
Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis



{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
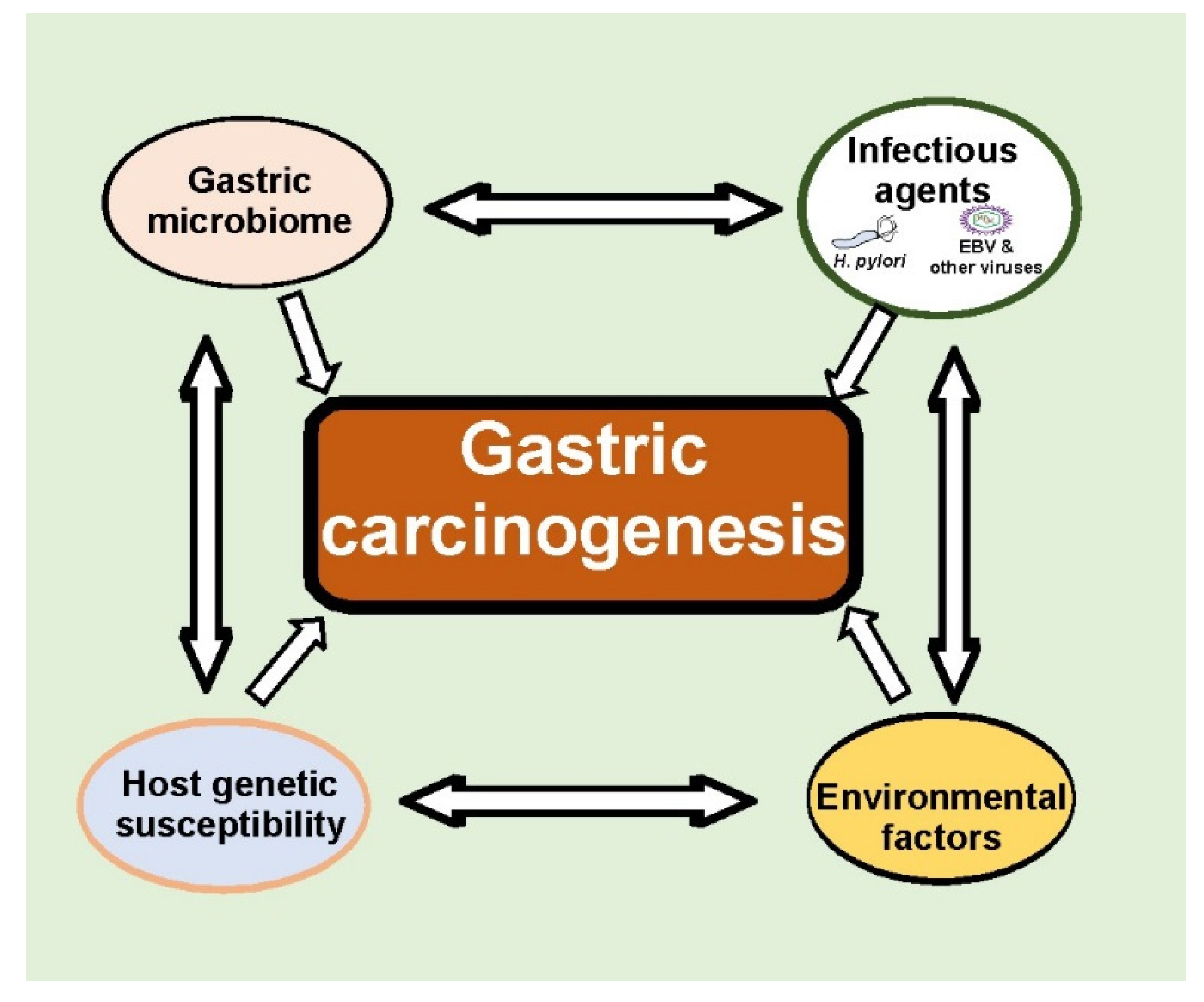
1. Introduction
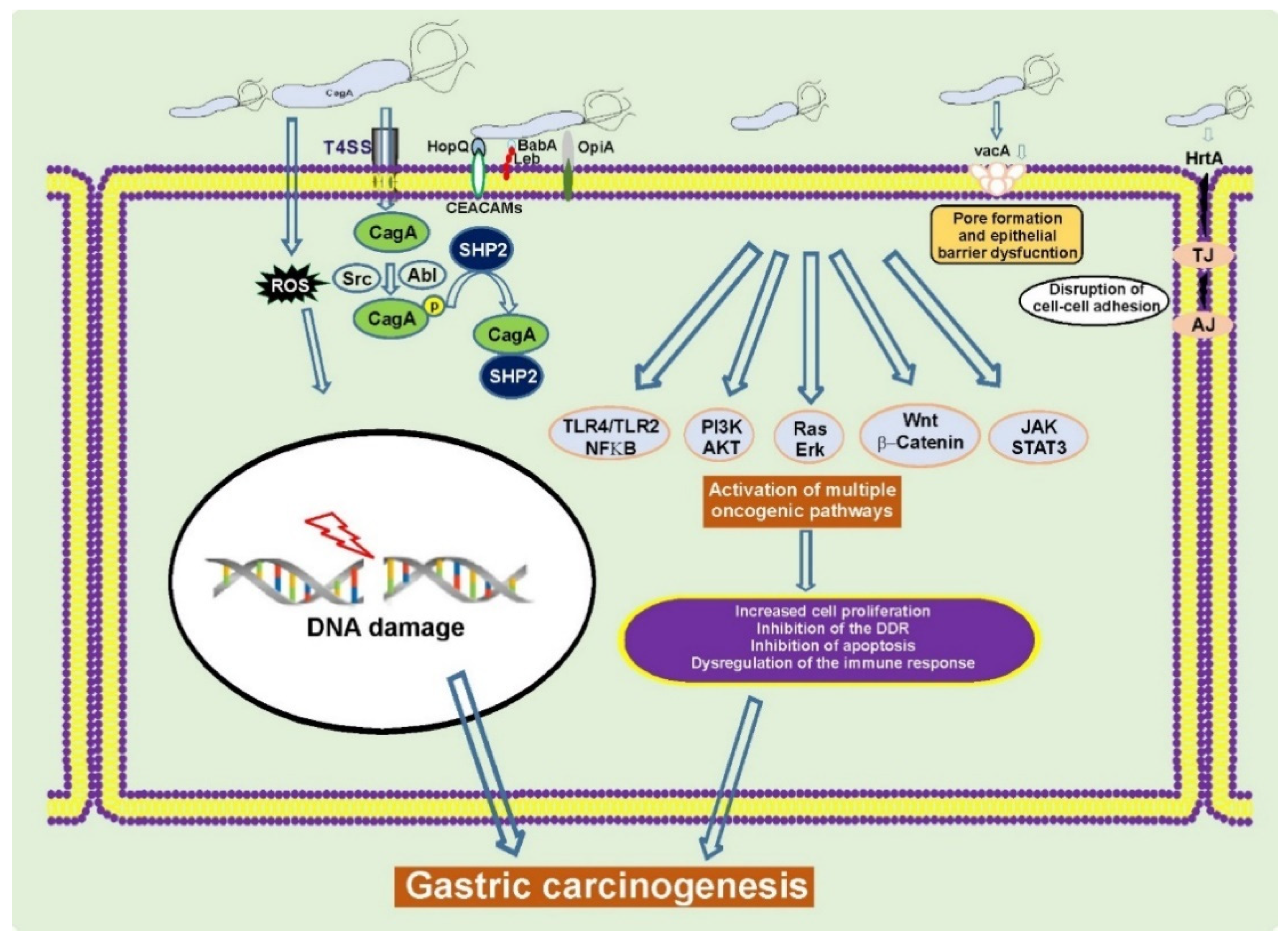
2. H. pylori and Gastric Cancer
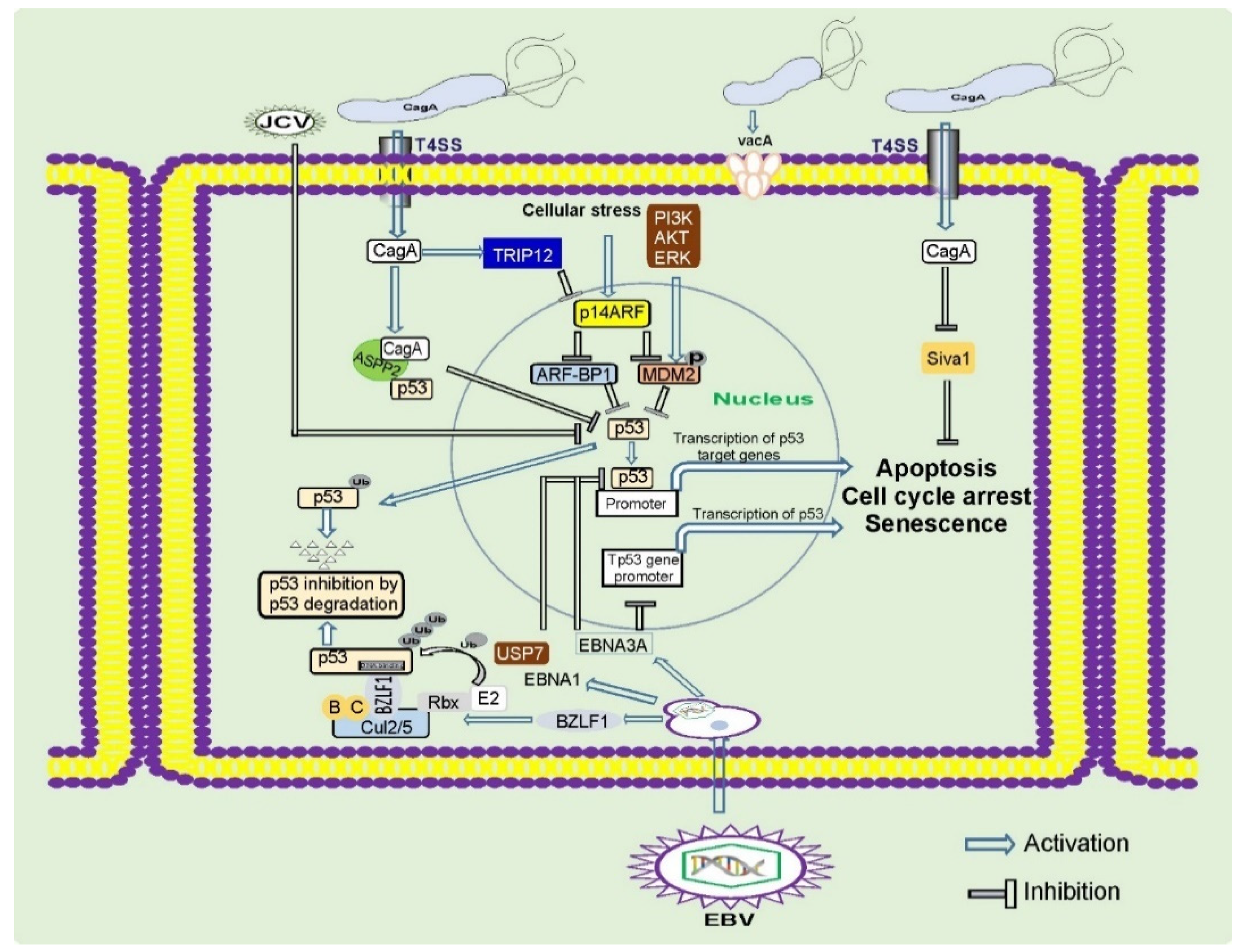
2.1. The cag Pathogenicity Island (cag PAI) and CagA Protein
2.2. VacA and Other Virulence Factors
3. Gastric Microbiota
4. EBV and Gastric Carcinogenesis
5. Other Carcinogenic Viruses
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System, 4th ed.; IARC: Lyon, France, 2010. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, G.; Hu, C. Molecular classification of gastric adenocarcinoma. Gastroenterol. Res. 2019, 12, 275–282. [Google Scholar] [CrossRef]
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983, 1, 1273–1275. [Google Scholar]
- Moss, S.F. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef]
- Correa, P. A human model of gastric carcinogenesis. Cancer Res. 1988, 48, 3554–3560. [Google Scholar] [PubMed]
- IARC working group on the evaluation of carcinogenic risks to humans. Schistosomes, liver flukes and Helicobacter pylori. Lyon, (FR): International Agency for Research on Cancer. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241.
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association between Helicobacter pylori eradication and gastric cancer incidence: A systematic review and meta-analysis. Gastroenterology 2016, 150, 1113–1124.e5. [Google Scholar] [CrossRef]
- Doorakkers, E.; Lagergren, J.; Engstrand, L.; Brusselaers, N. Helicobacter pylori eradication treatment and the risk of gastric adenocarcinoma in a Western population. Gut 2018, 67, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C.; Lam, S.K.; Wong, W.M.; Chen, J.S.; Zheng, T.T.; Feng, R.E.; Lai, K.C.; Hu, W.H.; Yuen, S.T.; Leung, S.Y.; et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. JAMA 2004, 291, 187–194. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Q.; Liang, X.; Liu, W.; Xiao, S.; Graham, D.Y.; Lu, H. Bismuth, lansoprazole, amoxicillin and metronidazole or clarithromycin as first-line Helicobacter pylori therapy. Gut 2015, 64, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. 1999, 79, 639–646. [Google Scholar] [PubMed]
- Saenz, J.B.; Vargas, N.; Mills, J.C. Tropism for spasmolytic polypeptide-expressing metaplasia allows Helicobacter pylori to expand its intragastric niche. Gastroenterology 2019, 156, 160–174.e7. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar]
- Naumann, M.; Sokolova, O.; Tegtmeyer, N.; Backert, S. Helicobacter pylori: A paradigm pathogen for subverting host cell signal transmission. Trends Microbiol. 2017, 25, 316–328. [Google Scholar] [CrossRef]
- Abadi, A.T.; Lee, Y.Y. Helicobacter pylori vacA as marker for gastric cancer and gastroduodenal diseases: One but not the only factor. J. Clin. Microbiol. 2014, 52, 4451. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vidal, Y.; Ponce-de-Leon, S.; Castillo-Rojas, G.; Barreto-Zuniga, R.; Torre-Delgadillo, A. High diversity of vacA and cagA Helicobacter pylori genotypes in patients with and without gastric cancer. PLoS ONE 2008, 3, e3849. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.T.; Crabtree, J.E. Immunology of Helicobacter pylori: Insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007, 133, 288–308. [Google Scholar] [CrossRef]
- Zaika, A.I.; Wei, J.; Noto, J.M.; Peek, R.M. Microbial regulation of p53 tumor suppressor. PLoS Pathog. 2015, 11, e1005099. [Google Scholar] [CrossRef]
- Stein, S.C.; Faber, E.; Bats, S.H.; Murillo, T.; Speidel, Y.; Coombs, N.; Josenhans, C. Helicobacter pylori modulates host cell responses by CagT4SS-dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog. 2017, 13, e1006514. [Google Scholar] [CrossRef]
- Varga, M.G.; Shaffer, C.L.; Sierra, J.C.; Suarez, G.; Piazuelo, M.B.; Whitaker, M.E.; Romero-Gallo, J.; Krishna, U.S.; Delgado, A.; Gomez, M.A.; et al. Pathogenic Helicobacter pylori strains translocate DNA and activate TLR9 via the cancer-associated cag type IV secretion system. Oncogene 2016, 35, 6262–6269. [Google Scholar] [CrossRef] [PubMed]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Memet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Naito, M.; Yamazaki, T.; Tsutsumi, R.; Higashi, H.; Onoe, K.; Yamazaki, S.; Azuma, T.; Hatakeyama, M. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology 2006, 130, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, J.; Gong, Y.; Yuan, Y. Association of CagA EPIYA-D or EPIYA-C phosphorylation sites with peptic ulcer and gastric cancer risks: A meta-analysis. Medicine 2017, 96, e6620. [Google Scholar] [CrossRef]
- Hatakeyama, M. Malignant Helicobacter pylori-associated diseases: Gastric cancer and MALT lymphoma. Adv. Exp. Med. Biol. 2019, 1149, 135–149. [Google Scholar] [CrossRef]
- Ren, S.; Higashi, H.; Lu, H.; Azuma, T.; Hatakeyama, M. Structural basis and functional consequence of Helicobacter pylori CagA multimerization in cells. J. Biol. Chem. 2006, 281, 32344–32352. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef]
- Huang, J.Q.; Zheng, G.F.; Sumanac, K.; Irvine, E.J.; Hunt, R.H. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology 2003, 125, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.I.; de Sousa, H.A.; Marcos-Pinto, R.; Dinis-Ribeiro, M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Peterson, T.S.; Kent, M.L.; Guillemin, K.H. pylori virulence factor CagA increases intestinal cell proliferation by Wnt pathway activation in a transgenic zebrafish model. Dis. Model. Mech. 2013, 6, 802–810. [Google Scholar] [CrossRef]
- Wandler, A.M.; Guillemin, K. Transgenic expression of the Helicobacter pylori virulence factor CagA promotes apoptosis or tumorigenesis through JNK activation in Drosophila. PLoS Pathog. 2012, 8, e1002939. [Google Scholar] [CrossRef]
- Wei, J.; Nagy, T.A.; Vilgelm, A.; Zaika, E.; Ogden, S.R.; Romero-Gallo, J.; Piazuelo, M.B.; Correa, P.; Washington, M.K.; El-Rifai, W.; et al. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 2010, 139, 1333–1343.e4. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Noto, J.M.; Wei, J.; Andl, C.; El-Rifai, W.; Peek, R.M.; Zaika, A.I. Helicobacter pylori bacteria alter the p53 stress response via ERK-HDM2 pathway. Oncotarget 2015, 6, 1531–1543. [Google Scholar] [CrossRef]
