
Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study

 ,
,  , , , , , , , , ,
, , , , , , , , , 
 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Study Population
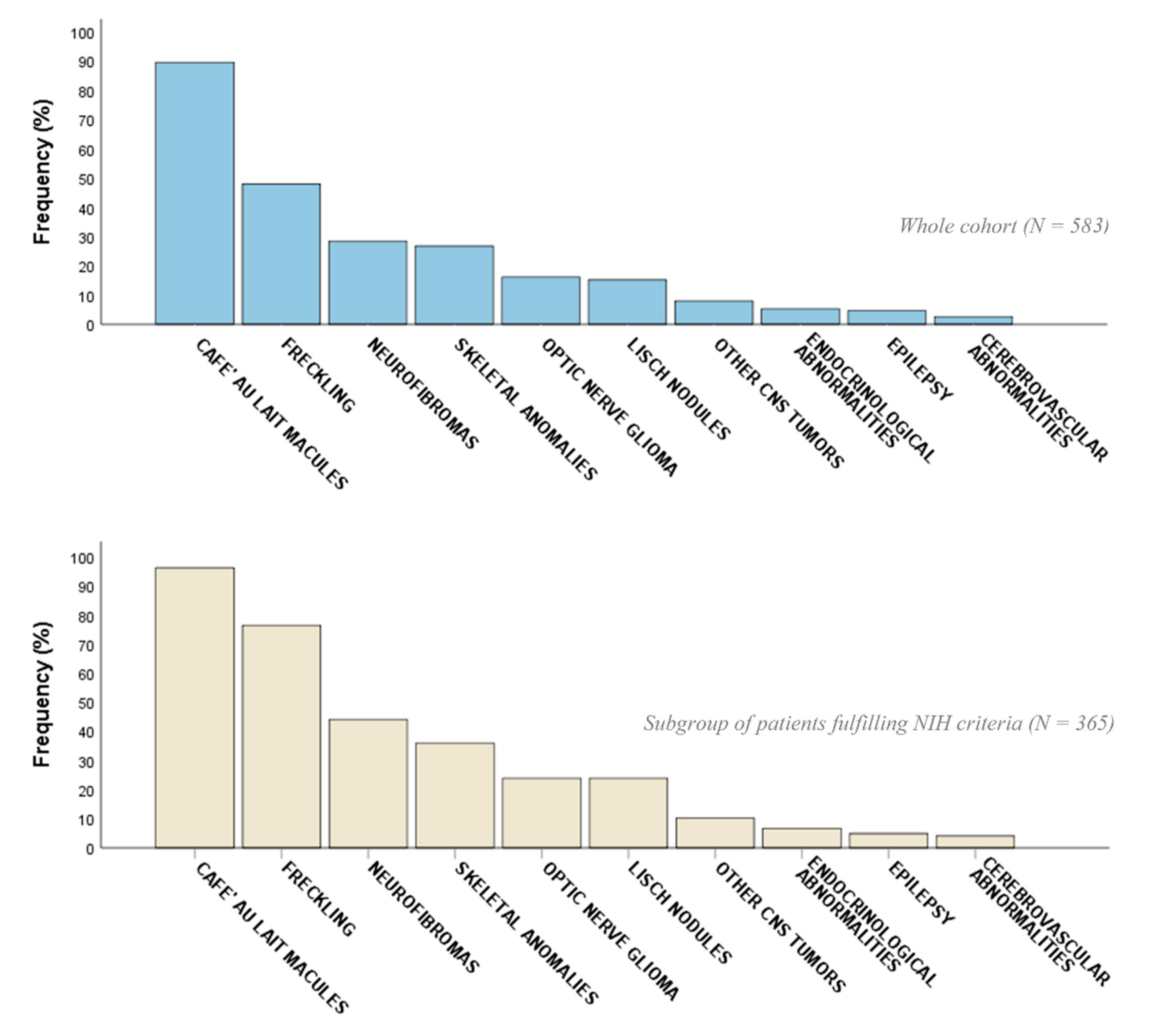
2.2. Phenotypic Characterization
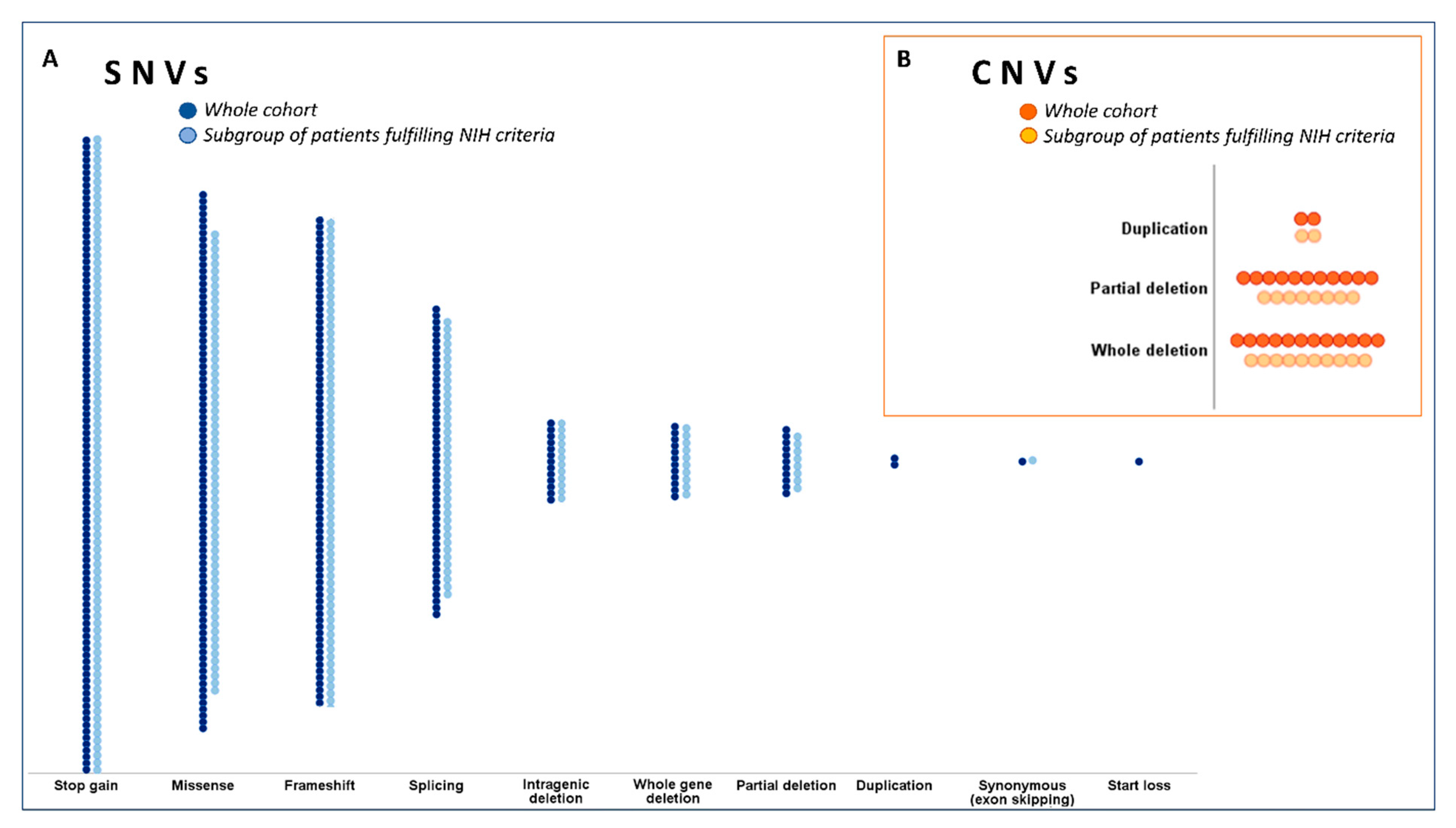
2.3. Genetic Data Analysis
2.4. Genotype–Phenotype Correlations
3. Discussion
4. Materials and Methods
4.1. Phenotypic Characterization
4.2. Genetic Testing
4.2.1. NGS Assay Design, Library Preparation, and Sequencing
4.2.2. Multiplex Ligation-Dependent Probe Amplification (MLPA)
4.2.3. RNA Extraction, RT-PCR
4.2.4. Sanger Sequencing
4.2.5. Variant Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Kallionpaa, R.A.; Uusitalo, E.; Leppavirta, J.; Poyhonen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018, 20, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Buchberg, A.M.; Cleveland, L.S.; Jenkins, N.A.; Copeland, N.G. Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway. Nature 1990, 347, 291–294. [Google Scholar] [CrossRef]
- Li, S.; Janosch, P.; Tanji, M.; Rosenfeld, G.C.; Waymire, J.C.; Mischak, H.; Kolch, W.; Sedivy, J.M. Regulation of Raf-1 kinase activity by the 14-3-3 family of proteins. EMBO J. 1995, 14, 685–696. [Google Scholar] [CrossRef]
- Trovó-Marqui, A.B.; Tajara, E.H. Neurofibromin: A general outlook. Clin. Genet. 2006, 70, 1–13. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Tabata, M.M.; Li, S.; Knight, P.; Bakker, A.; Sarin, K.Y. Phenotypic heterogeneity of neurofibromatosis type 1 in a large international registry. JCI Insight. 2020, 5, e136262. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Boucneau, J.; Lambert, J.; Messiaen, L.; Naeyaert, J.M. Pigment cell-related manifestations in neurofibromatosis type 1: An overview. Pigment Cell Res. 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.I.; Blakeley, J.O. The Diagnosis and Management of Neurofibromatosis Type 1. Med. Clin. N. Am. 2019, 103, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health consensus development conference statement: Neurofibromatosis. Bethesda, MD, USA, 1987. Neurofibromatosis 1988, 1, 172–178.
- Gutmann, D.H.; Aylsworth, A.; Carey, J.C.; Korf, B.; Marks, J.; Pyeritz, R.E.; Rubenstein, A.; Viskochil, D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997, 278, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Bottaro, G. Endocrine implications of neurofibromatosis 1 in childhood. Horm. Res Paediatr. 2015, 83, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Oderich, G.S.; Sullivan, T.M.; Bower, T.C.; Gloviczki, P.; Miller, D.V.; Babovic-Vuksanovic, D.; Macedo, T.A.; Stanson, A. Vascular abnormalities in patients with neurofibromatosis syndrome type I: Clinical spectrum, management, and results. J. Vasc. Surg. 2007, 46, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Baudou, E.; Nemmi, F.; Biotteau, M.; Maziero, S.; Assaiante, C.; Cignetti, F.; Vaugoyeau, M.; Audic, F.; Peran, P.; Chaix, Y. Are morphological and structural MRI characteristics related to specific cognitive impairments in neurofibromatosis type 1 (NF1) children? Eur. J. Paediatr. Neurol. 2020, 28, 89–100. [Google Scholar] [CrossRef]
- De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E.; Zampino, G.; Battaglia, A.; et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am. J. Hum. Genet. 2005, 77, 1092–1101. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum. Genom. 2011, 5, 623–690. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef]
- Frayling, I.M.; Mautner, V.F.; van Minkelen, R.; Kallionpaa, R.A.; Aktaş, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast cancer risk in neurofibromatosis type 1 is a function of the type of NF1 gene mutation: A new genotype-phenotype correlation. J. Med. Genet. 2019, 56, 209–219. [Google Scholar] [CrossRef]
- Peltonen, S.; Kallionpää, R.A.; Rantanen, M.; Uusitalo, E.; Lähteenmäki, P.M.; Pöyhönen, M.; Pitkäniemi, J.; Peltonen, J. Pediatric malignancies in neurofibromatosis type 1: A population-based cohort study. Int. J. Cancer 2019, 145, 2926–2932. [Google Scholar] [CrossRef]
