
Epigenetic Drift Association with Cancer Risk and Survival, and Modification by Sex

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Characteristics of Study Samples
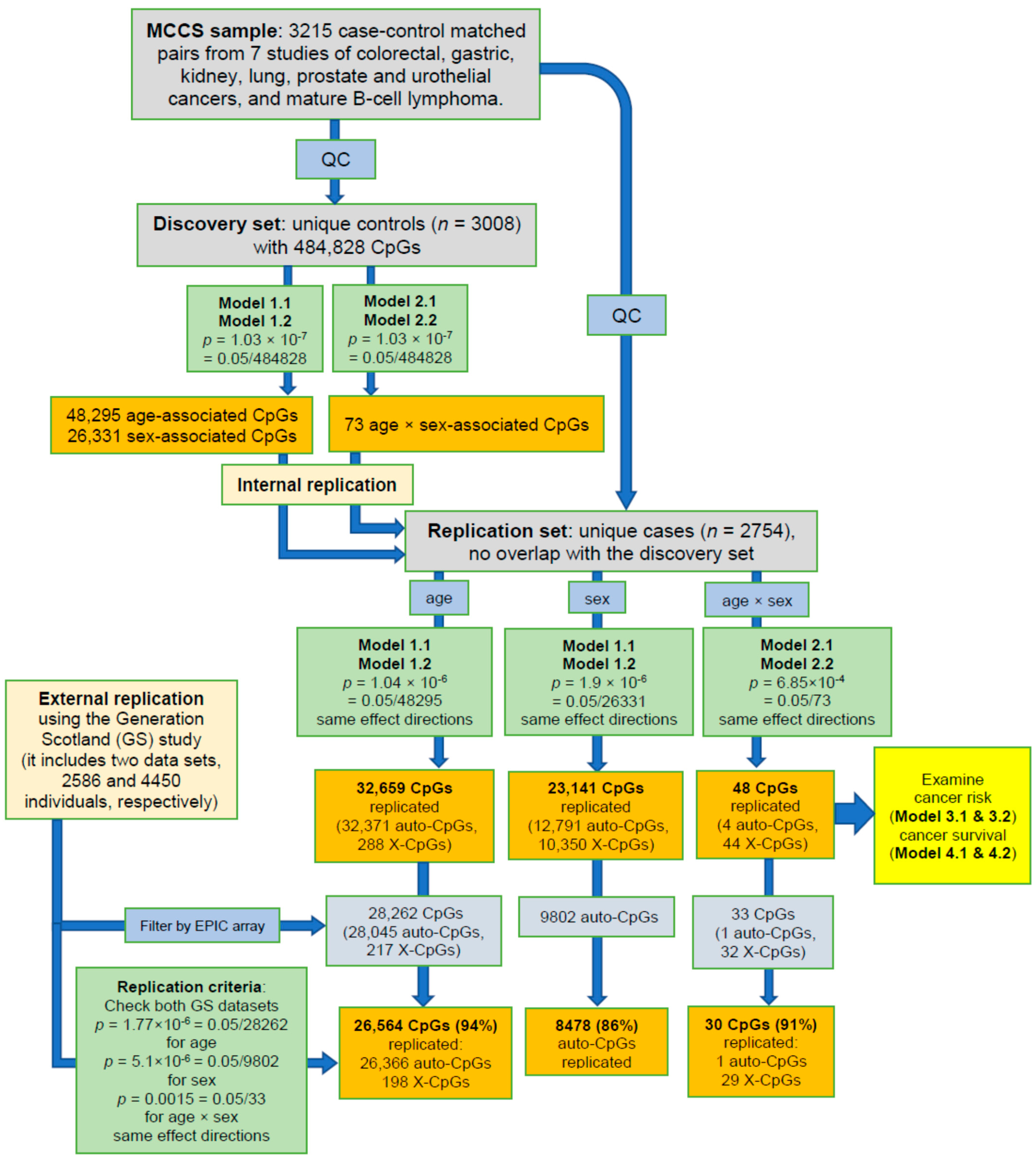
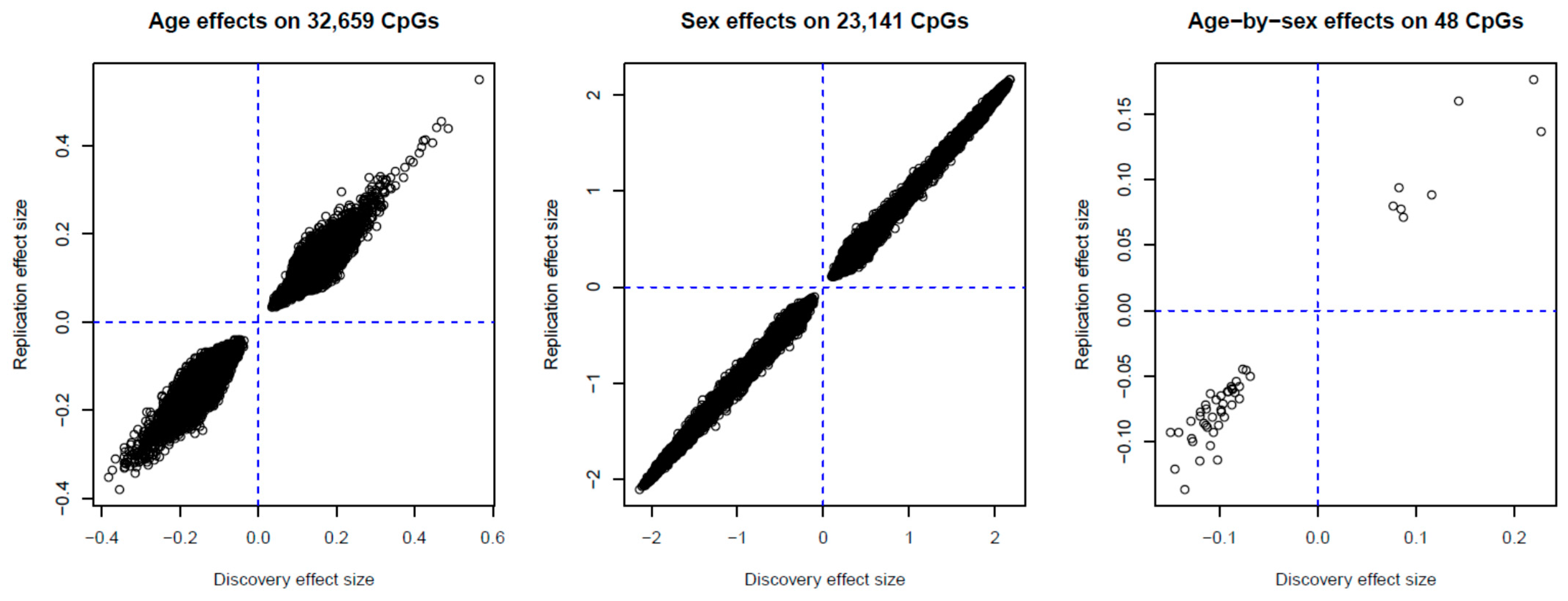
2.2. Identification of Age-, Sex- and Age-by-Sex-Associated Methylation CpGs
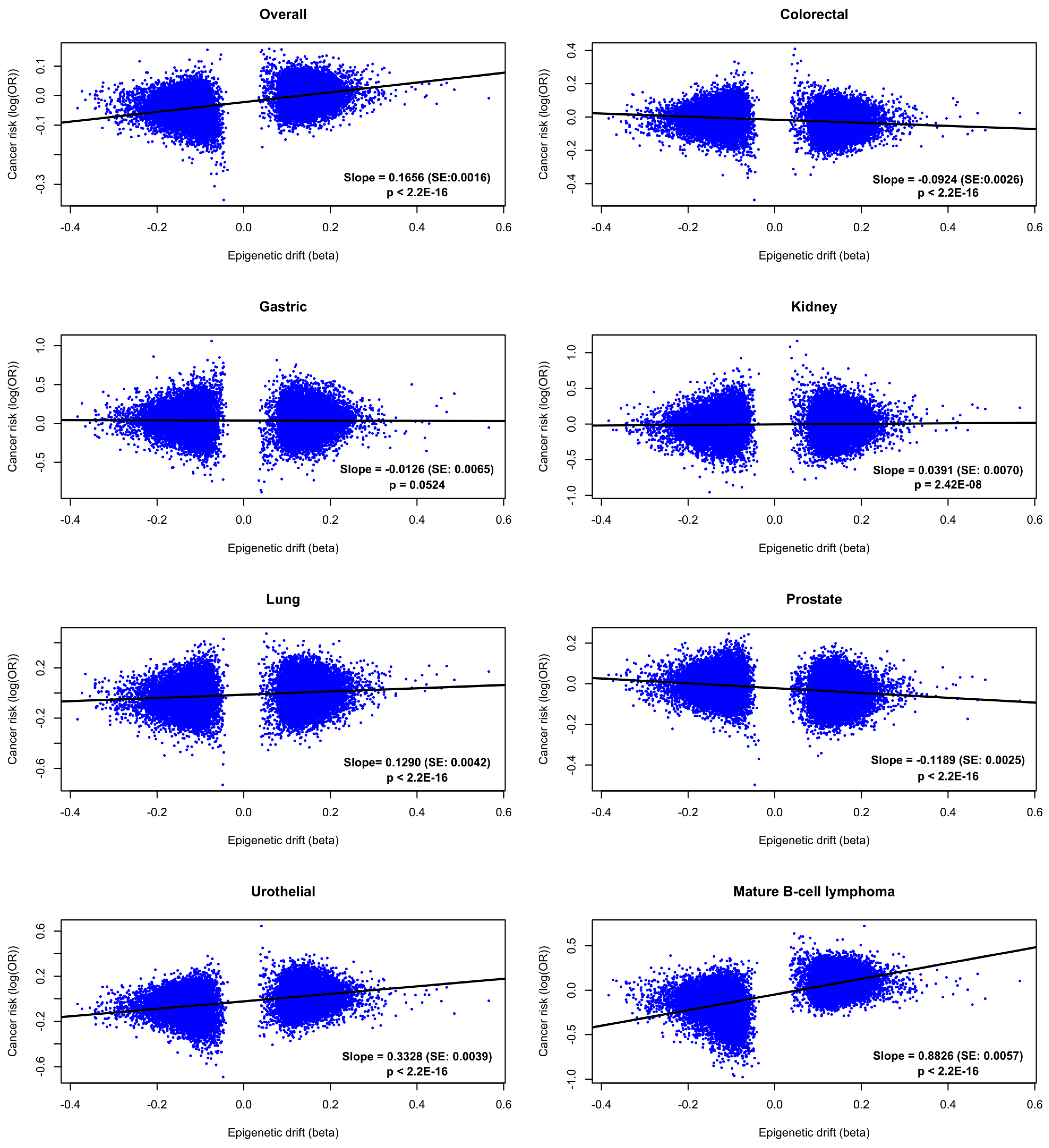
2.3. Cancer Risk
2.4. Cancer Survival
3. Discussion
4. Materials and Methods
4.1. Study Sample
4.2. Quality Control of DNA Methylation Data
4.3. Statistical Analyses
4.3.1. Discovery and Replication Sets
4.3.2. Age, Sex, and Age-By-Sex Associations
4.3.3. Cancer Risk and Survival Associations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- US Cancer Statistics Working Group. United States Cancer Statistics: 1999–2010 Incidence and Mortality Web-Based Report; US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute: Atlanta, GA, USA, 2013; p. 201. [Google Scholar]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef]
- Aunan, J.R.; Cho, W.C.; Søreide, K. The biology of aging and cancer: A brief overview of shared and divergent molecular hallmarks. Aging Dis. 2017, 8, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.B.; Dawsey, S.M.; Freedman, N.D.; Inskip, P.D.; Wichner, S.M.; Quraishi, S.M.; Devesa, S.S.; McGlynn, K.A. Sex disparities in cancer incidence by period and age. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1174–1182. [Google Scholar] [CrossRef]
- Cook, M.B.; McGlynn, K.A.; Devesa, S.S.; Freedman, N.D.; Anderson, W.F. Sex disparities in cancer mortality and survival. Cancer Epidemi. Biomark. Prev. 2011, 20, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Edgren, G.; Liang, L.; Adami, H.O.; Chang, E.T. Enigmatic sex disparities in cancer incidence. Eur. J. Epidemiol. 2012, 27, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shao, X.; Wang, X.; Liu, L.; Liang, H. Sex disparities in cancer. Cancer Lett. 2019, 466, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ramos, C.M.; Quackenbush, J.; DeMeo, D.L. Genome-Wide Sex and Gender Differences in Cancer. Front. Oncol. 2020, 10, 2486. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; et al. Sex differences in cancer mechanisms. Biol. Sex Differ. 2020, 11, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Majek, O.; Gondos, A.; Jansen, L.; Emrich, K.; Holleczek, B.; Katalinic, A.; Nennecke, A.; Eberle, A.; Brenner, H.; GEKID Cancer Survival Working Group. Sex differences in colorectal cancer survival: Population-based analysis of 164,996 colorectal cancer patients in Germany. PLoS ONE 2013, 8, e68077. [Google Scholar] [CrossRef]