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Natl. Acad. Sci. USA 2012, 109, E2543–E2550. [Google Scholar] [CrossRef]
- Wei, J.; Noto, J.M.; Zaika, E.; Romero-Gallo, J.; Piazuelo, M.B.; Schneider, B.; El-Rifai, W.; Correa, P.; Peek, R.M.; Zaika, A.I. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut 2015, 64, 1040–1048. [Google Scholar] [CrossRef]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14ARF tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, H.; Herschenhous, N.; Kuzushita, N.; Moss, S.F. Helicobacter pylori increases proteasome-mediated degradation of p27(kip1) in gastric epithelial cells. Cancer Res. 2003, 63, 4739–4746. [Google Scholar] [PubMed]
- Palrasu, M.; Zaika, E.; El-Rifai, W.; Garcia-Buitrago, M.; Piazuelo, M.B.; Wilson, K.T.; Peek, R.M., Jr.; Zaika, A.I. Bacterial CagA protein compromises tumor suppressor mechanisms in gastric epithelial cells. J. Clin. Investig. 2020, 130, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Danese, S.; Sgambato, A.; Ardito, R.; Zannoni, G.; Rinelli, A.; Vecchio, F.M.; Gentiloni-Silveri, N.; Cittadini, A.; Gasbarrini, G.; et al. Role of Helicobacter pylori CagA+ infection in determining oxidative DNA damage in gastric mucosa. Scand. J. Gastroenterol. 2002, 37, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Uchida, T.; Tsukamoto, Y.; Watada, M.; Yamaguchi, N.; Yamamoto, K.; Shiota, S.; Moriyama, M.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect. Immun. 2014, 82, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Gruter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Muller, A.H. pylori-Induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-kappaB target gene expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef]
- Raza, Y.; Khan, A.; Farooqui, A.; Mubarak, M.; Facista, A.; Akhtar, S.S.; Khan, S.; Kazi, J.I.; Bernstein, C.; Kazmi, S.U. Oxidative DNA damage as a potential early biomarker of Helicobacter pylori associated carcinogenesis. Pathol. Oncol. Res. 2014, 20, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Ma, D.; Yao, Y.; Lynch, J.P.; Morales, K.; Ziober, A.; Feldman, M.; Ota, H.; Sepulveda, A.R. H. pylori infection is associated with DNA damage of Lgr5-positive epithelial stem cells in the stomach of patients with gastric cancer. Dig. Dis. Sci. 2013, 58, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Xu, L.Y.; Yang, Z.; Cao, X.M.; Li, W.; Lu, N.H. Expression of gammaH2AX in various gastric pathologies and its association with Helicobacter pylori infection. Oncol. Lett. 2014, 7, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zamperone, A.; Cohen, D.; Stein, M.; Viard, C.; Musch, A. Inhibition of polarity-regulating kinase PAR1b contributes to Helicobacter pylori inflicted DNA Double Strand Breaks in gastric cells. Cell Cycle 2019, 18, 299–311. [Google Scholar] [CrossRef]
- Machado, A.M.; Desler, C.; Boggild, S.; Strickertsson, J.A.; Friis-Hansen, L.; Figueiredo, C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection affects mitochondrial function and DNA repair, thus, mediating genetic instability in gastric cells. Mech. Ageing Dev. 2013, 134, 460–466. [Google Scholar] [CrossRef]
- Kim, J.J.; Tao, H.; Carloni, E.; Leung, W.K.; Graham, D.Y.; Sepulveda, A.R. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002, 123, 542–553. [Google Scholar] [CrossRef]
- Machado, A.M.; Figueiredo, C.; Touati, E.; Maximo, V.; Sousa, S.; Michel, V.; Carneiro, F.; Nielsen, F.C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin. Cancer Res. 2009, 15, 2995–3002. [Google Scholar] [CrossRef]
- Park, D.I.; Park, S.H.; Kim, S.H.; Kim, J.W.; Cho, Y.K.; Kim, H.J.; Sohn, C.I.; Jeon, W.K.; Kim, B.I.; Cho, E.Y.; et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter 2005, 10, 179–184. [Google Scholar] [CrossRef]
- Strickertsson, J.A.; Desler, C.; Rasmussen, L.J. Impact of bacterial infections on aging and cancer: Impairment of DNA repair and mitochondrial function of host cells. Exp. Gerontol. 2014, 56, 164–174. [Google Scholar] [CrossRef]
- Vallejo-Flores, G.; Torres, J.; Sandoval-Montes, C.; Arevalo-Romero, H.; Meza, I.; Camorlinga-Ponce, M.; Torres-Morales, J.; Chavez-Rueda, A.K.; Legorreta-Haquet, M.V.; Fuentes-Panana, E.M. Helicobacter pylori CagA suppresses apoptosis through activation of AKT in a nontransformed epithelial cell model of glandular acini formation. BioMed Res. Int. 2015, 2015, 761501. [Google Scholar] [CrossRef]
- Lin, W.C.; Tsai, H.F.; Kuo, S.H.; Wu, M.S.; Lin, C.W.; Hsu, P.I.; Cheng, A.L.; Hsu, P.N. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010, 70, 5740–5748. [Google Scholar] [CrossRef]
- Mimuro, H.; Suzuki, T.; Nagai, S.; Rieder, G.; Suzuki, M.; Nagai, T.; Fujita, Y.; Nagamatsu, K.; Ishijima, N.; Koyasu, S.; et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2007, 2, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.M.; Piazuelo, M.B.; Chaturvedi, R.; Bartel, C.A.; Thatcher, E.J.; Delgado, A.; Romero-Gallo, J.; Wilson, K.T.; Correa, P.; Patton, J.G.; et al. Strain-specific suppression of microRNA-320 by carcinogenic Helicobacter pylori promotes expression of the antiapoptotic protein Mcl-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G786–G796. [Google Scholar] [CrossRef]
- Abu-Taleb, A.M.F.; Abdelattef, R.S.; Abdel-Hady, A.A.; Omran, F.H.; El-Korashi, L.A.; Abdel-Aziz El-Hady, H.; El-Gebaly, A.M. Prevalence of Helicobacter pylori cagA and iceA genes and their association with gastrointestinal diseases. Int. J. Microbiol. 2018, 2018, 4809093. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Blaser, M.J. Pathophysiology of Helicobacter pylori-induced gastritis and peptic ulcer disease. Am. J. Med. 1997, 102, 200–207. [Google Scholar] [CrossRef]