- Jouhilahti, E.M.; Peltonen, S.; Heape, A.M.; Peltonen, J. The pathoetiology of neurofibromatosis 1. Am. J. Pathol. 2011, 178, 1932–1939. [Google Scholar] [CrossRef]
- Knudson, A.J. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- De Schepper, S.; Maertens, O.; Callens, T.; Naeyaert, J.; Lambert, J.; Messiaen, L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J. Investig. Dermatol. 2008, 128, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- Eoli, M.; Saletti, V.; Finocchiaro, G. Neurological malignancies in neurofibromatosis type 1. Curr. Opin. Oncol. 2019, 31, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Kim, Y.M.; Seo, G.H.; Oh, A.; Yoon, H.M.; Ra, Y.S.; Kim, E.K.; Kim, H.; Heo, S.H.; Kim, G.H.; et al. Phenotype categorization of neurofibromatosis type I and correlation to NF1 mutation types. J. Hum. Genet. 2020, 65, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jordan, J.T.; Bredella, M.A.; Erdin, S.; Walker, J.A.; Vangel, M.; Harris, G.J.; Plotkin, S.R.; Cai, W. Correlation between NF1 genotype and imaging phenotype on whole-body MRI: NF1 radiogenomics. Neurology 2020, 94, e2521–e2531. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef]
- Dunning-Davies, B.M.; Parker, A.P. Annual review of children with neurofibromatosis type 1. Arch. Dis. Child. Educ. Pract. Ed. 2016, 101, 102–111. [Google Scholar] [CrossRef]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef]
- Wu-Chou, Y.H.; Hung, T.C.; Lin, Y.T.; Cheng, H.W.; Lin, J.L.; Lin, C.H.; Yu, C.C.; Chen, K.T.; Yeh, T.H.; Chen, Y.R. Genetic diagnosis of neurofibromatosis type 1: Targeted next-generation sequencing with Multiple Ligation-Dependent Probe Amplification analysis. J. Biomed. Sci. 2018, 25, 72. [Google Scholar] [CrossRef]
- Burkitt Wright, E.M.; Sach, E.; Sharif, S.; Quarrell, O.; Carroll, T.; Whitehouse, R.W.; Upadhyaya, M.; Huson, S.M.; Evans, D.G. Can the diagnosis of NF1 be excluded clinically? A lack of pigmentary findings in families with spinal neurofibromatosis demonstrates a limitation of clinical diagnosis. J. Med. Genet. 2013, 50, 606–613. [Google Scholar] [CrossRef]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar]
- Radtke, H.B.; Bergner, A.L.; Goetsch, A.L.; McGowan, C.; Panzer, K.; Cannon, A. Genetic Counseling for Neurofibromatosis 1, Neurofibromatosis 2, and Schwannomatosis-Practice Resource of the National Society of Genetic Counselors. J. Genet. Couns. 2020, 29, 692–714. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Vidaud, M.; Vidaud, D.; Wolkenstein, P. Neurofibromatosis type 1: From genotype to phenotype. J. Med. Genet. 2012, 49, 483–489. [Google Scholar] [CrossRef]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- Sabbagh, A.; Pasmant, E.; Laurendeau, I.; Parfait, B.; Barbarot, S.; Guillot, B.; Combemale, P.; Ferkal, S.; Vidaud, M.; Aubourg, P.; et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum. Mol. Genet. 2009, 18, 2768–2778. [Google Scholar] [CrossRef]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef][Green Version]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [PubMed]
- Pasmant, E.; Masliah-Planchon, J.; Lévy, P.; Laurendeau, I.; Ortonne, N.; Parfait, B.; Valeyrie-Allanore, L.; Leroy, K.; Wolkenstein, P.; Vidaud, M.; et al. Identification of genes potentially involved in the increased risk of malignancy in NF1-microdeleted patients. Mol. Med. 2011, 17, 79–87. [Google Scholar] [CrossRef]
- Ottenhoff, M.J.; Rietman, A.B.; Mous, S.E.; Plasschaert, E.; Gawehns, D.; Brems, H.; Oostenbrink, R.; ENCORE-NF1 Team; van Minkelen, R.; Nellist, M.; et al. Examination of the genetic factors underlying the cognitive variability associated with neurofibromatosis type 1. Genet. Med. 2020, 22, 889–897. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef]
- Santoro, C.; Maietta, A.; Giugliano, T.; Melis, D.; Perrotta, S.; Nigro, V.; Piluso, G. Arg(1809) substitution in neurofibromin: Further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur. J. Hum. Genet. 2015, 23, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Trevisson, E.; Morbidoni, V.; Forzan, M.; Daolio, C.; Fumini, V.; Parrozzani, R.; Cassina, M.; Midena, E.; Salviati, L.; Clementi, M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genom. Med. 2019, 7, e616. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Yu, T.; Xu, Y.; Yu, L.; Wang, J.; Wang, X.; Wang, J.; Shen, Y. Clinical Presentation and Novel Pathogenic Variants among 68 Chinese Neurofibromatosis 1 Children. Genes 2019, 10, 847. [Google Scholar] [CrossRef]
