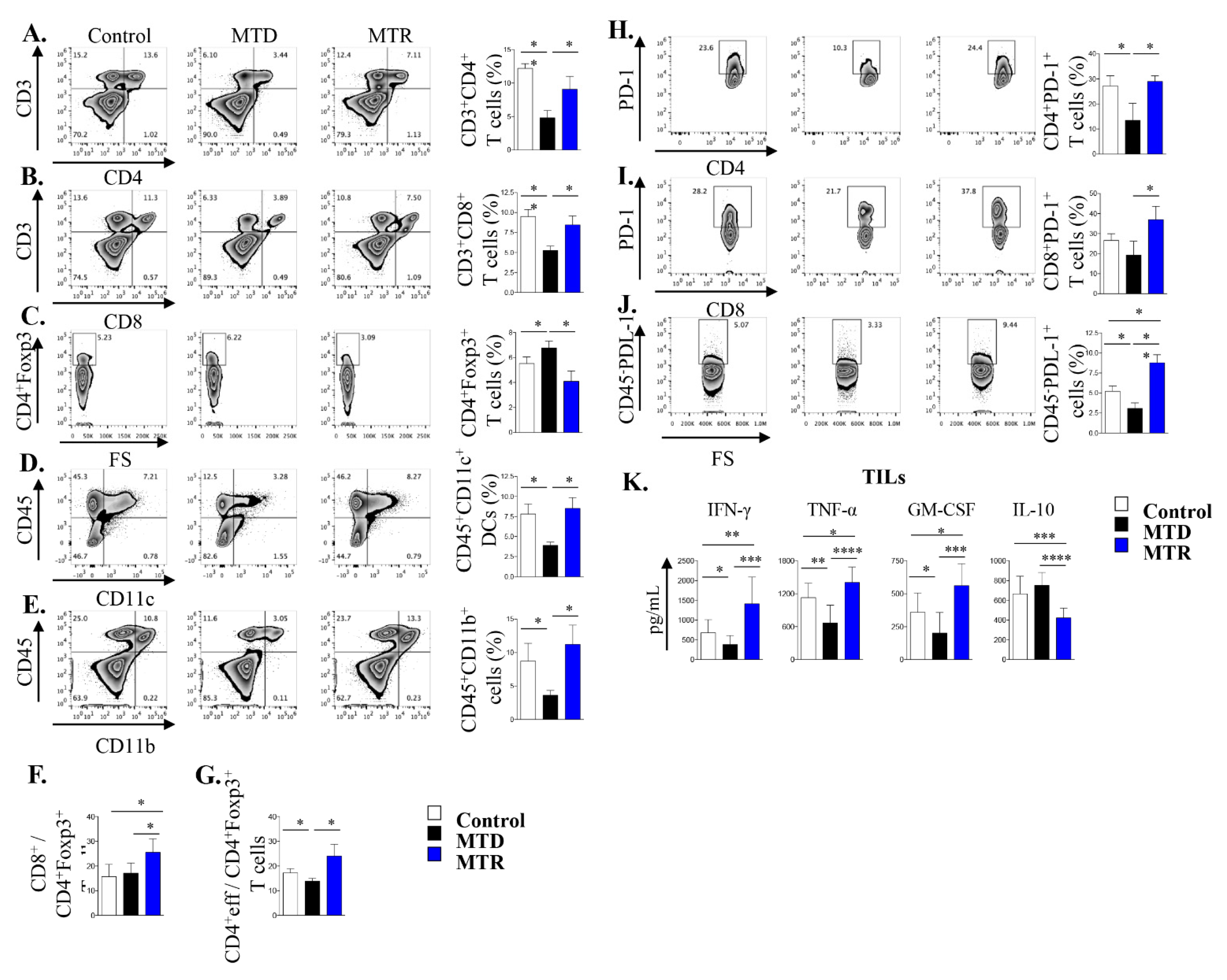
2.7. Treatment with MTR OralGem Chemotherapy Enhances Antitumor Effector Responses and Reduces Infiltration of CD4+Foxp3+ Treg Cells in the Syngeneic Mouse Lung Cancer Model
2.8. Co-Administration of MTR OralGem with Anti-PD1 Antibody Exhibits Increased Efficacy in the Syngeneic Lung Cancer Model
2.9. Increased Immunostimulatory Potential upon MTR OralGem and Anti-PD1 Co-Administration in the Syngeneic Lung Tumor Model
3. Discussion
4. Materials and Methods
4.1. Drugs and Synthesis
4.2. Cell Lines
4.3. Mice
4.4. Cell Growth Inhibition
4.5. Drug Doses and Schedules and Pharmacokinetic Analysis
4.6. In Vivo Toxicity Evaluation
4.7. Tumor Models
4.8. Tissue Immunostaining for Markers of Angiogenesis
4.9. Isolation of TILs
4.10. Flow-Cytometry Analysis
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Consent for Publication
Availability of Data and Materials
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Evans, T.; Gettinger, S.; Hensing, T.A.; Sequist, L.V.; Ireland, B.; Stinchcombe, T.E. Treatment of Stage IV Non-small Cell Lung Cancer. Chest 2013, 143, e341S–e368S. [Google Scholar] [CrossRef] [Green Version]
- Gesto, D.S.; Cerqueira, N.M.F.S.A.; Fernandes, A.P.; Ramos, M.J. Gemcitabine: A Critical Nucleoside for Cancer Therapy. Curr. Med. Chem. 2012, 19, 1076–1087. [Google Scholar] [CrossRef]
- Highlights of Prescribing Information. Available online: https://pi.lilly.com/us/gemzar.pdf (accessed on 1 May 2020).
- Veltkamp, S.A.; Beijnen, J.H.; Schellens, J.H.M. Prolonged Versus Standard Gemcitabine Infusion: Translation of Molecular Pharmacology to New Treatment Strategy. Oncology 2008, 13, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Castellano, D.; Paz-Ares, L.; Gravalos, C.; Diaz-Puente, M.; Hitt, R.; Alonso, S.; Cortes-Funes, H. Phase I-II Study of Gemcitabine and Fluorouracil as a Continuous Infusion in Patients With Pancreatic Cancer. J. Clin. Oncol. 1999, 17, 585. [Google Scholar] [CrossRef]
- Brand, R.; Capadano, M.; Tempero, M. A Phase I trial of weekly gemcitabine administered as a prolonged infusion in patients with pancreatic cancer and other solid tumors. Investig. New Drugs 1997, 15, 331–341. [Google Scholar] [CrossRef]
- Sul, J.; Blumenthal, G.M.; Jiang, X.; He, K.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non–Small Cell Lung Cancer Whose Tumors Express Programmed Death–Ligand 1. Oncology 2016, 21, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Camidge, D.; Liu, S.; Powderly, J.; Hodi, F.; Gettinger, S.; Heist, R.; Liu, B.; Wallin, J.; Funke, R.; et al. 513 Safety, activity and biomarkers of atezolizumab (MPDL3280A) with platinum-based chemotherapy (chemo) in non-small cell lung cancer (NSCLC): A Phase Ib study. Eur. J. Cancer 2015, 51, S107–S108. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in Combination with Platinum–Based Doublet Chemotherapy for First-Line Treatment of Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef]