- Figura, N.; Vindigni, C.; Covacci, A.; Presenti, L.; Burroni, D.; Vernillo, R.; Banducci, T.; Roviello, F.; Marrelli, D.; Biscontri, M.; et al. cagA positive and negative Helicobacter pylori strains are simultaneously present in the stomach of most patients with non-ulcer dyspepsia: Relevance to histological damage. Gut 1998, 42, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Tang, B.; Jia, Y.P.; Zhu, P.; Zhuang, Y.; Fang, Y.; Li, Q.; Wang, K.; Zhang, W.J.; Guo, G.; et al. Helicobacter pylori CagA protein negatively regulates autophagy and promotes inflammatory response via c-Met-PI3K/Akt-mTOR signaling pathway. Front. Cell. Infect. Microbiol. 2017, 7, 417. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Kashima, K.; Imanishi, J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 1997, 41, 442–451. [Google Scholar] [CrossRef]
- Wei, J.; O’Brien, D.; Vilgelm, A.; Piazuelo, M.B.; Correa, P.; Washington, M.K.; El-Rifai, W.; Peek, R.M.; Zaika, A. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 2008, 134, 1412–1423. [Google Scholar] [CrossRef]
- Bauer, B.; Pang, E.; Holland, C.; Kessler, M.; Bartfeld, S.; Meyer, T.F. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 2012, 11, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y.; et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol. Chem. 2004, 279, 17205–17216. [Google Scholar] [CrossRef]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; De Poire, E.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell. Microbiol. 2009, 11, 70–82. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Hartig, R.; Delahay, R.M.; Rohde, M.; Brandt, S.; Conradi, J.; Takahashi, S.; Smolka, A.J.; Sewald, N.; Backert, S. A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem. 2010, 285, 23515–23526. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wittelsberger, R.; Hartig, R.; Wessler, S.; Martinez-Quiles, N.; Backert, S. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe 2011, 9, 520–531. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Takahashi, A.; Azuma, T.; Higashi, H.; Hatakeyama, M. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol. Cell. Biol. 2006, 26, 261–276. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar] [CrossRef]
- Wang, W.C.; Wang, H.J.; Kuo, C.H. Two distinctive cell binding patterns by vacuolating toxin fused with glutathione S-transferase: One high-affinity m1-specific binding and the other lower-affinity binding for variant m forms. Biochemistry 2001, 40, 11887–11896. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Iwamoto, H.; Cover, T.L.; Shao, Z. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl. Acad. Sci. USA 1999, 96, 2001–2006. [Google Scholar] [CrossRef]
- Tombola, F.; Carlesso, C.; Szabo, I.; de Bernard, M.; Reyrat, J.M.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: Possible implications for the mechanism of cellular vacuolation. Biophys. J. 1999, 76, 1401–1409. [Google Scholar] [CrossRef]
- Pyburn, T.M.; Foegeding, N.J.; Gonzalez-Rivera, C.; McDonald, N.A.; Gould, K.L.; Cover, T.L.; Ohi, M.D. Structural organization of membrane-inserted hexamers formed by Helicobacter pylori VacA toxin. Mol. Microbiol. 2016, 102, 22–36. [Google Scholar] [CrossRef]
- Atherton, J.C.; Cao, P.; Peek, R.M., Jr.; Tummuru, M.K.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed]
- Boncristiano, M.; Paccani, S.R.; Barone, S.; Ulivieri, C.; Patrussi, L.; Ilver, D.; Amedei, A.; D’Elios, M.M.; Telford, J.L.; Baldari, C.T. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 2003, 198, 1887–1897. [Google Scholar] [CrossRef]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Torres, V.J.; Unutmaz, D.; Cover, T.L. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 2004, 101, 7727–7732. [Google Scholar] [CrossRef]
- Abdullah, M.; Greenfield, L.K.; Bronte-Tinkew, D.; Capurro, M.I.; Rizzuti, D.; Jones, N.L. VacA promotes CagA accumulation in gastric epithelial cells during Helicobacter pylori infection. Sci. Rep. 2019, 9, 38. [Google Scholar] [CrossRef]
- Van Doorn, L.J.; Figueiredo, C.; Sanna, R.; Pena, S.; Midolo, P.; Ng, E.K.; Atherton, J.C.; Blaser, M.J.; Quint, W.G. Expanding allelic diversity of Helicobacter pylori vacA. J. Clin. Microbiol. 1998, 36, 2597–2603. [Google Scholar] [CrossRef]
- Atherton, J.C.; Peek, R.M., Jr.; Tham, K.T.; Cover, T.L.; Blaser, M.J. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997, 112, 92–99. [Google Scholar] [CrossRef]
- Sinnett, C.G.; Letley, D.P.; Narayanan, G.L.; Patel, S.R.; Hussein, N.R.; Zaitoun, A.M.; Robinson, K.; Atherton, J.C. Helicobacter pylori vacA transcription is genetically-determined and stratifies the level of human gastric inflammation and atrophy. J. Clin. Pathol. 2016, 69, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, L.J.; Figueiredo, C.; Sanna, R.; Plaisier, A.; Schneeberger, P.; de Boer, W.; Quint, W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology 1998, 115, 58–66. [Google Scholar] [CrossRef]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.; Trust, T.J. Comparative genomics of Helicobacter pylori: Analysis of the outer membrane protein families. Infect. Immun. 2000, 68, 4155–4168. [Google Scholar] [CrossRef]
- Fujimoto, S.; Olaniyi Ojo, O.; Arnqvist, A.; Wu, J.Y.; Odenbreit, S.; Haas, R.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori BabA expression, gastric mucosal injury, and clinical outcome. Clin. Gastroenterol. Hepatol. 2007, 5, 49–58. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sonden, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Ishijima, N.; Suzuki, M.; Ashida, H.; Ichikawa, Y.; Kanegae, Y.; Saito, I.; Boren, T.; Haas, R.; Sasakawa, C.; Mimuro, H. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J. Biol. Chem. 2011, 286, 25256–25264. [Google Scholar] [CrossRef] [PubMed]
- Sheu, B.S.; Odenbreit, S.; Hung, K.H.; Liu, C.P.; Sheu, S.M.; Yang, H.B.; Wu, J.J. Interaction between host gastric Sialyl-Lewis X and H. pylori SabA enhances H. pylori density in patients lacking gastric Lewis B antigen. Am. J. Gastroenterol. 2006, 101, 36–44. [Google Scholar] [CrossRef]
- Azevedo, M.; Eriksson, S.; Mendes, N.; Serpa, J.; Figueiredo, C.; Resende, L.P.; Ruvoen-Clouet, N.; Haas, R.; Boren, T.; Le Pendu, J.; et al. Infection by Helicobacter pylori expressing the BabA adhesin is influenced by the secretor phenotype. J. Pathol. 2008, 215, 308–316. [Google Scholar] [CrossRef]