- Pinna, V.; Daniele, P.; Calcagni, G.; Mariniello, L.; Criscione, R.; Giardina, C.; Lepri, F.R.; Hozhabri, H.; Alberico, A.; Cavone, S.; et al. Prevalence, Type, and Molecular Spectrum of NF1 Mutations in Patients with Neurofibromatosis Type 1 and Congenital Heart Disease. Genes 2019, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Pros, E.; Gómez, C.; Martín, T.; Fábregas, P.; Serra, E.; Lázaro, C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Zhang, J.; Tong, H.; Fu, X.; Zhang, Y.; Liu, J.; Cheng, R.; Liang, J.; Peng, J.; Sun, Z.; Liu, H.; et al. Molecular Characterization of NF1 and Neurofibromatosis Type 1 Genotype-Phenotype Correlations in a Chinese Population. Sci. Rep. 2015, 5, 11291. [Google Scholar] [CrossRef]
- Ponti, G.; Martorana, D.; Pellacani, G.; Ruini, C.; Loschi, P.; Baccarani, A.; De Santis, G.; Pollio, A.; Neri, T.M.; Mandel, V.D.; et al. NF1 truncating mutations associated to aggressive clinical phenotype with elephantiasis neuromatosa and solid malignancies. Anticancer Res. 2014, 34, 3021–3030. [Google Scholar]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Alkindy, A.; Chuzhanova, N.; Kini, U.; Cooper, D.N.; Upadhyaya, M. Genotype-phenotype associations in neurofibromatosis type 1 (NF1): An increased risk of tumor complications in patients with NF1 splice-site mutations? Hum. Genom. 2012, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Kebudi, R.; Tuncer, S.; Upadhyaya, M.; Peksayar, G.; Spurlock, G.; Yazici, H. A novel mutation in the NF1 gene in two siblings with neurofibromatosis type 1 and bilateral optic pathway glioma. Pediatr. Blood. Cancer 2008, 50, 713–715. [Google Scholar] [CrossRef]
- Sharif, S.; Upadhyaya, M.; Ferner, R.; Majounie, E.; Shenton, A.; Baser, M.; Thakker, N.; Evans, D.G. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 2011, 48, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Melloni, G.; Eoli, M.; Cesaretti, C.; Bianchessi, D.; Ibba, M.C.; Esposito, S.; Scuvera, G.; Morcaldi, G.; Micheli, R.; Piozzi, E.; et al. Risk of Optic Pathway Glioma in Neurofibromatosis Type 1: No Evidence of Genotype-Phenotype Correlations in A Large Independent Cohort. Cancers 2019, 11, 1838. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Spurlock, G.; Kluwe, L.; Chuzhanova, N.; Bennett, E.; Thomas, N.; Guha, A.; Mautner, V. The spectrum of somatic and germline NF1 mutations in NF1 patients with spinal neurofibromas. Neurogenetics 2009, 10, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.; Müller, R.; Bartelt, B.; Wolf, M.; Kunzi-Rapp, K.; Hanemann, C.O.; Fahsold, R.; Hein, C.; Vogel, W.; Assum, G. Spinal neurofibromatosis without café-au-lait macules in two families with null mutations of the NF1 gene. Am. J. Hum. Genet. 2001, 69, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Tritto, V.; Ferrari, L.; Esposito, S.; Zuccotti, P.; Bianchessi, D.; Natacci, F.; Saletti, V.; Eoli, M.; Riva, P. Non-Coding RNA and Tumor Development in Neurofibromatosis Type 1: ANRIL Rs2151280 Is Associated with Optic Glioma Development and a Mild Phenotype in Neurofibromatosis Type 1 Patients. Genes 2019, 10, 892. [Google Scholar] [CrossRef]
- Bianchessi, D.; Morosini, S.; Saletti, V.; Ibba, M.C.; Natacci, F.; Esposito, S.; Cesaretti, C.; Riva, D.; Finocchiaro, G.; Eoli, M. 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol. Genet. Genom. Med. 2015, 3, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Stella, A.; Lastella, P.; Loconte, D.C.; Bukvic, N.; Varvara, D.; Patruno, M.; Bagnulo, R.; Lovaglio, R.; Bartolomeo, N.; Serio, G.; et al. Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes 2018, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef]