- Song, M.; Kang, D.; Yang, J.J.; Choi, J.Y.; Sung, H.; Lee, Y.; Yoon, H.S.; Choi, Y.; Kong, S.H.; Lee, H.J.; et al. Age and sex interactions in gastric cancer incidence and mortality trends in Korea. Gastric Cancer 2015, 18, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Chen, H.; Gu, W.; Gu, C.; Zhang, H.; Xu, J.; Zhu, Y.; Ye, D. Age-dependent association between sex and renal cell carcinoma mortality: A population-based analysis. Sci. Rep. 2015, 5, 9160. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wen, W.; Morgans, A.K.; Pao, W.; Shu, X.O.; Zheng, W. Disparities by race, age, and sex in the improvement of survival for major cancers: Results from the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program in the United States, 1990 to 2010. JAMA Oncol. 2015, 1, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Mamtani, R.; Wang, X.V.; Gyawali, B.; DiPaola, R.S.; Epperson, C.N.; Haas, N.B.; Dutcher, J.P. Association between age and sex and mortality after adjuvant therapy for renal cancer. Cancer 2019, 125, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Banumathy, G.; Adams, P.D. Aging by epigenetics—A consequence of chromatin damage? Exp. Cell Res. 2008, 314, 1909–1917. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- Liu, J.; Morgan, M.; Hutchison, K.; Calhoun, V.D. A study of the influence of sex on genome wide methylation. PLoS ONE 2010, 5, e10028. [Google Scholar] [CrossRef]
- Khramtsova, E.A.; Davis, L.K.; Stranger, B.E. The role of sex in the genomics of human complex traits. Nat. Rev. Genet. 2019, 20, 173–190. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Zheng, S.C.; Widschwendter, M.; Teschendorff, A.E. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 2016, 8, 705–719. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371. [Google Scholar] [CrossRef] [PubMed]
- Dugue, P.A.; Bassett, J.K.; Joo, J.E.; Baglietto, L.; Jung, C.H.; Wong, E.M.; Fiorito, G.; Schmidt, D.; Makalic, E.; Li, S.; et al. Association of DNA methylation-based biological age with health risk factors and overall and cause-specific mortality. Am. J. Epidemiol. 2018, 187, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Dugué, P.A.; Li, S.; Hopper, J.L.; Milne, R.L. Chapter 3—DNA Methylation-Based Measures of Biological Aging. In Epigenetics in Human Disease, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: London, UK, 2018; Volume 6, pp. 39–64. [Google Scholar]
- Dugué, P.A.; Bassett, J.K.; Joo, J.E.; Jung, C.H.; Ming Wong, E.; Moreno-Betancur, M.; Schmidt, D.; Makalic, E.; Li, S.; Severi, G.; et al. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int. J. Cancer 2018, 142, 1611–1619. [Google Scholar] [CrossRef]