- Gerhard, M.; Lehn, N.; Neumayer, N.; Boren, T.; Rad, R.; Schepp, W.; Miehlke, S.; Classen, M.; Prinz, C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. USA 1999, 96, 12778–12783. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.L.; Huang, H.L.; Huang, B.S.; Chen, P.C.; Chen, C.S.; Wang, H.L.; Lin, P.H.; Chieh, M.S.; Wu, J.J.; Yang, J.C.; et al. Combination of OipA, BabA, and SabA as candidate biomarkers for predicting Helicobacter pylori-related gastric cancer. Sci. Rep. 2016, 6, 36442. [Google Scholar] [CrossRef]
- Yu, J.; Leung, W.K.; Go, M.Y.; Chan, M.C.; To, K.F.; Ng, E.K.; Chan, F.K.; Ling, T.K.; Chung, S.C.; Sung, J.J. Relationship between Helicobacter pylori babA2 status with gastric epithelial cell turnover and premalignant gastric lesions. Gut 2002, 51, 480–484. [Google Scholar] [CrossRef]
- Prinz, C.; Schoniger, M.; Rad, R.; Becker, I.; Keiditsch, E.; Wagenpfeil, S.; Classen, M.; Rosch, T.; Schepp, W.; Gerhard, M. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 2001, 61, 1903–1909. [Google Scholar]
- Lehours, P.; Menard, A.; Dupouy, S.; Bergey, B.; Richy, F.; Zerbib, F.; Ruskone-Fourmestraux, A.; Delchier, J.C.; Megraud, F. Evaluation of the association of nine Helicobacter pylori virulence factors with strains involved in low-grade gastric mucosa-associated lymphoid tissue lymphoma. Infect. Immun. 2004, 72, 880–888. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Ojo, O.; Fujimoto, S.; Odenbreit, S.; Haas, R.; Gutierrez, O.; El-Zimaity, H.M.; Reddy, R.; Arnqvist, A.; Graham, D.Y. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 2006, 55, 775–781. [Google Scholar] [CrossRef]
- Chen, M.Y.; He, C.Y.; Meng, X.; Yuan, Y. Association of Helicobacter pylori babA2 with peptic ulcer disease and gastric cancer. World J. Gastroenterol. 2013, 19, 4242–4251. [Google Scholar] [CrossRef]
- Zhao, Q.; Song, C.; Wang, K.; Li, D.; Yang, Y.; Liu, D.; Wang, L.; Zhou, N.; Xie, Y. Prevalence of Helicobacter pylori babA, oipA, sabA, and homB genes in isolates from Chinese patients with different gastroduodenal diseases. Med. Microbiol. Immunol. 2020, 209, 565–577. [Google Scholar] [CrossRef]
- Bartpho, T.S.; Wattanawongdon, W.; Tongtawee, T.; Paoin, C.; Kangwantas, K.; Dechsukhum, C. Precancerous gastric lesions with Helicobacter pylori vacA (+)/babA2(+)/oipA (+) genotype increase the risk of gastric cancer. BioMed Res. Int. 2020, 2020, 7243029. [Google Scholar] [CrossRef]
- Liu, J.; He, C.; Chen, M.; Wang, Z.; Xing, C.; Yuan, Y. Association of presence/absence and on/off patterns of Helicobacter pylori oipA gene with peptic ulcer disease and gastric cancer risks: A meta-analysis. BMC Infect. Dis. 2013, 13, 555. [Google Scholar] [CrossRef]
- Cao, P.; Lee, K.J.; Blaser, M.J.; Cover, T.L. Analysis of hopQ alleles in East Asian and Western strains of Helicobacter pylori. FEMS Microbiol. Lett. 2005, 251, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Leylabadlo, H.E.; Yekani, M.; Ghotaslou, R. Helicobacter pylori hopQ alleles (type I and II) in gastric cancer. Biomed. Rep. 2016, 4, 601–604. [Google Scholar] [CrossRef]
- Jung, S.W.; Sugimoto, M.; Graham, D.Y.; Yamaoka, Y. homB status of Helicobacter pylori as a novel marker to distinguish gastric cancer from duodenal ulcer. J. Clin. Microbiol. 2009, 47, 3241–3245. [Google Scholar] [CrossRef]
- Kang, J.; Jones, K.R.; Jang, S.; Olsen, C.H.; Yoo, Y.J.; Merrell, D.S.; Cha, J.H. The geographic origin of Helicobacter pylori influences the association of the homB gene with gastric cancer. J. Clin. Microbiol. 2012, 50, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.T.B.; Rafiei, A.; Ajami, A.; Hosseini, V.; Taghvaei, T.; Jones, K.R.; Merrell, D.S. Helicobacter pylori homB, but not cagA, is associated with gastric cancer in Iran. J. Clin. Microbiol. 2011, 49, 3191–3197. [Google Scholar] [CrossRef]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Wong, G.L.; To, K.F.; Wong, V.W.; Lai, L.H.; Chow, D.K.; Lau, J.Y.; Sung, J.J.; Ding, C. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS ONE 2009, 4, e7985. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Backhed, F.; Nyren, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Matsumoto, A.; Tanaka, H.; Matsumura, I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett. 2018, 414, 147–152. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Chen, X.H.; Wang, A.; Chu, A.N.; Gong, Y.H.; Yuan, Y. Mucosa-associated microbiota in gastric cancer tissues compared with non-cancer tissues. Front. Microbiol. 2019, 10, 1261. [Google Scholar] [CrossRef]
- Aebischer, T.; Fischer, A.; Walduck, A.; Schlotelburg, C.; Lindig, M.; Schreiber, S.; Meyer, T.F.; Bereswill, S.; Gobel, U.B. Vaccination prevents Helicobacter pylori-induced alterations of the gastric flora in mice. FEMS Immunol. Med. Microbiol. 2006, 46, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Contreras, A.; Goldfarb, K.C.; Godoy-Vitorino, F.; Karaoz, U.; Contreras, M.; Blaser, M.J.; Brodie, E.L.; Dominguez-Bello, M.G. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011, 5, 574–579. [Google Scholar] [CrossRef]
- Noto, J.M.; Rose, K.L.; Hachey, A.J.; Delgado, A.G.; Romero-Gallo, J.; Wroblewski, L.E.; Schneider, B.G.; Shah, S.C.; Cover, T.L.; Wilson, K.T.; et al. Carcinogenic Helicobacter pylori strains selectively dysregulate the in vivo gastric proteome, which may be associated with stomach cancer progression. Mol. Cell. Proteom. 2019, 18, 352–371. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Matsuki, T.; Asahara, T.; Zaman, C.; Hanawa, T.; Yonezawa, H.; Kurata, S.; Woo, T.D.; Nomoto, K.; Kamiya, S. Comparative analysis of gastric bacterial microbiota in Mongolian gerbils after long-term infection with Helicobacter pylori. Microb. Pathog. 2012, 53, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Q.; Monstein, H.J.; Nilsson, L.E.; Petersson, F.; Borch, K. Profiling and identification of eubacteria in the stomach of Mongolian gerbils with and without Helicobacter pylori infection. Helicobacter 2003, 8, 149–157. [Google Scholar] [CrossRef]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori persistence: Biology and disease. J. Clin. Investig. 2004, 113, 321–333. [Google Scholar] [CrossRef]
- Sanduleanu, S.; Jonkers, D.; De Bruine, A.; Hameeteman, W.; Stockbrugger, R.W. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: Differential findings in gastric juice and gastric mucosa. Aliment. Pharmacol. Ther. 2001, 15, 379–388. [Google Scholar] [CrossRef]