- Calì, F.; Chiavetta, V.; Ruggeri, G.; Piccione, M.; Selicorni, A.; Palazzo, D.; Bonsignore, M.; Cereda, A.; Elia, M.; Failla, P.; et al. Mutation spectrum of NF1 gene in Italian patients with neurofibromatosis type 1 using Ion Torrent PGM™ platform. Eur. J. Med. Genet. 2017, 60, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Rothman, K.J. Statistics in nonrandomized studies. Epidemiology 1990, 1, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Perneger, T.V. What’s wrong with Bonferroni adjustments. BMJ 1998, 316, 1236–1238. [Google Scholar] [CrossRef]
- Streiner, D.L.; Norman, G.R. Correction for multiple testing: Is there a resolution? Chest 2011, 140, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Ranstam, J. Multiple P-values and Bonferroni correction. Osteoarthr. Cartil. 2016, 24, 763–764. [Google Scholar] [CrossRef]
- Assunto, A.; Ferrara, U.; De Luca, A.; Pivonello, C.; Lombardo, L.; Piscitelli, A.; Tortora, C.; Pinna, V.; Daniele, P.; Pivonello, R.; et al. Isoform-specific NF1 mRNA levels correlate with disease severity in Neurofibromatosis type 1. Orphanet. J. Rare Dis. 2019, 14, 261. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Fulfilling the NIH Criteria for Diagnosis | Genetic Confirmation | |||||||
|---|---|---|---|---|---|---|---|---|
| Total (n = 583) | Yes (Group A) (n = 365) | No (Group B) (n = 218) | p | Yes (n = 351) | No (n= 232) | p | ||
| Age, years | 16.8 ± 11.91 14.0 (1.0–73.0) | 18.5 ± 12.86 15.0 (1.0–71.0) | 14.1 ± 9.55 12.5 (2.0–73.0) | <0.001 ** | 17.5 ± 12.59 15.0 (2.0–71.0) | 15.7 ± 10.74 14.0 (1.0–73.0) | 0.18 | |
| Age classes | 0–12 years | 247 (42.4) | 138 (37.8) | 109 (50.0) | - | 145 (41.3) | 102 (44.0) | - |
| 13–18 years | 310 (53.2) | 207 (56.7) | 103 (47.2) | 0.009 ** | 189 (53.8) | 121 (52.2) | 0.74 | |
| 19–44 years | 26 (4.5) | 20 (5.5) | 6 (2.7) | - | 17 (4.8) | 9 (3.9) | - | |
| Sex, males | 309 (53.0) | 182 (49.9) | 127 (58.3) | 0.049 ** | 181 (51.6) | 128 (55.2) | 0.39 | |
| Positive family history * | 134 (23.0) | 104 (28.5) | 30 (13.8) | 0.001 ** | 108 (30.8) | 26 (11.2) | <0.001 ** | |
| Status | De novo | 60 (10.3) | 48 (13.2) | 12 (5.5) | - | 60 (17.1) | 0 (0.0) | - |
| Maternal | 39 (6.7) | 30 (8.2) | 9 (4.1) | <0.001 ** | 37 (10.5) | 2 (0.9) | <0.001 ** | |
| Paternal | 48 (8.2) | 35 (9.6) | 13 (6.0) | - | 46 (13.1) | 2 (0.9) | - | |
| Not available | 436 (74.8) | 252 (69.0) | 184 (84.4) | - | 208 (59.3) | 228 (98.3) | - | |
| Number of NIH criteria satisfied | 2.3 ± 1.41 2.0 (1.0–7.0) | 3.1 ± 1.21 3.0 (2.0–7.0) | 1.0 ± 0.00 1.0 (1.0–1.0) | <0.001 ** | 2.8 ± 1.40 3.0 (1.0–7.0) | 1.6 ± 1.04 1.0 (1.0–7.0) | <0.001 ** | |
| Deceased | 3 (0.5) | 3 (0.8) | 0 (0.0) | 0.30 | 3 (0.9) | 0 (0.0) | 0.16 | |
| Genetic confirmation | 351 (60.3) | 287 (78.6) | 64 (29.4) | <0.001 ** | - | - | - | |
| Fulfilling NIH criteria for diagnosis | 365 (62.6) | - | - | - | 287 (81.8) | 78 (33.6) | <0.001 ** | |
| Fulfilling the NIH Criteria for Diagnosis | Genetic Confirmation | ||||||
|---|---|---|---|---|---|---|---|
| Total (n = 583) | Yes (n = 365) (Group A) | No (n = 218) (Group B) | p | Yes (n = 351) | No (n = 232) | p | |
| CALMs | 522 (89.5) | 351 (96.2) | 171 (78.4) | <0.001 ** | 316 (90.0) | 206 (88.8) | 0.63 |
| Freckling | 280 (48.0) | 279 (76.4) | 1 (0.5) | <0.001 ** | 222 (63.2) | 58 (25.0) | <0.001 ** |
| Other neurological findings (e.g., abnormal tone/DTRs, tremor, ataxia) | 188 (32.1) | 136 (37.3) | 52 (23.9) | 0.001 ** | 130 (37.0) | 58 (25.0) | 0.002 ** |
| Neurofibromas (cNFs, sNFs) | 137 (23.5) | 134 (36.7) | 3 (1.4) | <0.001 ** | 108 (30.8) | 29 (12.5) | <0.001 ** |
| Common neurological features † | 120 (20.6) | 85 (23.3) | 35 (16.1) | 0.032 ** | 83 (23.6) | 37 (15.9) | 0.024 ** |
| OPG | 94 (16.1) | 87 (23.8) | 7 (3.2) | <0.001 ** | 72 (20.5) | 22 (9.5) | 0.009 ** |
| Lisch nodules | 89 (15.3) | 87 (23.8) | 2 (0.9) | <0.001 ** | 74 (21.1) | 15 (6.5) | <0.001 ** |
| Scoliosis | 89 (15.3) | 77 (21.1) | 12 (5.5) | <0.001 ** | 63 (17.9) | 26 (11.2) | 0.027 ** |
| Neurofibromas (pNFs) | 76 (13.0) | 75 (20.5) | 1 (0.5) | <0.001 ** | 68 (19.4) | 8 (3.4) | <0.001 ** |
| Other skeletal alterations (e.g., osteoporosis, vertebral malformations) | 55 (9.4) | 44 (12.1) | 11 (5.0) | 0.005 ** | 39 (11.1) | 16 (6.9) | 0.09 |
| Neuroimaging abnormalities (e.g., Chiari malformation, tethered cord) | 36 (6.2) | 29 (7.9) | 7 (3.2) | 0.07 | 24 (6.8) | 12 (5.2) | 0.84 |