- Dugué, P.A.; Bassett, J.K.; Wong, E.M.; Joo, J.E.; Li, S.; Yu, C.; Schmidt, D.F.; Makalic, E.; Doo, N.W.; Buchanan, D.D.; et al. Biological aging measures based on blood DNA methylation and risk of cancer: A prospective study. JNCI Cancer Spectr. 2021, 5, pkaa109. [Google Scholar] [CrossRef]
- Chung, M.; Ruan, M.; Zhao, N.; Koestler, D.C.; De Vivo, I.; Kelsey, K.T.; Michaud, D.S. DNA methylation ageing clocks and pancreatic cancer risk: Pooled analysis of three prospective nested case-control studies. Epigenetics 2021, 1–11. [Google Scholar] [CrossRef]
- Ambatipudi, S.; Horvath, S.; Perrier, F.; Cuenin, C.; Hernandez-Vargas, H.; Le Calvez-Kelm, F.; Durand, G.; Byrnes, G.; Ferrari, P.; Bouaoun, L.; et al. DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur. J. Cancer 2017, 75, 299–307. [Google Scholar] [CrossRef]
- Gào, X.; Zhang, Y.; Boakye, D.; Li, X.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Whole blood DNA methylation aging markers predict colorectal cancer survival: A prospective cohort study. Clin. Epigenet. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- McCartney, D.L.; Zhang, F.; Hillary, R.F.; Zhang, Q.; Stevenson, A.J.; Walker, R.M.; Bermingham, M.L.; Boutin, T.; Morris, S.W.; Campbell, A.; et al. An epigenome-wide association study of sex-specific chronological ageing. Genome Med. 2020, 12, 1–11. [Google Scholar] [CrossRef]
- Yusipov, I.; Bacalini, M.G.; Kalyakulina, A.; Krivonosov, M.; Pirazzini, C.; Gensous, N.; Ravaioli, F.; Milazzo, M.; Giuliani, C.; Vedunova, M.; et al. Age-related DNA methylation changes are sex-specific: A comprehensive assessment. Aging 2020, 12, 24057. [Google Scholar] [CrossRef]
- Li, S.; Lund, J.B.; Christensen, K.; Baumbach, J.; Mengel-From, J.; Kruse, T.; Li, W.; Mohammadnejad, A.; Pattie, A.; Marioni, R.E.; et al. Exploratory analysis of age and sex dependent DNA methylation patterns on the X-chromosome in whole blood samples. Genome Med. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.E.; Novakovic, B.; Cruickshank, M.; Doyle, L.W.; Craig, J.M.; Saffery, R. Human active X-specific DNA methylation events showing stability across time and tissues. Eur. J. Hum. Genet. 2014, 22, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Castillo-Morales, A.; Jiang, M.; Zhu, Y.; Hu, L.; Urrutia, A.O.; Kong, X.; Hurst, L.D. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol. Biol. Evol. 2013, 30, 2588–2601. [Google Scholar] [CrossRef] [PubMed]