- Weinberg, F.; Gadgeel, S. Combination pembrolizumab plus chemotherapy: A new standard of care for patients with advanced non-small-cell lung cancer. Lung Cancer Targets Ther. 2019, 10, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay, Ş.F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Francia, G.; Shaked, Y.; Hashimoto, K.; Sun, J.; Yin, M.; Cesta, C.; Xu, P.; Man, S.; Hackl, C.; Stewart, J.; et al. Low-Dose Metronomic Oral Dosing of a Prodrug of Gemcitabine (LY2334737) Causes Antitumor Effects in the Absence of Inhibition of Systemic Vasculogenesis. Mol. Cancer Ther. 2012, 11, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, S.E.; Durland–Busbice, S.; Shepard, R.L.; Donoho, G.P.; Starling, J.J.; Wickremsinhe, E.R.; Perkins, E.J.; Dantzig, A.H. Efficacy of Low-Dose Oral Metronomic Dosing of the Prodrug of Gemcitabine, LY2334737, in Human Tumor Xenografts. Mol. Cancer Ther. 2013, 12, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Bocci, G.; Nicolaou, K.C.; Kerbel, R.S. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002, 62, 6938–6943. [Google Scholar]
- Hamano, Y.; Sugimoto, H.; Soubasakos, M.A.; Kieran, M.; Olsen, B.R.; Lawler, J.; Sudhakar, A.; Kalluri, R. Thrombospondin-1 Associated with Tumor Microenvironment Contributes to Low-Dose Cyclophosphamide-Mediated Endothelial Cell Apoptosis and Tumor Growth Suppression. Cancer Res. 2004, 64, 1570–1574. [Google Scholar] [CrossRef] [Green Version]
- Bocci, G.; Kerbel, R.S. Pharmacokinetics of metronomic chemotherapy: A neglected but crucial aspect. Nat. Rev. Clin. Oncol. 2016, 13, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Francia, G. (Eds.) Metronomic Chemotherapy: Pharmacology and Clinical Applications; Springer: New York, NY, USA, 2014. [Google Scholar]
- Bender, D.M.; Bao, J.; Dantzig, A.H.; Diseroad, W.D.; Law, K.L.; Magnus, N.A.; Peterson, J.A.; Perkins, E.J.; Pu, Y.J.; Reutzel-Edens, S.M.; et al. Synthesis, Crystallization, and Biological Evaluation of an Orally Active Prodrug of Gemcitabine. J. Med. Chem. 2009, 52, 6958–6961. [Google Scholar] [CrossRef]