| Endocrinological abnormalities (e.g., thyreopathy, Addison’s disease, PP) | 31 (5.3) | 24 (6.6) | 7 (3.2) | 0.08 | 22 (6.3) | 9 (3.9) | 0.21 |
| Headache | 27 (4.6) | 23 (6.3) | 4 (1.8) | 0.012 ** | 21 (6.0) | 6 (2.6) | 0.06 |
| Epilepsy | 27 (4.6) | 18 (4.9) | 9 (4.1) | 0.64 | 18 (5.1) | 9 (3.9) | 0.48 |
| Gliomas other than OPG (e.g., pilocytic astrocytoma, glioblastoma) | 24 (4.1) | 22 (6.0) | 2 (0.9) | 0.008 ** | 21 (6.0) | 3 (1.3) | 0.018 ** |
| Non-glioma brain tumors (e.g., amartoma, meningioma, lipoma) | 24 (4.1) | 15 (4.1) | 9 (4.1) | 0.66 | 16 (4.6) | 8 (3.4) | 0.87 |
| Severe learning disability | 17 (2.9) | 14 (3.8) | 3 (1.4) | 0.09 | 13 (3.7) | 4 (1.7) | 0.16 |
| Sphenoid bone dysplasia | 15 (2.6) | 14 (3.8) | 1 (0.5) | 0.026 ** | 13 (3.7) | 2 (0.9) | 0.08 |
| Cerebrovascular abnormalities (ectasia, aneurysms, hypoplasia) | 15 (2.6) | 15 (4.1) | 0 (0.0) | 0.005 ** | 10 (2.8) | 5 (2.2) | 0.90 |
| Moyamoya syndrome | 15 (2.6) | 14 (3.8) | 1 (0.5) | 0.026 ** | 13 (3.7) | 2 (0.9) | 0.08 |
| Tibial dysplasia | 13 (2.2) | 11 (3.0) | 2 (0.9) | 0.21 | 11 (3.1) | 2 (0.9) | 0.08 |
| Noonan-like dysmorphic features | 13 (2.2) | 8 (2.2) | 5 (2.3) | 0.95 | 12 (3.4) | 1 (0.4) | 0.017 ** |
| Behavioral abnormalities (ADHD, ARFID, ASD) | 12 (2.1) | 12 (3.3) | 0 (0.0) | 0.007 ** | 11 (3.1) | 1 (0.4) | 0.024 ** |
| Severe DD/ID | 12 (2.1) | 7 (1.9) | 5 (2.3) | 0.77 | 9 (2.6) | 3 (1.3) | 0.29 |
| Lymphatic and vascular abnormalities (e.g., angioma, cavernoma, LD) | 11 (1.9) | 8 (2.2) | 3 (1.4) | 0.48 | 7 (2.0) | 4 (1.7) | 0.82 |
| Pectus excavatum | 10 (1.7) | 10 (2.7) | 0 (0.0) | 0.013 ** | 9 (2.6) | 1 (0.4) | 0.05 |
| Renal malformations/disease (multicystic/duplex kidney, hydronephrosis) | 7 (1.2) | 3 (0.8) | 4 (1.8) | 0.17 | 4 (1.1) | 3 (1.3) | 0.63 |
| Variants in cerebrovascular anatomy | 7 (1.2) | 7 (1.9) | 0 (0.0) | 0.06 | 5 (1.4) | 2 (0.9) | 0.55 |
| Hypertension | 7 (1.2) | 6 (1.6) | 1 (0.5) | 0.20 | 5 (1.4) | 2 (0.9) | 0.55 |
| Ocular abnormalities (coloboma, glaucoma) | 4 (0.7) | 3 (0.8) | 1 (0.5) | 0.99 | 4 (1.1) | 0 (0.0) | 0.16 |
| Spinal tumors (astrocytoma) | 3 (0.5) | 2 (0.5) | 1 (0.5) | 0.99 | 2 (0.6) | 1 (0.4) | 0.99 |
| Pulmonic stenosis | 3 (0.5) | 2 (0.5) | 1 (0.5) | 0.99 | 3 (0.9) | 0 (0.0) | 0.16 |
| Other cardiac abnormalities (valvulopathies, arrhythmia) | 3 (0.5) | 2 (0.5) | 1 (0.5) | 0.99 | 1 (0.3) | 2 (0.9) | 0.57 |
| Renovascular abnormalities | 1 (0.2) | 1 (0.3) | 0 (0.0) | 0.99 | 0 (0.0) | 1 (0.4) | 0.34 |
| Fulfilling the NIH Criteria for Diagnosis | ||||||
|---|---|---|---|---|---|---|
| Total (n = 351) | Yes (n = 287) (Group A) | No (n = 64) (Group B) | Univariate | Multivariate | ||
| Variant types | Stop-gain | 100 (28.5) | 87 (30.3) | 13 (20.3) | 0.11 * | NS |
| Missense | 85 (24.2) | 63 (22.0) | 22 (34.4) | 0.036 ** | NS | |
| Frameshift | 77 (21.9) | 66 (23.0) | 11 (17.2) | 0.31 | - | |
| Splicing | 49 (14.0) | 38 (13.2) | 11 (17.2) | 0.41 | - | |
| Whole gene deletion | 12 (3.4) | 10 (3.5) | 2 (3.1) | 0.029 ** | 0.04 (0.01–0.29); 0.002 | |
| Partial deletion | 11 (3.1) | 8 (2.8) | 3 (4.7) | 0.001 ** | 0.02 (0.01–0.14), <0.001 | |
| Intragenic deletion | 13 (3.7) | 12 (4.2) | 1 (1.6) | 0.32 | - | |
| Duplication | 2 (0.6) | 2 (0.7) | 0 (0.0) | 0.99 | - | |
| Start loss | 1 (0.3) | 0 (0.0) | 1 (1.6) | 0.42 | - | |
| Synonymous (exon skipping) † | 1 (0.3) | 1 (0.3) | 0 (0.0) | 0.42 | - | |
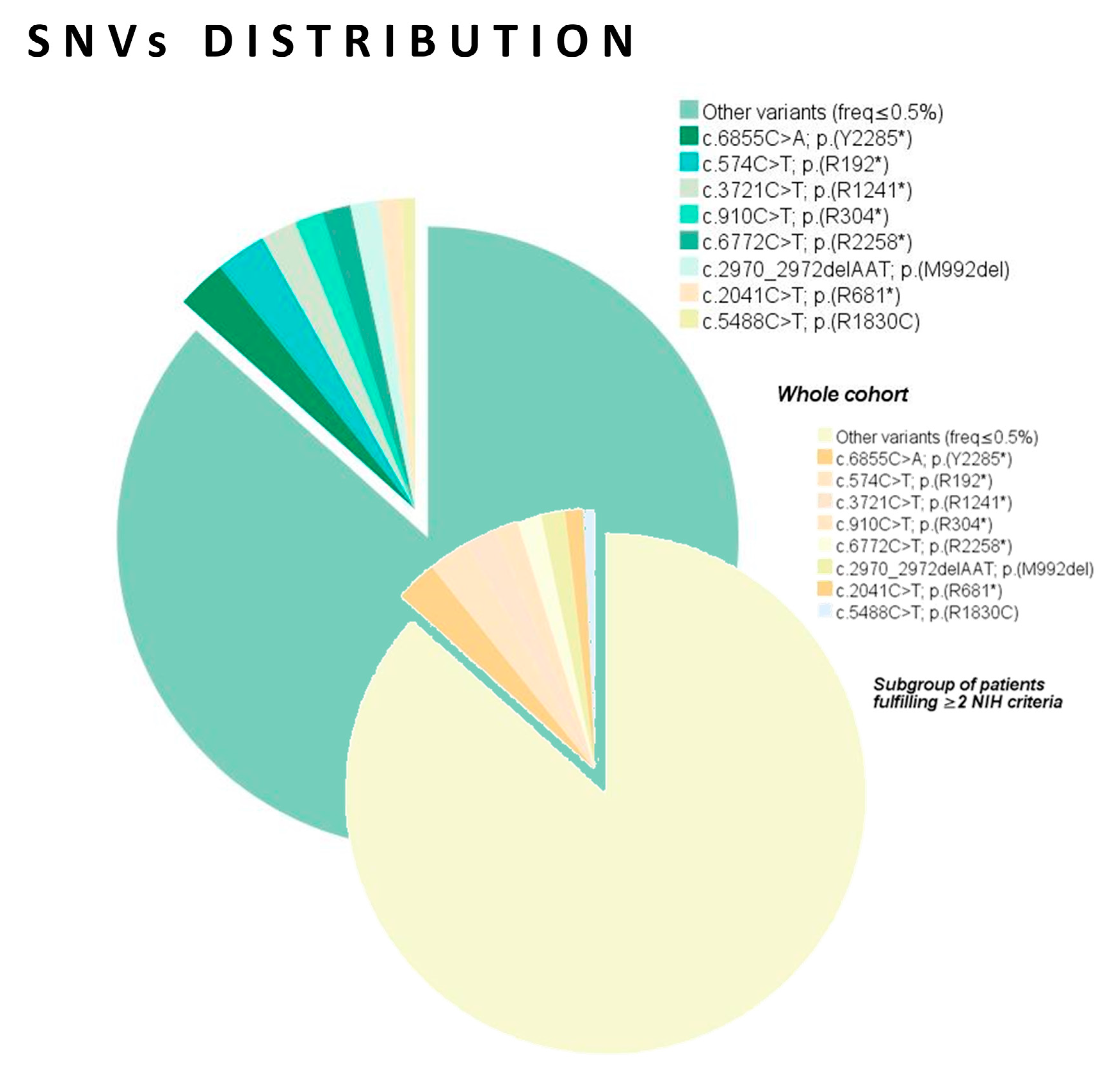
| Recurrent SNVs (NM_001042492.3; NP_001035957.1) | Wild type | 23 (6.6) | 18 (6.3) | 5 (7.8) | 0.99 | - |
| c.574C>T; p.(R192*) | 7 (2.0) | 7 (2.4) | 0 (0.0) | 0.20 * | NS | |
| c.6855C>A; p.(Y2285*) | 7 (2.0) | 7 (2.4) | 0 (0.0) | 0.20 * | NS | |
| c.3721C>T; p.(R1241*) | 5 (1.4) | 5 (1.7) | 0 (0.0) | 0.59 | - | |
| c.5488C>T; p.(R1830C) c.6772C>T; p.(R2258*) | 5 (1.4) 4 (1.1) | 2 (0.7) 4 (1.4) | 3 (4.7) 0 (0.0) | 0.044 ** 0.20 * | 0.14 (0.02–0.87); 0.035 NS | |
| c.910C>T; p.(R304*) | 4 (1.1) | 4 (1.4) | 0 (0.0) | 0.99 | - | |
| c.2041C>T; p.(R681*) | 4 (1.1) | 3 (1.0) | 1 (1.6) | 0.56 | - | |
| c.2970_2972delAAT; p.(M992del) | 4 (1.1) | 4 (1.4) | 0 (0.0) | 0.99 | - | |