- Khongsti, S.; Lamare, F.A.; Shunyu, N.B.; Ghosh, S.; Maitra, A.; Ghosh, S. Whole genome DNA methylation profiling of oral cancer in ethnic population of Meghalaya, North East India reveals novel genes. Genomics 2018, 110, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Wang, M.C.; Zhang, F.H.; Kong, X. An integrated analysis of genome-wide DNA methylation and gene expression data in hepatocellular carcinoma. FEBS Open Bio 2018, 8, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Yao, L.; Xu, H.W.; Tang, W.T.; Fu, J.H.; Hu, X.F.; Cui, L.; Xu, X.M. Identification of MXRA5 as a novel biomarker in colorectal cancer. Oncol. Lett. 2013, 5, 544–548. [Google Scholar] [CrossRef]
- Xiong, D.; Li, G.; Li, K.; Xu, Q.; Pan, Z.; Ding, F.; Vedell, P.; Liu, P.; Cui, P.; Hua, X.; et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis 2012, 33, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Sauermann, M.; Sahin, Ö.; Sültmann, H.; Hahne, F.; Blaszkiewicz, S.; Majety, M.; Zatloukal, K.; Füzesi, L.; Poustka, A.; Wiemann, S.; et al. Reduced expression of vacuole membrane protein 1 affects the invasion capacity of tumor cells. Oncogene 2008, 27, 1320–1326. [Google Scholar] [CrossRef]
- Qian, Q.; Zhou, H.; Chen, Y.; Shen, C.; He, S.; Zhao, H.; Wang, L.; Wan, D.; Gu, W. VMP1 related autophagy and apoptosis in colorectal cancer cells: VMP1 regulates cell death. Biochem. Biophys. Res. Commun. 2014, 443, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Chen, L.; Zhang, X.; Zhan, J.; Chen, J. TMEM49-related apoptosis and metastasis in ovarian cancer and regulated cell death. Mol. Cell. Biochem. 2016, 416, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Greenland, S. Quantifying biases in causal models: Classical confounding vs collider-stratification bias. Epidemiology 2003, 14, 300–306. [Google Scholar] [CrossRef]
- Pearce, N.; Richiardi, L. Commentary: Three worlds collide: Berkson’s bias, selection bias and collider bias. Int. J. Epidemiol. 2014, 43, 521–524. [Google Scholar] [CrossRef]
- Milne, R.L.; Fletcher, A.S.; MacInnis, R.J.; Hodge, A.M.; Hopkins, A.H.; Bassett, J.K.; Bruinsma, F.J.; Lynch, B.M.; Dugué, P.A.; Jayasekara, H.; et al. Cohort profile: The Melbourne collaborative cohort study (health 2020). Int. J. Epidemiol. 2017, 46, 1757–1757i. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- McCartney, D.; Zhang, F.; Hillary, R.; Zhang, Q.; Stevenson, A.; Walker, R.; Bermingham, M.; Boutin, T.; Morris, S.; Campbell, A.; et al. Generation Scotland Age x Sex Epigenome Wide Association Study Summary Statistics; University of Edinburgh: Edinburgh, UK, 2019. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Dugue, P.A.; Jung, C.H.; Joo, J.E.; Wang, X.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; Severi, G.; Southey, M.C.; et al. Smoking and blood DNA methylation: An epigenome-wide association study and assessment of reversibility. Epigenetics Off. J. DNA Methylation Soc. 2020, 15, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Dugue, P.A.; Wilson, R.; Lehne, B.; Jayasekara, H.; Wang, X.; Jung, C.H.; Joo, J.E.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addict. Biol. 2021, 26, e12855. [Google Scholar] [CrossRef]
- Geurts, Y.M.; Dugue, P.A.; Joo, J.E.; Makalic, E.; Jung, C.H.; Guan, W.; Nguyen, S.; Grove, M.L.; Wong, E.M.; Hodge, A.M.; et al. Novel associations between blood DNA methylation and body mass index in middle-aged and older adults. Int. J. Obes. 2018, 42, 887–896. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | MCCS Sample | |
|---|---|---|
| Cancer Cases (n = 2754) | Controls (n = 3008) | |
| Age at blood draw, median (IQR) | 61.1 | 61.1 |
| (54.2–66.0) | (54.3–65.8) | |
| Sex: | ||
| male, n (%) | 1846 (67%) | 2067 (68.7%) |
| female, n (%) | 908 (33%) | 941 (31.3%) |
| Country of birth: | ||
| Australia/New Zealand, N (%) | 1847 (67.1%) | 2011 (66.9%) |
| Greece, n (%) | 291 (10.6%) | 322 (10.7%) |
| Italy, n (%) | 430 (15.6%) | 478 (15.9%) |
| UK/other, n (%) | 186 (6.8%) | 197 (6.5%) |
| Blood sample type: | ||
| dried blood spots, n (%) | 1880 (68.3%) | 2053 (68.3%) |
| peripheral blood mononuclear cells, N (%) | 706 (25.6%) | 767 (25.5%) |
| buffy coats, n (%) | 168 (6.1%) | 188 (6.3%) |
| Smoking status: | ||
| current, n (%) | 417 (15.1%) | 429 (14.3%) |
| former, n (%) | 1128 (41%) | 1198 (39.8%) |
| never, n (%) | 1209 (43.9%) | 1381 (45.9%) |
| Smoking pack-years, median (IQR) | 4 | 2.4 |
| (0–30.1) | (0–27) | |
| Body mass index (kg/m2), median (IQR) | 26.9 | 26.8 |
| (24.5–29.7) | (24.5–29.4) | |
| Alcohol consumption (g/day), median (IQR) | 5.2 | 4.3 |
| (0–19) | (0–19) | |
| CpG | CHR | MAPINFO | Strand | Gene | Gene Feature | Discovery Set in MCCS | Replication Set in MCCS | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | SE | p-Value | Effect | SE | p-Value | ||||||
| cg12054453 | 17 | 57915717 | F | TMEM49 | Body | 0.22 | 0.03 | 1.81 × 10−12 | 0.18 | 0.03 | 5.56 × 10−8 |
| cg06758848 | X | 153603268 | F | FLNA | TSS1500 | 0.14 | 0.02 | 9.77 × 10−15 | 0.16 | 0.02 | 3.41 × 10−16 |
| cg13775533 | X | 99663073 | F | PCDH19 | 1stExon | −0.14 | 0.02 | 2.55 × 10−14 | −0.09 | 0.02 | 8.69 × 10−7 |
| cg09761247 | X | 148585951 | R | IDS | Body | 0.23 | 0.03 | 6.57 × 10−13 | 0.14 | 0.03 | 2.96 × 10−5 |