- Shaked, Y.; Pham, E.; Hariharan, S.; Magidey, K.; Beyar-Katz, O.; Xu, P.; Man, S.; Wu, F.T.; Miller, V.; Andrews, D.; et al. Evidence Implicating Immunological Host Effects in the Efficacy of Metronomic Low-Dose Chemotherapy. Cancer Res. 2016, 76, 5983–5993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnis, E.; Alishekevitz, D.; Gingis-Veltski, S.; Bril, R.; Fremder, E.; Voloshin, T.; Raviv, Z.; Karban, A.; Shaked, Y. Anti-Bv8 Antibody and Metronomic Gemcitabine Improve Pancreatic Adenocarcinoma Treatment Outcome Following Weekly Gemcitabine Therapy. Neoplasia 2014, 16, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumor regression in an implanted glioma model. Cancer Lett. 2014, 353, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Irvin, C.G.; Bates, J.H.T. Measuring the lung function in the mouse: The challenge of size. Respir. Res. 2003, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, D.; Li, Z.; Jiao, D.; Jin, L.; Cong, J.; Zheng, X.; Xu, L. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Deshmukh, S.K.; Tyagi, N.; Khan, M.A.; Srivastava, S.K.; Al-Ghadhban, A.; Dugger, K.; Carter, J.E.; Singh, S.; Singh, A.P. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.B.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Gnoni, A.; Silvestris, N.; Licchetta, A.; Santini, D.; Scartozzi, M.; Ria, R.; Pisconti, S.; Petrelli, F.; Vacca, A.; Lorusso, V. Metronomic chemotherapy from rationale to clinical studies: A dream or reality? Crit. Rev. Oncol. 2015, 95, 46–61. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2006, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions with Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Vyas, D.; Laput, G.; Vyas, A. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nars, M.S.; Kaneno, R. Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int. J. Cancer 2012, 132, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Simsek, C.; Esin, E.; Yalcin, S. Metronomic Chemotherapy: A Systematic Review of the Literature and Clinical Experience. J. Oncol. 2019, 2019, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 1–17. [Google Scholar] [CrossRef]
- Chen, Q.; Xia, R.; Zheng, W.; Zhang, L.; Li, P.; Sun, X.; Shi, J. Metronomic paclitaxel improves the efficacy of PD-1 monoclonal antibodies in breast cancer by transforming the tumor immune microenvironment. Am. J. Transl. Res. 2020, 12, 519–530. [Google Scholar]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Ho, T.T.B.; Nasti, A.; Seki, A.; Komura, T.; Inui, H.; Kozaka, T.; Kitamura, Y.; Shiba, K.; Yamashita, T.; Yamashita, T.; et al. Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J. Immunother. Cancer 2020, 8, e001367. [Google Scholar] [CrossRef]
- Du, B.; Wen, X.; Wang, Y.; Lin, M.; Lai, J. Gemcitabine and checkpoint blockade exhibit synergistic anti-tumor effects in a model of murine lung carcinoma. Int. Immunopharmacol. 2020, 86, 106694. [Google Scholar] [CrossRef]