| Miscellaneous (freq ≤ 0.5%) | 288 (82.1) | 233 (81.2) | 55 (85.9) | - | - | |
| MLPA category | Normal | 23 (6.6) | 21 (7.3) | 2 (3.1) | - | - |
| Partial deletion | 11 (3.1) | 8 (2.8) | 3 (4.7) | - | - | |
| Whole gene deletion | 12 (3.4) | 10 (3.5) | 2 (3.1) | - | - | |
| Duplication | 2 (0.6) | 2 (0.7) | 0 (0.0) | - | - | |
| Not executed | 303 (86.3) | 246 (85.7) | 57 (89.1) | - | - | |
| No (n = 65) | Yes (n = 222) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 23 (35.4) | 92 (41.4) | 0.013 ** | Ref. |
| 13–18 years | 34 (52.3) | 123 (55.4) | NS | ||
| 19–44 years | 8 (12.3) | 7 (3.2) | 0.22 (0.07–0.69); 0.010 | ||
| Sex, males | - | 32 (49.2) | 110 (49.5) | 0.96 | - |
| Family history | - | 30 (46.2) | 58 (26.1) | 0.002 ** | 0.41 (0.23–0.73); 0.003 |
| Status | De novo | 8 (12.3) | 40 (18.0) | 0.021 ** | - |
| Maternal | 12 (18.5) | 17 (7.7) | - | ||
| Paternal | 11 (16.9) | 23 (10.4) | NS | ||
| Not available | 34 (52.3) | 142 (64.0) | - | ||
| Duplication | - | 0 (0.0) | 2 (0.9) | 0.44 | - |
| Partial deletions | - | 2 (3.1) | 6 (2.7) | 0.87 | - |
| Whole gene deletions | - | 0 (0.0) | 10 (4.5) | 0.08 * | NS |
| Splicing variants | - | 13 (20.0) | 25 (11.3) | 0.07 * | NS |
| Missense variants | - | 17 (26.2) | 46 (20.7) | 0.35 | - |
| Stop-gain variants | - | 15 (23.1) | 72 (32.4) | 0.15 * | NS |
| Frameshift variants | - | 16 (24.6) | 50 (22.5) | 0.72 | - |
| Intragenic deletions | - | 2 (3.1) | 10 (4.5) | 0.61 | - |
| c.574C>T; p.(R192*) | - | 0 (0.0) | 7 (3.2) | 0.15 * | NS |
| c.6855C>A; p.(Y2285*) | - | 1 (1.5) | 6 (2.7) | 0.59 | - |
| c.3721C>T; p.(R1241*) | - | 2 (3.1) | 3 (1.4) | 0.32 | - |
| c.6772C>T; p.(R2258*) | - | 1 (1.5) | 3 (1.4) | 0.99 | - |
| c.910C>T; p.(R304*) | - | 0 (0.0) | 4 (1.8) | 0.58 | - |
| c.2041C>T; p.(R681*) | - | 1 (1.5) | 2 (0.9) | 0.54 | - |
| c.5488C>T; p.(R1830C) | - | 1 (1.5) | 1 (0.5) | 0.40 | - |
| c.2970_2972delAAT; p.(M992del) | - | 1 (1.5) | 3 (1.4) | 0.99 | - |
| No (n = 219) | Yes (n = 68) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 93 (42.5) | 22 (32.4) | 0.33 | - |
| 13–18 years | 115 (52.5) | 42 (61.8) | - | ||
| 19–44 years | 11 (5.0) | 4 (5.9) | - | ||
| Sex, males | - | 107 (48.9) | 35 (51.5) | 0.71 | - |
| Family history | - | 71 (32.4) | 17 (25.0) | 0.25 | - |
| Status | De novo | 40 (18.3) | 8 (11.8) | 0.10 * | - |
| Maternal | 23 (10.5) | 6 (8.8) | - | ||
| Paternal | 30 (13.7) | 4 (5.9) | NS | ||
| Not available | 126 (57.5) | 50 (73.5) | - | ||
| Duplication | - | 1 (0.5) | 1 (1.5) | 0.38 | - |
| Partial deletions | - | 7 (3.2) | 1 (1.5) | 0.45 | - |
| Whole gene deletions | - | 6 (2.7) | 4 (5.9) | 0.22 | - |
| Splicing variants | - | 28 (12.8) | 10 (14.7) | 0.68 | - |
| Missense variants | - | 57 (26.0) | 6 (8.8) | 0.003 ** | 0.28 (0.11–0.67); 0.005 |
| Stop-gain variants | - | 58 (26.5) | 29 (42.6) | 0.011 ** | NS |
| Frameshift variants | - | 51 (23.3) | 15 (22.1) | 0.83 | - |
| Intragenic deletions | - | 10 (4.6) | 2 (2.9) | 0.56 | - |
| c.574C>T; p.(R192*) | - | 3 (1.4) | 4 (5.9) | 0.035 ** | NS |
| c.6855C>A; p.(Y2285*) | - | 4 (1.8) | 3 (4.4) | 0.23 | - |
| c.3721C>T; p.(R1241*) | - | 3 (1.4) | 2 (2.9) | 0.39 | - |
| c.6772C>T; p.(R2258*) | - | 3 (1.4) | 1 (1.5) | 0.95 | - |
| c.910C>T; p.(R304*) | - | 3 (1.4) | 1 (1.5) | 0.95 | - |
| c.2041C>T; p.(R681*) | - | 2 (0.9) | 1 (1.5) | 0.69 | - |
| c.5488C>T; p.(R1830C) | - | 2 (0.9) | 0 (0.0) | 0.43 | - |
| c.2970_2972delAAT; p.(M992del) | - | 4 (1.8) | 0 (0.0) | 0.58 | - |
| No (n = 213) | Yes (n = 74) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 93 (43.7) | 22 (29.7) | 0.002 ** | Ref. |
| 13–18 years | 105 (49.3) | 52 (70.3) | 2.37 (1.31–4.28); 0.004 | ||
| 19–44 years | 15 (7.0) | 0 (0.0) | NS | ||
| Sex, males | - | 108 (50.7) | 34 (45.9) | 0.48 | - |
| Family history | - | 74 (34.7) | 14 (18.9) | 0.011 ** | 0.40 (0.21–0.78); 0.007 |
| Status | De novo | 34 (16.0) | 14 (18.9) | 0.13 * | - |
| Maternal | 25 (11.7) | 4 (5.4) | - | ||
| Paternal | 29 (13.6) | 5 (6.8) | NS | ||
| Not available | 125 (58.7) | 51 (68.9) | - | ||
| Duplication | - | 2 (0.9) | 0 (0.0) | 0.40 | - |
| Partial deletions | - | 5 (2.3) | 3 (4.1) | 0.43 | - |