| cg08783090 | X | 151143125 | R | GABRE | 1stExon | −0.12 | 0.02 | 7.85 × 10−13 | −0.08 | 0.02 | 3.27 × 10−6 |
| cg25888700 | X | 21676344 | R | KLHL34 | 5′UTR | −0.11 | 0.02 | 2.96 × 10−12 | −0.08 | 0.02 | 3.68 × 10−7 |
| cg01828474 | X | 21676593 | R | KLHL34 | TSS200 | −0.11 | 0.02 | 8.17 × 10−12 | −0.09 | 0.02 | 1.45 × 10−7 |
| cg17036062 | X | 25034037 | F | ARX | 1stExon | −0.09 | 0.01 | 9.43 × 10−12 | −0.06 | 0.01 | 1.20 × 10−5 |
| cg25988710 | X | 72667379 | R | CDX4 | 1stExon | −0.11 | 0.02 | 2.47 × 10−11 | −0.07 | 0.02 | 3.18 × 10−5 |
| cg06775759 | X | 21676994 | R | KLHL34 | TSS1500 | −0.10 | 0.02 | 4.24 × 10−11 | −0.09 | 0.02 | 2.92 × 10−8 |
| cg03671371 | X | 36975540 | R | - | - | −0.11 | 0.02 | 4.72 × 10−11 | −0.09 | 0.02 | 1.34 × 10−6 |
| cg08814148 | X | 118407645 | F | - | - | −0.14 | 0.02 | 1.86 × 10−10 | −0.14 | 0.02 | 1.57 × 10−10 |
| cg16108684 | X | 125300035 | F | DCAF12L2 | TSS200 | −0.12 | 0.02 | 3.66 × 10−10 | −0.08 | 0.02 | 4.02 × 10−5 |
| cg24823082 | X | 38660587 | F | MID1IP1 | TSS200 | −0.15 | 0.02 | 4.31 × 10−10 | −0.09 | 0.03 | 2.79 × 10−4 |
| cg25156485 | X | 64887827 | R | MSN | Body | 0.08 | 0.01 | 5.02 × 10−10 | 0.08 | 0.01 | 1.97 × 10−10 |
| cg04424215 | X | 111325143 | R | TRPC5 | 5′UTR | −0.13 | 0.02 | 6.53 × 10−10 | −0.08 | 0.02 | 1.18 × 10−4 |
| cg25528646 | X | 151143302 | R | GABRE | TSS200 | −0.09 | 0.01 | 8.18 × 10−10 | −0.06 | 0.02 | 4.62 × 10−5 |
| cg24931094 | X | 119737891 | R | MCTS1 | 5′UTR | 0.09 | 0.01 | 1.31 × 10−9 | 0.07 | 0.01 | 1.12 × 10−6 |
| cg12537796 | X | 106515818 | R | - | - | −0.12 | 0.02 | 1.86 × 10−9 | −0.09 | 0.02 | 8.22 × 10−6 |
| cg25127732 | X | 21676692 | F | KLHL34 | TSS1500 | −0.08 | 0.01 | 2.71 × 10−9 | −0.05 | 0.01 | 3.56 × 10−4 |
| cg03202526 | X | 139587311 | F | SOX3 | TSS200 | −0.07 | 0.01 | 3.62 × 10−9 | −0.05 | 0.01 | 1.52 × 10−5 |
| cg11194545 | X | 105066793 | F | NRK | 5′UTR | −0.10 | 0.02 | 4.06 × 10−9 | −0.08 | 0.02 | 9.14 × 10−7 |
| cg06779802 | X | 107979401 | F | IRS4 | 1stExon | −0.08 | 0.01 | 7.73 × 10−9 | −0.07 | 0.01 | 1.30 × 10−6 |
| cg22606540 | X | 100914483 | R | ARMCX2 | 5′UTR | −0.10 | 0.02 | 1.23 × 10−8 | −0.07 | 0.02 | 3.86 × 10−5 |
| cg21729122 | X | 25034488 | F | ARX | TSS1500 | −0.10 | 0.02 | 1.34 × 10−8 | −0.08 | 0.02 | 1.55 × 10−5 |
| cg02295369 | X | 117959263 | R | ZCCHC12 | Body | −0.09 | 0.02 | 3.41 × 10−8 | −0.06 | 0.02 | 2.28 × 10−4 |
| cg23208903 | X | 30327487 | F | NR0B1 | 5′UTR | −0.09 | 0.02 | 3.82 × 10−8 | −0.06 | 0.02 | 2.10 × 10−4 |
| cg14586560 | X | 133118453 | R | GPC3 | Body | −0.08 | 0.01 | 4.43 × 10−8 | −0.06 | 0.01 | 8.19 × 10−5 |