- Mowat, C.; Williams, C.; Gillen, D.; Hossack, M.; Gilmour, D.; Carswell, A.; Wirz, A.; Preston, T.; McColl, K.E. Omeprazole, Helicobacter pylori status, and alterations in the intragastric milieu facilitating bacterial N-nitrosation. Gastroenterology 2000, 119, 339–347. [Google Scholar] [CrossRef]
- Khosravi, Y.; Dieye, Y.; Poh, B.H.; Ng, C.G.; Loke, M.F.; Goh, K.L.; Vadivelu, J. Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients. Sci. World J. 2014, 2014, 610421. [Google Scholar] [CrossRef]
- Tan, M.P.; Kaparakis, M.; Galic, M.; Pedersen, J.; Pearse, M.; Wijburg, O.L.; Janssen, P.H.; Strugnell, R.A. Chronic Helicobacter pylori infection does not significantly alter the microbiota of the murine stomach. Appl. Environ. Microbiol. 2007, 73, 1010–1013. [Google Scholar] [CrossRef]
- Eun, C.S.; Kim, B.K.; Han, D.S.; Kim, S.Y.; Kim, K.M.; Choi, B.Y.; Song, K.S.; Kim, Y.S.; Kim, J.F. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 2014, 19, 407–416. [Google Scholar] [CrossRef]
- Aviles-Jimenez, F.; Vazquez-Jimenez, F.; Medrano-Guzman, R.; Mantilla, A.; Torres, J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci. Rep. 2014, 4, 4202. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Dicksved, J.; Lindberg, M.; Rosenquist, M.; Enroth, H.; Jansson, J.K.; Engstrand, L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J. Med. Microbiol. 2009, 58, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019, 40, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Lin, J.T.; Ho, H.J.; Lai, Z.L.; Wang, C.B.; Tang, S.L.; Wu, C.Y. Gastric microbiota and predicted gene functions are altered after subtotal gastrectomy in patients with gastric cancer. Sci. Rep. 2016, 6, 20701. [Google Scholar] [CrossRef]
- Fox, J.G.; Li, X.; Cahill, R.J.; Andrutis, K.; Rustgi, A.K.; Odze, R.; Wang, T.C. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology 1996, 110, 155–166. [Google Scholar] [CrossRef]
- Fox, J.G.; Rogers, A.B.; Ihrig, M.; Taylor, N.S.; Whary, M.T.; Dockray, G.; Varro, A.; Wang, T.C. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003, 63, 942–950. [Google Scholar]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220.e4. [Google Scholar] [CrossRef]
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut 2014, 63, 54–63. [Google Scholar] [CrossRef]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Muthupalani, S.; Takaishi, S.; Yang, P.; Wang, T.C.; Fox, J.G. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009, 69, 8166–8174. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Ge, Z.; Wang, T.C.; Fox, J.G. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008, 68, 3540–3548. [Google Scholar] [CrossRef] [PubMed]
- Rolig, A.S.; Cech, C.; Ahler, E.; Carter, J.E.; Ottemann, K.M. The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect. Immun. 2013, 81, 1382–1389. [Google Scholar] [CrossRef]
- Leach, S.A.; Thompson, M.; Hill, M. Bacterially catalysed N-nitrosation reactions and their relative importance in the human stomach. Carcinogenesis 1987, 8, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Stockbruegger, R.W. Bacterial overgrowth as a consequence of reduced gastric acidity. Scand. J. Gastroenterol. Suppl. 1985, 111, 7–16. [Google Scholar] [CrossRef]
- Lemke, L.B.; Ge, Z.; Whary, M.T.; Feng, Y.; Rogers, A.B.; Muthupalani, S.; Fox, J.G. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates proinflammatory H. pylori-induced gastric pathology. Infect. Immun. 2009, 77, 2147–2158. [Google Scholar] [CrossRef]
- Ge, Z.; Feng, Y.; Muthupalani, S.; Eurell, L.L.; Taylor, N.S.; Whary, M.T.; Fox, J.G. Coinfection with Enterohepatic Helicobacter species can ameliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 mice. Infect. Immun. 2011, 79, 3861–3871. [Google Scholar] [CrossRef]
- Chen, Y.H.; Tsai, W.H.; Wu, H.Y.; Chen, C.Y.; Yeh, W.L.; Chen, Y.H.; Hsu, H.Y.; Chen, W.W.; Chen, Y.W.; Chang, W.W.; et al. Probiotic Lactobacillus spp. act against Helicobacter pylori-induced inflammation. J. Clin. Med. 2019, 8, 90. [Google Scholar] [CrossRef]
- Kabir, A.M.; Aiba, Y.; Takagi, A.; Kamiya, S.; Miwa, T.; Koga, Y. Prevention of Helicobacter pylori infection by lactobacilli in a gnotobiotic murine model. Gut 1997, 41, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Zaman, C.; Osaki, T.; Hanawa, T.; Yonezawa, H.; Kurata, S.; Kamiya, S. Analysis of the microbial ecology between Helicobacter pylori and the gastric microbiota of Mongolian gerbils. J. Med. Microbiol. 2014, 63, 129–137. [Google Scholar] [CrossRef][Green Version]
- Schmitz, J.M.; Durham, C.G.; Schoeb, T.R.; Soltau, T.D.; Wolf, K.J.; Tanner, S.M.; McCracken, V.J.; Lorenz, R.G. Helicobacter felis—Associated gastric disease in microbiota-restricted mice. J. Histochem. Cytochem. 2011, 59, 826–841. [Google Scholar] [CrossRef]
- Sakamoto, I.; Igarashi, M.; Kimura, K.; Takagi, A.; Miwa, T.; Koga, Y. Suppressive effect of Lactobacillus gasseri OLL 2716 (LG21) on Helicobacter pylori infection in humans. J. Antimicrob. Chemother. 2001, 47, 709–710. [Google Scholar] [CrossRef] [PubMed]
- De Klerk, N.; Maudsdotter, L.; Gebreegziabher, H.; Saroj, S.D.; Eriksson, B.; Eriksson, O.S.; Roos, S.; Linden, S.; Sjolinder, H.; Jonsson, A.B. Lactobacilli reduce Helicobacter pylori attachment to host gastric epithelial cells by inhibiting adhesion gene expression. Infect. Immun. 2016, 84, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Scholte, L.L.S.; Pascoal-Xavier, M.A.; Nahum, L.A. Helminths and cancers from the evolutionary perspective. Front. Med. 2018, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Whary, M.T.; Sundina, N.; Bravo, L.E.; Correa, P.; Quinones, F.; Caro, F.; Fox, J.G. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: Possible implications for gastric carcinogenesis. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Whary, M.T.; Muthupalani, S.; Ge, Z.; Feng, Y.; Lofgren, J.; Shi, H.N.; Taylor, N.S.; Correa, P.; Versalovic, J.; Wang, T.C.; et al. Helminth co-infection in Helicobacter pylori infected INS-GAS mice attenuates gastric premalignant lesions of epithelial dysplasia and glandular atrophy and preserves colonization resistance of the stomach to lower bowel microbiota. Microbes Infect. 2014, 16, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Fuenmayor-Boscan, A.; Hernandez-Rincon, I.; Arismendi-Morillo, G.; Mengual, E.; Rivero, Z.; Romero, G.; Lizarzabal, M.; Alvarez-Mon, M. Changes in the severity of gastric mucosal inflammation associated with Helicobacter pylori in humans coinfected with intestinal helminths. Indian J. Gastroenterol. 2020, 39, 186–195. [Google Scholar] [CrossRef]