| Whole gene deletions | - | 6 (2.8) | 4 (5.4) | 0.30 | - |
| Splicing variants | - | 28 (13.1) | 10 (13.5) | 0.94 | - |
| Missense variants | - | 46 (21.6) | 17 (23.0) | 0.81 | - |
| Stop-gain variants | - | 64 (30.0) | 23 (31.1) | 0.87 | - |
| Frameshift variants | - | 54 (25.4) | 12 (16.2) | 0.11 * | NS |
| Intragenic deletions | - | 8 (3.8) | 4 (5.4) | 0.54 | - |
| c.574C>T; p.(R192*) | - | 5 (2.3) | 2 (2.7) | 0.86 | - |
| c.6855C>A; p.(Y2285*) | - | 3 (1.4) | 4 (5.4) | 0.06 * | 6.03 (0.98–36.94); 0.05 |
| c.3721C>T; p.(R1241*) | - | 5 (2.3) | 0 (0.0) | 0.33 | - |
| c.6772C>T; p.(R2258*) | - | 3 (1.4) | 1 (1.4) | 0.99 | - |
| c.910C>T; p.(R304*) | - | 3 (1.4) | 1 (1.4) | 0.99 | - |
| c.2041C>T; p.(R681*) | - | 2 (0.9) | 1 (1.4) | 0.76 | - |
| c.5488C>T; p.(R1830C) | - | 2 (0.9) | 0 (0.0) | 0.99 | - |
| c.2970_2972delAAT; p.(M992del) | - | 3 (1.4) | 1 (1.4) | 0.99 | - |
| No (n = 249) | Yes (n = 38) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 98 (39.4) | 17 (44.7) | 0.28 | - |
| 13–18 years | 136 (54.6) | 21 (55.3) | - | ||
| 19–44 years | 15 (6.0) | 0 (0.0) | - | ||
| Sex, males | - | 123 (49.4) | 19 (50.0) | 0.95 | - |
| Family history (first degree) | - | 75 (30.1) | 13 (34.2) | 0.61 | - |
| Status | De novo | 44 (17.7) | 4 (10.5) | 0.75 | - |
| Maternal | 25 (10.0) | 4 (10.5) | - | ||
| Paternal | 29 (11.6) | 5 (13.2) | - | ||
| Not available | 151 (60.6) | 25 (65.8) | - | ||
| Duplication | - | 2 (0.8) | 0 (0.0) | 0.99 | - |
| Partial deletions | - | 7 (2.8) | 1 (2.6) | 0.95 | - |
| Whole gene deletions | - | 7 (2.8) | 3 (7.9) | 0.11 * | 4.09 (0.98–17.08); 0.05 |
| Splicing variants | - | 36 (14.5) | 2 (5.3) | 0.12 * | NS |
| Missense variants | - | 57 (22.9) | 6 (15.8) | 0.33 | - |
| Stop-gain variants | - | 78 (31.3) | 9 (23.7) | 0.34 | - |
| Frameshift variants | - | 51 (20.5) | 15 (39.51) | 0.010 ** | 2.81 (1.34–5.87); 0.006 |
| Intragenic deletions | - | 10 (4.0) | 2 (5.3) | 0.72 | - |
| c.574C>T; p.(R192*) | - | 6 (2.4) | 1 (2.6) | 0.99 | - |
| c.6855C>A; p.(Y2285*) | - | 7 (2.8) | 0 (0.0) | 0.60 | - |
| c.3721C>T; p.(R1241*) | - | 4 (1.6) | 1 (2.6) | 0.51 | - |
| c.6772C>T; p.(R2258*) | - | 3 (1.2) | 1 (2.6) | 0.44 | - |
| c.910C>T; p.(R304*) | - | 4 (1.6) | 0 (0.0) | 0.99 | - |
| c.2041C>T; p.(R681*) | - | 3 (1.2) | 0 (0.0) | 0.99 | - |
| c.5488C>T; p.(R1830C) | - | 2 (0.8) | 0 (0.0) | 0.99 | - |
| c.2970_2972delAAT; p.(M992del) | - | 3 (1.2) | 1 (2.6) | 0.44 | - |
| No (n = 244) † | Yes (n = 23) † | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 100 (41.0) | 7 (30.4) | 0.29 | - |
| 13–18 years | 133 (54.5) | 16 (69.6) | - | ||
| 19–44 years | 11 (4.5) | 0 (0.0) | - | ||
| Sex, males | - | 123 (50.4) | 10 (43.5) | 0.53 | - |
| Family history (first degree) | - | 76 (31.1) | 5 (21.7) | 0.35 | - |
| Status | De novo | 43 (17.6) | 4 (17.4) | 0.32 | - |
| Maternal | 23 (9.4) | 5 (21.7) | - | ||
| Paternal | 31 (12.7) | 2 (8.7) | - | ||
| Not available | 147 (60.2) | 12 (52.2) | - | ||
| Duplication | - | 1 (0.4) | 0 (0.0) | 0.99 | - |
| Partial deletions | - | 8 (3.3) | 0 (0.0) | 0.99 | - |
| Whole gene deletions | - | 10 (4.1) | 0 (0.0) | 0.99 | - |
| Splicing variants | - | 31 (12.7) | 4 (17.4) | 0.52 | - |
| Missense variants | - | 56 (23.0) | 3 (13.0) | 0.27 | - |
| Stop-gain variants | - | 70 (28.7) | 9 (39.1) | 0.29 | - |
| Frameshift variants | - | 57 (23.4) | 6 (26.1) | 0.77 | - |
| Intragenic deletions | - | 10 (4.1) | 1 (4.3) | 0.99 | - |
| c.574C>T; p.(R192*) | - | 6 (2.5) | 0 (0.0) | 0.99 | - |
| c.6855C>A; p.(Y2285*) | - | 6 (2.5) | 0 (0.0) | 0.99 | - |
| c.3721C>T; p.(R1241*) | - | 3 (1.2) | 2 (8.7) | 0.06 * | 7.65 (1.21–48.36); 0.031 |
| c.6772C>T; p.(R2258*) | - | 4 (1.6) | 0 (0.0) | 0.99 | - |
| c.910C>T; p.(R304*) | - | 3 (1.2) | 0 (0.0) | 0.99 | - |
| c.2041C>T; p.(R681*) | - | 3 (1.2) | 0 (0.0) | 0.99 | - |
| c.5488C>T; p.(R1830C) | - | 2 (0.8) | 0 (0.0) | 0.99 | - |
| c.2970_2972delAAT; p.(M992del) | - | 3 (1.2) | 0 (0.0) | 0.99 | - |
| No (n = 267) | Yes (n = 20) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 108 (40.4) | 7 (35.0) | 0.43 | Ref. |
| 13–18 years | 144 (53.9) | 13 (65.0) | - | ||
| 19–44 years | 15 (5.6) | 0 (0.0) | - | ||