| cg21040569 | X | 137793665 | F | FGF13 | Body | −0.08 | 0.02 | 5.24 × 10−8 | −0.05 | 0.02 | 5.55 × 10−4 |
| cg09407917 | X | 130192151 | R | FLJ30058 | TSS200 | −0.09 | 0.02 | 6.52 × 10−8 | −0.07 | 0.02 | 2.52 × 10−5 |
| CpG | CHR | MAPINFO | Strand | Gene | Gene Feature | Model 3.1 | Model 3.2 | Specific Cancer | ||
|---|---|---|---|---|---|---|---|---|---|---|
| OR (95%CI) | p-Value | OR (95%CI) | p-Value | |||||||
| cg25119261 | 6 | 33081351 | R | HLA-DPB2 | Body | 0.80 (0.73–0.87) | 1.63 × 10−7 | 0.80 (0.73–0.87) | 1.52 × 10−7 | Overall |
| cg05497216 | 16 | 89408076 | R | ANKRD11 | 5′UTR | 0.83 (0.78–0.89) | 3.04 × 10−7 | 0.83 (0.78–0.89) | 4.94 × 10−7 | |
| cg05772125 | 16 | 31539169 | R | AHSP | TSS200 | 0.40 (0.29–0.57) | 2.20 × 10−7 | 0.39 (0.27–0.55) | 1.59 × 10−7 | Mature B-cell lymphoma |
| cg04771285 | 12 | 117557630 | R | - | - | 0.49 (0.37–0.64) | 3.28 × 10−7 | 0.49 (0.37–0.65) | 6.49 × 10−7 | |
| cg09046979 | 16 | 28333134 | R | SBK1 | 3′UTR | 0.61 (0.50–0.74) | 7.92 × 10−7 | 0.60 (0.49–0.73) | 4.80 × 10−7 | |
| cg11876705 | 19 | 38918253 | R | RASGRP4 | TSS1500 | 0.44 (0.32–0.61) | 9.00 × 10−7 | 0.45 (0.32–0.62) | 1.44 × 10−6 | |
| cg06774893 | 16 | 68011109 | R | DPEP3 | Body | 0.46 (0.34–0.63) | 1.26 × 10−6 | 0.43 (0.31–0.59) | 2.55 × 10−7 | |
| cg22361106 | 1 | 33909498 | R | - | - | 0.46 (0.34–0.63) | 1.31 × 10−6 | 0.44 (0.32–0.61) | 7.69 × 10−7 | |
| CpG | CHR | MAPINFO | Strand | Gene | Gene Feature | Model 4.1 | Model 4.2 | ||
|---|---|---|---|---|---|---|---|---|---|
| HR (95%CI) | p Value | HR (95%CI) | p Value | ||||||
| cg26427498 | 7 | 105987258 | R | - | - | 0.78 (0.74–0.82) | 8.35 × 10−19 | 0.80 (0.76–0.85) | 1.21 × 10−14 |
| cg26470501 | 19 | 45252955 | F | BCL3 | Body | 0.80 (0.76–0.84) | 6.87 × 10−18 | 0.84 (0.80–0.89) | 6.68 × 10−11 |
| cg25143652 | 20 | 62168670 | R | PTK6 | 1stExon | 0.76 (0.72–0.81) | 1.56 × 10−16 | 0.81 (0.76–0.87) | 3.20 × 10−10 |
| cg01127300 | 22 | 38614796 | F | - | - | 0.82 (0.78–0.86) | 5.19 × 10−16 | 0.87 (0.83–0.91) | 1.07 × 10−8 |
| cg08857221 | 1 | 37941361 | R | ZC3H12A | Body | 0.76 (0.71–0.81) | 1.34 × 10−15 | 0.78 (0.73–0.84) | 4.82 × 10−11 |
| cg19572487 | 17 | 38476024 | R | RARA | 5′UTR | 0.81 (0.77–0.86) | 4.51 × 10−14 | 0.87 (0.82–0.92) | 4.85 × 10−7 |
| cg02773019 | 3 | 135684688 | F | PPP2R3A | 5′UTR | 1.21 (1.15–1.27) | 6.22 × 10−14 | 1.20 (1.14–1.26) | 7.34 × 10−13 |
| cg15114651 | 19 | 47289410 | F | SLC1A5 | TSS1500 | 0.77 (0.72–0.83) | 7.16 × 10−14 | 0.83 (0.78–0.89) | 2.30 × 10−7 |
| cg07069636 | 16 | 30671749 | F | - | - | 0.79 (0.74–0.84) | 1.04 × 10−13 | 0.85 (0.79–0.90) | 2.95 × 10−7 |