- Fattahi, S.; Kosari-Monfared, M.; Ghadami, E.; Golpour, M.; Khodadadi, P.; Ghasemiyan, M.; Akhavan-Niaki, H. Infection-associated epigenetic alterations in gastric cancer: New insight in cancer therapy. J. Cell Physiol. 2018, 233, 9261–9270. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. Gastric cancer: Genome damaged by bugs. Oncogene 2020, 39, 3427–3442. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Lieberman, P.M. Virology. Epstein-Barr virus turns 50. Science 2014, 343, 1323–1325. [Google Scholar] [CrossRef]
- Cardenas-Mondragon, M.G.; Carreon-Talavera, R.; Camorlinga-Ponce, M.; Gomez-Delgado, A.; Torres, J.; Fuentes-Panana, E.M. Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS ONE 2013, 8, e62850. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8; IARC: Lyon, France, 1997; Volume 70. [Google Scholar]
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Camargo, M.C.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein(-)Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef]
- Neyts, J.; Sadler, R.; De Clercq, E.; Raab-Traub, N.; Pagano, J.S. The antiviral agent cidofovir [(S)-1-(3-hydroxy-2-phosphonyl-methoxypropyl)cytosine] has pronounced activity against nasopharyngeal carcinoma grown in nude mice. Cancer Res. 1998, 58, 384–388. [Google Scholar]
- Yoshizaki, T.; Wakisaka, N.; Kondo, S.; Murono, S.; Shimizu, Y.; Nakashima, M.; Tsuji, A.; Furukawa, M. Treatment of locally recurrent Epstein-Barr virus-associated nasopharyngeal carcinoma using the anti-viral agent cidofovir. J. Med. Virol. 2008, 80, 879–882. [Google Scholar] [CrossRef]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein(-)Barr Virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef]
- Kang, B.W.; Baek, D.W.; Kang, H.; Baek, J.H.; Kim, J.G. Novel Therapeutic Approaches for Epstein-Barr Virus Associated Gastric Cancer. Anticancer Res. 2019, 39, 4003–4010. [Google Scholar] [CrossRef]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef]
- Gratama, J.W.; Ernberg, I. Molecular epidemiology of Epstein-Barr virus infection. Adv. Cancer Res. 1995, 67, 197–255. [Google Scholar]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.; Deitsch, K.W.; Duca, K.A.; Torgbor, C. The link between Plasmodium falciparum malaria and endemic Burkitt’s lymphoma-new insight into a 50-year-old enigma. PLoS Pathog. 2016, 12, e1005331. [Google Scholar] [CrossRef]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Z.; Han, L.; Liu, S.; Luo, B. Epstein-Barr virus infection in gastric remnant carcinoma and recurrent gastric carcinoma in Qingdao of Northern China. PLoS ONE 2016, 11, e0148342. [Google Scholar] [CrossRef]
- Choi, M.G.; Jeong, J.Y.; Kim, K.M.; Bae, J.M.; Noh, J.H.; Sohn, T.S.; Kim, S. Clinical significance of gastritis cystica profunda and its association with Epstein-Barr virus in gastric cancer. Cancer 2012, 118, 5227–5233. [Google Scholar] [CrossRef]
- Burke, A.P.; Yen, T.S.; Shekitka, K.M.; Sobin, L.H. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod. Pathol. 1990, 3, 377–380. [Google Scholar]
- Shibata, D.; Weiss, L.M. Epstein-Barr virus-associated gastric adenocarcinoma. Am. J. Pathol. 1992, 140, 769–774. [Google Scholar] [PubMed]
- Dong, M.; Wang, H.Y.; Zhao, X.X.; Chen, J.N.; Zhang, Y.W.; Huang, Y.; Xue, L.; Li, H.G.; Du, H.; Wu, X.Y.; et al. Expression and prognostic roles of PIK3CA, JAK2, PD-L1, and PD-L2 in Epstein-Barr virus-associated gastric carcinoma. Hum. Pathol. 2016, 53, 25–34. [Google Scholar] [CrossRef]
- Camargo, M.C.; Bowlby, R.; Chu, A.; Pedamallu, C.S.; Thorsson, V.; Elmore, S.; Mungall, A.J.; Bass, A.J.; Gulley, M.L.; Rabkin, C.S. Validation and calibration of next-generation sequencing to identify Epstein-Barr virus-positive gastric cancer in The Cancer Genome Atlas. Gastric Cancer 2016, 19, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Sadato, D.; Ogawa, M.; Hirama, C.; Hishima, T.; Horiguchi, S.I.; Harada, Y.; Shimoyama, T.; Itokawa, M.; Ohashi, K.; Oboki, K. Potential prognostic impact of EBV RNA-seq reads in gastric cancer: A reanalysis of The Cancer Genome Atlas cohort. FEBS Open Bio 2020, 10, 455–467. [Google Scholar] [CrossRef]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein–Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Swan, K.; Zhang, X.; Cao, S.; Brett, Z.; Drury, S.; Strong, M.J.; Fewell, C.; Puetter, A.; Wang, X.; et al. Secreted Oral Epithelial Cell Membrane Vesicles Induce Epstein-Barr Virus Reactivation in Latently Infected B Cells. J. Virol. 2016, 90, 3469–3479. [Google Scholar] [CrossRef]
- Luo, B.; Wang, Y.; Wang, X.F.; Liang, H.; Yan, L.P.; Huang, B.H.; Zhao, P. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinomas. World J. Gastroenterol. 2005, 11, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Imai, S.; Tokunaga, M.; Koizumi, S.; Uchizawa, M.; Okamoto, K.; Osato, T. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: Unique viral latency in the tumour cells. Br. J. Cancer 1996, 74, 625–631. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef]
- Hino, R.; Uozaki, H.; Inoue, Y.; Shintani, Y.; Ushiku, T.; Sakatani, T.; Takada, K.; Fukayama, M. Survival advantage of EBV-associated gastric carcinoma: Survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 2008, 68, 1427–1435. [Google Scholar] [CrossRef]
- Pal, A.D.; Basak, N.P.; Banerjee, A.S.; Banerjee, S. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis 2014, 35, 1592–1601. [Google Scholar] [CrossRef]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef]
- Sivachandran, N.; Dawson, C.W.; Young, L.S.; Liu, F.F.; Middeldorp, J.; Frappier, L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J. Virol. 2012, 86, 60–68. [Google Scholar] [CrossRef]