| Sex, males | - | 135 (50.6) | 7 (35.0) | 0.18 * | NS |
| Family history (first degree) | - | 85 (31.8) | 3 (15.0) | 0.12 * | NS |
| Status | De novo | 42 (15.7) | 6 (30.0) | 0.39 | - |
| Maternal | 28 (10.5) | 1 (5.0) | - | ||
| Paternal | 32 (12.0) | 2 (10.0) | - | ||
| Not available | 165 (61.8) | 11 (55.0) | - | ||
| Duplication | - | 2 (0.7) | 0 (0.0) | 0.99 | - |
| Partial deletions | - | 8 (3.0) | 0 (0.0) | 0.99 | - |
| Whole gene deletions | - | 10 (3.7) | 0 (0.0) | 0.99 | - |
| Splicing variants | - | 37 (13.9) | 1 (5.0) | 0.26 | NS |
| Missense variants | - | 58 (21.7) | 5 (25.0) | 0.73 | - |
| Stop-gain variants | - | 79 (29.6) | 8 (40.0) | 0.33 | - |
| Frameshift variants | - | 62 (23.2) | 4 (20.0) | 0.74 | - |
| Intragenic deletions | - | 10 (3.7) | 2 (10.0) | 0.15 * | NS |
| c.574C>T; p.(R192*) | - | 7 (2.6) | 0 (0.0) | 0.99 | - |
| c.6855C>A; p.(Y2285*) | - | 5 (1.9) | 2 (10.0) | 0.08 * | 5.82 (1.06–32.13); 0.043 |
| c.3721C>T; p.(R1241*) | - | 5 (1.9) | 0 (0.0) | 0.99 | - |
| c.6772C>T; p.(R2258*) | - | 4 (1.5) | 0 (0.0) | 0.99 | - |
| c.910C>T; p.(R304*) | - | 4 (1.5) | 0 (0.0) | 0.99 | - |
| c.2041C>T; p.(R681*) | - | 3 (1.1) | 0 (0.0) | 0.99 | - |
| c.5488C>T; p.(R1830C) | - | 2 (0.7) | 0 (0.0) | 0.99 | - |
| c.2970_2972delAAT; p.(M992del) | - | 4 (1.5) | 0 (0.0) | 0.99 | - |
| No (n = 179) | Yes (n = 108) | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| Age classes | 0–12 years | 93 (52.0) | 22 (20.4) | <0.001 ** | Ref. |
| 13–18 years | 86 (48.0) | 71 (65.7) | 3.36 (1.91–5.91); <0.001 | ||
| 19–44 years | 0 (0.0) | 15 (13.9) | NS | ||
| Sex, males | - | 91 (50.8) | 51 (47.2) | 0.55 | - |
| Family history (first degree) | - | 59 (33.0) | 29 (26.9) | 0.28 | - |
| Status | De novo | 35 (19.6) | 13 (12.0) | 0.007 ** | - |
| Maternal | 22 (12.3) | 7 (6.5) | - | ||
| Paternal | 26 (14.5) | 8 (7.4) | NS | ||
| Not available | 96 (53.6) | 80 (74.1) | - | ||
| Duplication | - | 2 (1.1) | 0 (0.0) | 0.53 | - |
| Partial deletions | - | 5 (2.8) | 3 (2.8) | 0.99 | - |
| Whole gene deletions | - | 5 (2.8) | 5 (4.6) | 0.41 | - |
| Splicing variants | - | 21 (11.7) | 17 (15.7) | 0.33 | - |
| Missense variants | - | 48 (26.8) | 15 (13.9) | 0.010 ** | 0.44 (0.22–0.90); 0.024 |
| Stop-gain variants | - | 50 (27.9) | 37 (34.3) | 0.26 | - |
| Frameshift variants | - | 40 (22.3) | 26 (24.1) | 0.74 | - |
| Intragenic deletions | - | 7 (3.9) | 5 (4.6) | 0.77 | - |
| c.574C>T; p.(R192*) | - | 4 (2.2) | 3 (2.8) | 0.77 | - |
| c.6855C>A; p.(Y2285*) | - | 5 (2.8) | 2 (1.9) | 0.62 | - |
| c.3721C>T; p.(R1241*) | - | 2 (1.1) | 3 (2.8) | 0.37 | - |
| c.6772C>T; p.(R2258*) | - | 4 (2.2) | 0 (0.0) | 0.30 | - |
| c.910C>T; p.(R304*) | - | 3 (1.7) | 1 (0.9) | 0.99 | - |
| c.2041C>T; p.(R681*) | - | 2 (1.1) | 1 (0.9) | 0.99 | - |
| c.5488C>T; p.(R1830C) | - | 1 (0.6) | 1 (0.9) | 0.99 | - |
| c.2970_2972delAAT; p.(M992del) | - | 3 (1.7) | 1 (0.9) | 0.99 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, M.; Schiavetti, I.; Madia, F.; Chelleri, C.; Piccolo, G.; Accogli, A.; Riva, A.; Salpietro, V.; Bocciardi, R.; Morcaldi, G.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers 2021, 13, 1879. https://doi.org/10.3390/cancers13081879
Scala M, Schiavetti I, Madia F, Chelleri C, Piccolo G, Accogli A, Riva A, Salpietro V, Bocciardi R, Morcaldi G, et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers. 2021; 13(8):1879. https://doi.org/10.3390/cancers13081879
Chicago/Turabian StyleScala, Marcello, Irene Schiavetti, Francesca Madia, Cristina Chelleri, Gianluca Piccolo, Andrea Accogli, Antonella Riva, Vincenzo Salpietro, Renata Bocciardi, Guido Morcaldi, and et al. 2021. "Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study" Cancers 13, no. 8: 1879. https://doi.org/10.3390/cancers13081879
APA StyleScala, M., Schiavetti, I., Madia, F., Chelleri, C., Piccolo, G., Accogli, A., Riva, A., Salpietro, V., Bocciardi, R., Morcaldi, G., Di Duca, M., Caroli, F., Verrico, A., Milanaccio, C., Viglizzo, G., Traverso, M., Baldassari, S., Scudieri, P., Iacomino, M., ... Zara, F. (2021). Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers, 13(8), 1879. https://doi.org/10.3390/cancers13081879