| cg15962267 | 5 | 138612986 | F | SNHG4 | Body | 0.77 (0.72–0.83) | 1.54 × 10−13 | 0.81 (0.75–0.87) | 3.70 × 10−8 |
| cg02584867 | 19 | 1861099 | R | KLF16 | Body | 0.83 (0.79–0.87) | 1.56 × 10−13 | 0.85 (0.81–0.89) | 5.84 × 10−11 |
| cg09287933 | 6 | 33384473 | R | CUTA | Body | 0.75 (0.70–0.81) | 1.56 × 10−13 | 0.77 (0.72–0.84) | 8.99 × 10−11 |
| cg03519879 | 14 | 74227499 | R | C14orf43 | 5′UTR | 0.77 (0.72–0.83) | 2.46 × 10−13 | 0.82 (0.77–0.88) | 3.72 × 10−8 |
| cg12170787 | 19 | 1130965 | R | SBNO2 | Body | 0.76 (0.71–0.82) | 5.32 × 10−13 | 0.80 (0.74–0.86) | 5.26 × 10−9 |
| cg07148697 | 6 | 31323253 | R | HLA-B | Body | 0.76 (0.71–0.82) | 5.67 × 10−13 | 0.77 (0.72–0.84) | 3.32 × 10−11 |
| cg26283141 | 6 | 33240471 | F | VPS52 | TSS1500 | 0.82 (0.77–0.86) | 9.04 × 10−13 | 0.84 (0.79–0.89) | 9.42 × 10−10 |
| cg25264101 | 19 | 14064374 | F | PODNL1 | TSS200 | 0.84 (0.80–0.88) | 1.29 × 10−12 | 0.86 (0.82–0.90) | 3.27 × 10−10 |
| cg05352838 | 6 | 33384391 | R | CUTA | 3′UTR | 0.73 (0.67–0.80) | 2.44 × 10−12 | 0.76 (0.69–0.83) | 1.48 × 10−9 |
| cg19513004 | 6 | 34206683 | F | HMGA1 | 5′UTR | 0.80 (0.75–0.85) | 2.70 × 10−12 | 0.83 (0.78–0.88) | 2.68 × 10−9 |
| cg02003183 | 14 | 103415882 | F | CDC42BPB | Body | 1.21 (1.14–1.27) | 3.43 × 10−12 | 1.17 (1.11–1.23) | 8.61 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Wong, E.M.; Joo, J.E.; Hodge, A.M.; Makalic, E.; Schmidt, D.; Buchanan, D.D.; Severi, G.; Hopper, J.L.; English, D.R.; et al. Epigenetic Drift Association with Cancer Risk and Survival, and Modification by Sex. Cancers 2021, 13, 1881. https://doi.org/10.3390/cancers13081881
Yu C, Wong EM, Joo JE, Hodge AM, Makalic E, Schmidt D, Buchanan DD, Severi G, Hopper JL, English DR, et al. Epigenetic Drift Association with Cancer Risk and Survival, and Modification by Sex. Cancers. 2021; 13(8):1881. https://doi.org/10.3390/cancers13081881
Chicago/Turabian StyleYu, Chenglong, Ee Ming Wong, Jihoon Eric Joo, Allison M. Hodge, Enes Makalic, Daniel Schmidt, Daniel D. Buchanan, Gianluca Severi, John L. Hopper, Dallas R. English, and et al. 2021. "Epigenetic Drift Association with Cancer Risk and Survival, and Modification by Sex" Cancers 13, no. 8: 1881. https://doi.org/10.3390/cancers13081881
APA StyleYu, C., Wong, E. M., Joo, J. E., Hodge, A. M., Makalic, E., Schmidt, D., Buchanan, D. D., Severi, G., Hopper, J. L., English, D. R., Giles, G. G., Southey, M. C., & Dugué, P.-A. (2021). Epigenetic Drift Association with Cancer Risk and Survival, and Modification by Sex. Cancers, 13(8), 1881. https://doi.org/10.3390/cancers13081881