- Chavez-Calvillo, G.; Martin, S.; Hamm, C.; Sztuba-Solinska, J. The structure-to-function relationships of gammaherpesvirus-encoded long non-coding RNAs and their contributions to viral pathogenesis. Noncoding RNA 2018, 4, 24. [Google Scholar] [CrossRef]
- Namba-Fukuyo, H.; Funata, S.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Mano, Y.; Fukayama, M.; Aburatani, H.; Kaneda, A. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget 2016, 7, 81512–81526. [Google Scholar] [CrossRef]
- Wang, J.; Liu, W.; Zhang, X.; Zhang, Y.; Xiao, H.; Luo, B. LMP2A induces DNA methylation and expression repression of AQP3 in EBV-associated gastric carcinoma. Virology 2019, 534, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Geddert, H.; zur Hausen, A.; Gabbert, H.E.; Sarbia, M. EBV-infection in cardiac and non-cardiac gastric adenocarcinomas is associated with promoter methylation of p16, p14 and APC, but not hMLH1. Cell. Oncol. 2011, 34, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liang, Q.; Cheung, K.F.; Kang, W.; Dong, Y.; Lung, R.W.; Tong, J.H.; To, K.F.; Sung, J.J.; Yu, J. Somatostatin receptor 1, a novel EBV-associated CpG hypermethylated gene, contributes to the pathogenesis of EBV-associated gastric cancer. Br. J. Cancer 2013, 108, 2557–2564. [Google Scholar] [CrossRef]
- He, D.; Zhang, Y.W.; Zhang, N.N.; Zhou, L.; Chen, J.N.; Jiang, Y.; Shao, C.K. Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Med. Oncol. 2015, 32, 92. [Google Scholar] [CrossRef]
- Yu, J.; Liang, Q.; Wang, J.; Wang, K.; Gao, J.; Zhang, J.; Zeng, Y.; Chiu, P.W.; Ng, E.K.; Sung, J.J. REC8 functions as a tumor suppressor and is epigenetically downregulated in gastric cancer, especially in EBV-positive subtype. Oncogene 2017, 36, 182–193. [Google Scholar] [CrossRef]
- Ushiku, T.; Chong, J.M.; Uozaki, H.; Hino, R.; Chang, M.S.; Sudo, M.; Rani, B.R.; Sakuma, K.; Nagai, H.; Fukayama, M. p73 gene promoter methylation in Epstein-Barr virus-associated gastric carcinoma. Int. J. Cancer 2007, 120, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.T.; Chang, Y.T.; Chen, S.C.; Chuang, Y.C.; Chen, Y.R.; Lin, C.S.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005, 24, 2635–2646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gutsch, D.; Kenney, S. Functional and physical interaction between p53 and BZLF1: Implications for Epstein-Barr virus latency. Mol. Cell. Biol. 1994, 14, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Gossai, A.; Waterboer, T.; Nelson, H.H.; Michel, A.; Willhauck-Fleckenstein, M.; Farzan, S.F.; Hoen, A.G.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; et al. Seroepidemiology of human polyomaviruses in a US population. Am. J. Epidemiol. 2016, 183, 61–69. [Google Scholar] [CrossRef]
- Lundstig, A.; Dillner, J. Serological diagnosis of human polyomavirus infection. Adv. Exp. Med. Biol. 2006, 577, 96–101. [Google Scholar] [CrossRef]
- Del Valle, L.; White, M.K.; Enam, S.; Pina Oviedo, S.; Bromer, M.Q.; Thomas, R.M.; Parkman, H.P.; Khalili, K. Detection of JC virus DNA sequences and expression of viral T antigen and agnoprotein in esophageal carcinoma. Cancer 2005, 103, 516–527. [Google Scholar] [CrossRef]
- Hori, R.; Murai, Y.; Tsuneyama, K.; Abdel-Aziz, H.O.; Nomoto, K.; Takahashi, H.; Cheng, C.M.; Kuchina, T.; Harman, B.V.; Takano, Y. Detection of JC virus DNA sequences in colorectal cancers in Japan. Virchows Arch. 2005, 447, 723–730. [Google Scholar] [CrossRef]
- Murai, Y.; Zheng, H.C.; Abdel Aziz, H.O.; Mei, H.; Kutsuna, T.; Nakanishi, Y.; Tsuneyama, K.; Takano, Y. High JC virus load in gastric cancer and adjacent non-cancerous mucosa. Cancer Sci. 2007, 98, 25–31. [Google Scholar] [CrossRef]
- Shin, S.K.; Li, M.S.; Fuerst, F.; Hotchkiss, E.; Meyer, R.; Kim, I.T.; Goel, A.; Boland, C.R. Oncogenic T-antigen of JC virus is present frequently in human gastric cancers. Cancer 2006, 107, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Ksiaa, F.; Ziadi, S.; Mokni, M.; Korbi, S.; Trimeche, M. The presence of JC virus in gastric carcinomas correlates with patient’s age, intestinal histological type and aberrant methylation of tumor suppressor genes. Mod. Pathol. 2010, 23, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, S.; Yamamoto, H.; Nosho, K.; Taniguchi, H.; Adachi, Y.; Sasaki, S.; Arimura, Y.; Imai, K.; Shinomura, Y. Genetic and epigenetic characteristics of gastric cancers with JC virus T-antigen. World J. Gastroenterol. 2009, 15, 5579–5585. [Google Scholar] [CrossRef] [PubMed]
- Baez, C.F.; Brandao Varella, R.; Villani, S.; Delbue, S. Human Polyomaviruses: The battle of large and small tumor antigens. Virol. Res. Treat. 2017, 8. [Google Scholar] [CrossRef]
- Caracciolo, V.; Macaluso, M.; D’Agostino, L.; Montanari, M.; Scheff, J.; Reiss, K.; Khalili, K.; Giordano, A. Cross-talk between T-Ag presence and pRb family and p53/p73 signaling in mouse and human medulloblastoma. J. Cell. Biochem. 2010, 110, 182–190. [Google Scholar] [CrossRef]
- Enam, S.; Del Valle, L.; Lara, C.; Gan, D.D.; Ortiz-Hidalgo, C.; Palazzo, J.P.; Khalili, K. Association of human polyomavirus JCV with colon cancer: Evidence for interaction of viral T-antigen and beta-catenin. Cancer Res. 2002, 62, 7093–7101. [Google Scholar]
- Al Mana, H.; Yassine, H.M.; Younes, N.N.; Al-Mohannadi, A.; Al-Sadeq, D.W.; Alhababi, D.; Nasser, E.A.; Nasrallah, G.K. The current status of Cytomegalovirus (CMV) prevalence in the MENA region: A systematic review. Pathogens 2019, 8, 213. [Google Scholar] [CrossRef]