- Boven, E.; Schipper, H.; Erkelens, C.A.; Hatty, S.A.; Pinedo, H.M. The influence of the schedule and the dose of gemcitabine on the anti-tumour efficacy in experimental human cancer. Br. J. Cancer 1993, 68, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Schrag, D. The Price Tag on Progres—Chemotherapy for Colorectal Cancer. N. Engl. J. Med. 2004, 351, 317–319. [Google Scholar] [CrossRef]
- Bocci, G.; Tuccori, M.; Emmenegger, U.; Liguori, V.; Falcone, A.; Kerbel, R.S.; Del Tacca, M. Cyclophosphamide-methotrexate ‘metronomic’ chemotherapy for the palliative treatment of metastatic breast cancer. A comparative pharmacoeconomic evaluation. Ann. Oncol. 2005, 16, 1243–1252. [Google Scholar] [CrossRef]
- Fujifilm Collaborates with Merck & Co., Inc., Kenilworth, N.J., U.S.A. in a Clinical Trial for Advanced Solid Tumor in Combination Therapy of FF-10832, a Liposome Drug Candidate with KEYTRUDA® (Pembrolizumab). Available online: https://www.fujifilm.com/news/n200526.html (accessed on 1 June 2020).
- Karampelas, T.; Argyros, O.; Sayyad, N.; Spyridaki, K.; Pappas, C.; Morgan, K.; Kolios, G.; Millar, R.P.; Liapakis, G.; Tzakos, A.G.; et al. GnRH-Gemcitabine Conjugates for the Treatment of Androgen-Independent Prostate Cancer: Pharmacokinetic Enhancements Combined with Targeted Drug Delivery. Bioconjug. Chem. 2014, 25, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Depoy, E.; Gitlin, L.N. Statistical Analysis for Experimental-Type Designs. In Introduction to Research; Elsevier: Amsterdam, The Netherlands, 2016; pp. 282–310. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skavatsou, E.; Semitekolou, M.; Morianos, I.; Karampelas, T.; Lougiakis, N.; Xanthou, G.; Tamvakopoulos, C. Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers 2021, 13, 1901. https://doi.org/10.3390/cancers13081901
Skavatsou E, Semitekolou M, Morianos I, Karampelas T, Lougiakis N, Xanthou G, Tamvakopoulos C. Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers. 2021; 13(8):1901. https://doi.org/10.3390/cancers13081901
Chicago/Turabian StyleSkavatsou, Eleni, Maria Semitekolou, Ioannis Morianos, Theodoros Karampelas, Nikolaos Lougiakis, Georgina Xanthou, and Constantin Tamvakopoulos. 2021. "Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC" Cancers 13, no. 8: 1901. https://doi.org/10.3390/cancers13081901
APA StyleSkavatsou, E., Semitekolou, M., Morianos, I., Karampelas, T., Lougiakis, N., Xanthou, G., & Tamvakopoulos, C. (2021). Immunotherapy Combined with Metronomic Dosing: An Effective Approach for the Treatment of NSCLC. Cancers, 13(8), 1901. https://doi.org/10.3390/cancers13081901