- Lv, Y.L.; Han, F.F.; An, Z.L.; Jia, Y.; Xuan, L.L.; Gong, L.L.; Zhang, W.; Ren, L.L.; Yang, S.; Liu, H.; et al. Cytomegalovirus infection is a risk factor in gastrointestinal cancer: A cross-sectional and meta-analysis study. Intervirology 2020, 63, 10–16. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, G.; Xu, J.; Sun, X.; Chen, W.; Jin, J.; Hu, C.; Zhang, P.; Shen, X.; Xue, X. Human cytomegalovirus detection in gastric cancer and its possible association with lymphatic metastasis. Diagn. Microbiol. Infect. Dis. 2017, 88, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hu, C.; Wang, P.; Chen, J.; Wu, T.; Chen, W.; Ye, L.; Zhu, G.; Zhang, L.; Xue, X.; et al. Latent infection of human cytomegalovirus is associated with the development of gastric cancer. Oncol. Lett. 2014, 8, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Nikbakhsh, N.; Taheri, H.; Ghadami, E.; Kosari-Monfared, M.; Amirbozorgi, G.; Asouri, M.; Pilehchian-Langroudi, M.; Ranaee, M.; Samadani, A.A.; et al. Prevalence of multiple infections and the risk of gastric adenocarcinoma development at earlier age. Diagn. Microbiol. Infect. Dis. 2018, 92, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kosari-Monfared, M.; Nikbakhsh, N.; Fattahi, S.; Ghadami, E.; Ranaei, M.; Taheri, H.; Amjadi-Moheb, F.; Godazandeh, G.A.; Shafaei, S.; Pilehchian-Langroudi, M.; et al. CTNNBIP1 downregulation is associated with tumor grade and viral infections in gastric adenocarcinoma. J. Cell. Physiol. 2019, 234, 2895–2904. [Google Scholar] [CrossRef]
- Cui, H.; Jin, Y.; Chen, F.; Ni, H.; Hu, C.; Xu, Y.; Xuan, H.; Hu, D.; Deng, W.; Zhang, Y.; et al. Clinicopathological evidence of hepatitis B virus infection in the development of gastric adenocarcinoma. J. Med. Virol. 2020, 92, 71–77. [Google Scholar] [CrossRef]
- Baghbanian, M.; Hoseini Mousa, S.A.; Doosti, M.; Moghimi, M. Association between gastric pathology and Hepatitis B virus infection in patients with or without Helicobacter pylori. Asian Pac. J. Cancer Prev. 2019, 20, 2177–2180. [Google Scholar] [CrossRef]
- Chen, C.W.; Cheng, J.S.; Chen, T.D.; Le, P.H.; Ku, H.P.; Chang, M.L. The irreversible HCV-associated risk of gastric cancer following interferon-based therapy: A joint study of hospital-based cases and nationwide population-based cohorts. Ther. Adv. Gastroenterol. 2019, 12, 1756284819855732. [Google Scholar] [CrossRef]
- Kai, K.; Miyoshi, A.; Kitahara, K.; Masuda, M.; Takase, Y.; Miyazaki, K.; Noshiro, H.; Tokunaga, O. Analysis of extrahepatic multiple primary malignancies in patients with hepatocellular carcinoma according to viral infection status. Int. J. Hepatol. 2012, 2012, 495950. [Google Scholar] [CrossRef]
- Kocoglu, H.; Karaca, M.; Tural, D.; Hocaoglu, E.; Okuturlar, Y.; Fetullahoglu, Z.; Gunaldi, M.; Ciftci, R.; Tuna, S.; Yucil, O.K.; et al. Hepatitis B and C rates are significantly increased in certain solid tumors: A large retrospective study. J. Cancer Res. Ther. 2018, 14, S774–S778. [Google Scholar] [CrossRef]
- Song, C.; Lv, J.; Liu, Y.; Chen, J.G.; Ge, Z.; Zhu, J.; Dai, J.; Du, L.B.; Yu, C.; Guo, Y.; et al. Associations between Hepatitis B virus infection and risk of all cancer types. JAMA Netw. Open 2019, 2, e195718. [Google Scholar] [CrossRef]
- Huang, C.F.; Lai, H.C.; Chen, C.Y.; Tseng, K.C.; Kuo, H.T.; Hung, C.H.; Wang, J.H.; Chen, J.J.; Lee, P.L.; Chien, R.N.; et al. Extrahepatic malignancy among patients with chronic Hepatitis C after antiviral therapy: A real-world nationwide Study on Taiwanese chronic Hepatitis C cohort (T-COACH). Am. J. Gastroenterol. 2020. [Google Scholar] [CrossRef]
- Kang, J.S.; Lee, S.H.; Lee, S.; Kim, G.H.; Park, Y.J.; Han, I.S.; Lee, J.E.; Lee, S.O.; Moon, C. Role of upper gastrointestinal endoscopy in patients with human immunodeficiency virus infection in the era of combination antiretroviral therapy. Infect. Chemother. 2019, 51, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Schierhout, G.; McGregor, S.; Gessain, A.; Einsiedel, L.; Martinello, M.; Kaldor, J. Association between HTLV-1 infection and adverse health outcomes: A systematic review and meta-analysis of epidemiological studies. Lancet Infect. Dis. 2020, 20, 133–143. [Google Scholar] [CrossRef]
- Falasca, F.; Maida, P.; Gaeta, A.; Verzaro, S.; Mezzaroma, I.; Fantauzzi, A.; Donato, G.; Bonci, E.; Castilletti, C.; Antonelli, G.; et al. Detection and quantification of EBV, HHV-6 and CMV DNA in the gastrointestinal tract of HIV-positive patients. Infection 2014, 42, 1033–1037. [Google Scholar] [CrossRef]
- Kayamba, V.; Monze, M.; Asombang, A.W.; Zyambo, K.; Kelly, P. Serological response to Epstein-Barr virus early antigen is associated with gastric cancer and human immunodeficiency virus infection in Zambian adults: A case-control study. Pan Afr. Med. J. 2016, 23, 45. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yamasaki, K.; Tsuji, K.; Shirahama, S. Human T lymphotropic virus type 1 infection and gastric cancer development in Japan. J. Infect. Dis. 2008, 198, 10–15. [Google Scholar] [CrossRef]
- Bozdayi, G.; Dinc, B.; Avcikucuk, H.; Turhan, N.; Altay-Kocak, A.; Ozkan, S.; Ozin, Y.; Bostanci, B. Is human papillomavirus and Helicobacter pylori related in gastric lesions? Clin. Lab. 2019, 65. [Google Scholar] [CrossRef]
- Zeng, Z.M.; Luo, F.F.; Zou, L.X.; He, R.Q.; Pan, D.H.; Chen, X.; Xie, T.T.; Li, Y.Q.; Peng, Z.G.; Chen, G. Human papillomavirus as a potential risk factor for gastric cancer: A meta-analysis of 1917 cases. OncoTargets Ther. 2016, 9, 7105–7114. [Google Scholar] [CrossRef]
- De Souza, C.R.T.; Almeida, M.C.A.; Khayat, A.S.; da Silva, E.L.; Soares, P.C.; Chaves, L.C.; Burbano, R.M.R. Association between Helicobacter pylori, Epstein-Barr virus, human papillomavirus and gastric adenocarcinomas. World J. Gastroenterol. 2018, 24, 4928–4938. [Google Scholar] [CrossRef]
- Snietura, M.; Waniczek, D.; Piglowski, W.; Kopec, A.; Nowakowska-Zajdel, E.; Lorenc, Z.; Muc-Wierzgon, M. Potential role of human papilloma virus in the pathogenesis of gastric cancer. World J. Gastroenterol. 2014, 20, 6632–6637. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palrasu, M.; Zaika, E.; El-Rifai, W.; Que, J.; Zaika, A.I. Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis. Cancers 2021, 13, 1878. https://doi.org/10.3390/cancers13081878
Palrasu M, Zaika E, El-Rifai W, Que J, Zaika AI. Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis. Cancers. 2021; 13(8):1878. https://doi.org/10.3390/cancers13081878
Chicago/Turabian StylePalrasu, Manikandan, Elena Zaika, Wael El-Rifai, Jianwen Que, and Alexander I. Zaika. 2021. "Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis" Cancers 13, no. 8: 1878. https://doi.org/10.3390/cancers13081878
APA StylePalrasu, M., Zaika, E., El-Rifai, W., Que, J., & Zaika, A. I. (2021). Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis. Cancers, 13(8), 1878. https://doi.org/10.3390/cancers13081878